Key Points
rVWF:rFVIII is safe and hemostatically effective in severe VWD patients for a variety of bleeding symptoms.
rVWF induces sustained stabilization of endogenous FVIII, which could obviate the need for rFVIII after the first infusion.
Abstract
This phase 3 trial evaluated the safety and hemostatic efficacy of a recombinant von Willebrand factor (rVWF) for treatment of bleeds in severe von Willebrand disease (VWD). rVWF was initially administered together with recombinant factor VIII (rFVIII) and subsequently alone, as long as hemostatic factor VIII activity (FVIII:C) levels were maintained. Pharmacokinetics (PK) were evaluated in a randomized cross-over design (rVWF vs rVWF:rFVIII at 50 IU VWF:ristocetin cofactor activity [RCo]/kg). Bleed control for all treated bleeds (N = 192 bleeds in 22 subjects) was rated good or excellent (96.9% excellent; 119 of 122 minor, 59 of 61 moderate, and 6 of 7 major bleeds) on a 4-point scale (4 = none to 1 = excellent). A single infusion was effective in 81.8% of bleeds. Treatment success, defined as the number of subjects with a mean efficacy rating of <2.5, was 100%. The PK profile of rVWF was not influenced by rFVIII (mean VWF:RCo terminal half-life: 22.6 hours for rVWF and 22.5 hours for rVWF:rFVIII). FVIII:C levels increased rapidly after rVWF alone, with hemostatic levels achieved within 6 hours and sustained through 72 hours after infusion. Eight adverse effects (AEs; 6 nonserious AEs in 4 subjects and 2 serious AEs [chest discomfort and increased heart rate, without cardiac symptomatology] concurrently in 1 subject) were associated with rVWF. There were no thrombotic events or severe allergic reactions. No VWF or FVIII inhibitors, anti-VWF binding antibodies, or antibodies against host cell proteins were detected. These results show that rVWF was safe and effective in treating bleeds in VWD patients and stabilizes endogenous FVIII:C, which may eliminate the need for rFVIII after the first infusion. This trial was registered at www.clinicaltrials.gov as #NCT01410227.
Introduction
von Willebrand factor (VWF) is a large multimeric plasma glycoprotein with a dual role in hemostasis by mediating platelet adhesion and aggregation and stabilizing procoagulant factor VIII (FVIII).1-3 The 178-kb VWF gene is located on the short arm of chromosome 12 (12p12), encoding a protein with a molecular weight ranging from 500 to 20 000 kDa.4,5 VWF deficiencies and abnormalities caused by mutations of the VWF gene result in von Willebrand disease (VWD), the most common inherited bleeding disorder.6
VWD can be classified into 6 types (1, 2A, 2B, 2M, 2N, and 3) according to distinct genotypic, clinical, and laboratory phenotypic characteristics.7 Clinically, VWD is characterized by mucocutaneous bleeding and less frequently by hemarthrosis and soft tissue hematomas, depending on the severity of the associated FVIII deficiency. VWD patients, especially those with type 3 and some type 2 variants, have an increased risk of experiencing potentially life-threatening bleeding. Effective treatment requires correction of the dual defect of hemostasis associated with VWD: (1) abnormal platelet adhesion and aggregation and (2) defective secondary hemostasis. Desmopressin (1-desamino-8-d-arginine-vasopressin [DDAVP]) is used to transiently correct the FVIII/VWF levels by inducing the release of VWF from endothelial cellular compartments. However, in patients with type 3 VWD and in severe forms of types 1 and 2 VWD, DDAVP may not always be effective8 and may be associated with tachyphylaxis in some DDAVP-responsive patients when closely spaced infusions are administered, such as during major surgery. Moreover, DDAVP treatment is contraindicated in VWD patients with cardiovascular disease, and fluid restriction is necessary to avoid hyponatremia and the risk of seizures, especially in young children.
Plasma-derived VWF/FVIII factor concentrates, initially developed for the treatment of hemophilia A, contain large amounts of VWF and are the treatment of choice for DDAVP-unresponsive or intolerant patients.9-13 Several high purity plasma-derived VWF/FVIII products are available on the market that are effective in the treatment and prevention of bleeds in VWF patients. However, plasma-derived products are limited by plasma donor availability, and the theoretical risk of pathogen transmission cannot be excluded despite viral screening and attenuation measures that have been instituted to minimize such concerns.14,15 Plasma-derived VWF/FVIII products also vary in composition depending on the source plasma and the manufacturing process.16-19 This variability could lead to excessive FVIII levels in VWD patients, with inherent risk of venous thromboembolism. Furthermore, these concentrates show variable deficiency of ultralarge multimers (ULM) of VWF due to proteolysis. In addition, plasma-derived VWF/FVIII products contain extraneous plasma proteins that may engender allergic responses, which are sometimes severe and may limit their use.20
Human VWF produced by recombinant technology could offer an important alternative for treatment of VWD by overcoming limitations associated with plasma-derived VWF concentrates.21 A human recombinant VWF (rVWF), vonicog alfa, which is synthesized by a genetically engineered CHO cell line coexpressing the VWF and the FVIII genes, has been developed to advance the standard of care for patients with VWD and make the treatment independent from plasma supply.22
Vonicog alfa is the first rVWF manufactured in the absence of animal or other human plasma proteins, rendering theoretical the risk of transmission of adventitious agents and other blood-borne pathogens such as prions. In contrast to plasma-derived coagulation factors, no therapeutic protein produced by a recombinant cell line has ever resulted in virus transmission, which is primarily due to the ability to control the recombinant cell line manufacturing environment.
Ultralarge and high-molecular-weight multimers (HMWMs) of VWF are essential for effective platelet plug formation.18 Plasma-derived VWF concentrates variably lack ULM due to in vivo exposure to plasma ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif, member 13) and proteolytic cleavage by granulocyte elastases.16-19 In contrast to VWF concentrates derived from plasma, rVWF has no exposure to the human protease ADAMTS13 during the production process and therefore contains intact HMWM and ULM.22 The presence of ULM and the higher purity also contribute to a greater specific activity of rVWF measured as VWF:ristocetin cofactor activity (VWF:RCo) relative to VWF:antigen (VWF:Ag).
The pharmacokinetics of rVWF:rFVIII in patients with type 3 VWF have been evaluated in a phase 1 clinical trial, showing a tendency for a longer terminal half-life of rVWF VWF:RCo compared with a plasma-derived VWF:FVIII concentrate and stabilization of endogenous FVIII activity (FVIII:C).23 ULMs present in rVWF were rapidly proteolyzed by endogenous ADAMTS13, and no thrombotic events were observed. The current trial was conducted to further assess the pharmacokinetics (PK) of rVWF with and without rFVIII infusion and evaluate the safety and hemostatic efficacy of rVWF for the treatment of bleeds in patients with severe type 1, 2, or 3 VWD when administered initially together with rFVIII and subsequently alone provided adequate levels of FVIII:C had been achieved.
Patients, materials, and methods
Subject population
Male and female patients with type 3 or severe type 1 and type 2A (VWF:RCo < 20 IU/dL), type 2B (as diagnosed by genotype), type 2N (FVIII:C < 10 IU/dL and historically documented genetics), type 2M, or type 3 (VWF:Ag ≤ 3 IU/dL), aged 18 to 65 years, and treated for ≥1 bleed with a VWF concentrate within 12 months prior to enrollment were eligible for inclusion. Patients with a history of VWF or FVIII inhibitors, immunologic disorders, or thromboembolic events were excluded.
Trial design and conduct
This phase 3 prospective clinical trial was designed to assess the PK, safety, and hemostatic efficacy of rVWF in the treatment of bleeding episodes in adults with severe VWD. All subjects provided written informed consent prior to any screening procedures. Approval to conduct the trial was granted by the institutional review boards or independent ethics committees of all participating sites. The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice.
Subjects
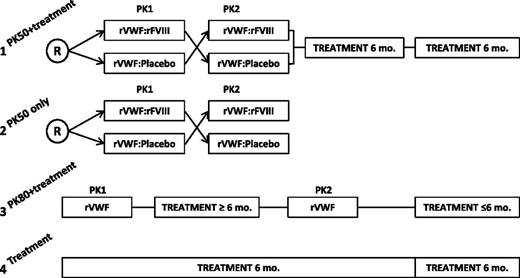
Subjects were enrolled into 1 of 4 arms to receive rVWF for PK assessment and/or 6 months of on-demand bleed treatment (Figure 1). Enrollment was conducted across all 4 treatment arms in parallel. Subjects were allocated to a treatment arm at the investigator’s discretion as long as enrollment was still open in the respective arm (it was planned to include ≥7 subjects with type 3 VWD in each PK 50 arm, ∼12 subjects with severe VWD in the PK80 + bleed treatment arm [of which ≥6 were to have type 3 VWD], and ∼7 subjects with severe VWD of any type in the arm for bleed treatment only).
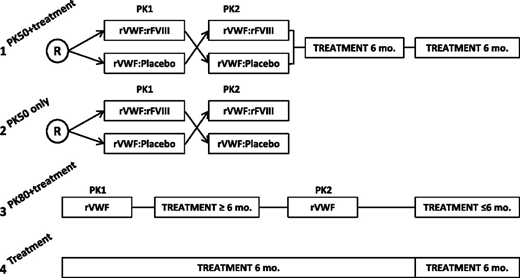
Study design. Subjects were enrolled into 1 of 4 treatment arms: (1) PK50 + treatment: crossover PK at a dose of 50 IU/kg VWF:RCo followed by 12 months of on-demand bleed treatment; (2) PK50 only: crossover PK at a dose of 50 IU/kg VWF:RCo; (3) PK80 + treatment: PK at a dose of 80 IU/kg VWF:RCo repeated after 6 months, followed by 6 months of on-demand bleed treatment; and (4) treatment only: 12 months of on-demand treatment. R, randomized.
Study design. Subjects were enrolled into 1 of 4 treatment arms: (1) PK50 + treatment: crossover PK at a dose of 50 IU/kg VWF:RCo followed by 12 months of on-demand bleed treatment; (2) PK50 only: crossover PK at a dose of 50 IU/kg VWF:RCo; (3) PK80 + treatment: PK at a dose of 80 IU/kg VWF:RCo repeated after 6 months, followed by 6 months of on-demand bleed treatment; and (4) treatment only: 12 months of on-demand treatment. R, randomized.
PK
PK of rVWF was assessed at 50 IU/kg VWF:RCo and 80 IU/kg VWF:RCo. The doses of 50 and 80 U/kg VWF:RCo were selected for PK evaluation as these doses represent the lower and upper ends of the dose ranges described previously for pdVWF concentrate replacement therapy in patients with severe VWD.11,24,25 PK of rVWF (50 IU/kg VWF:RCo) administered together with rFVIII (ADVATE; Baxalta) or placebo (saline) was assessed in a randomized, single-blinded crossover design. Subjects enrolled in the PK50 arms were randomized 1:1 according to a central randomization list to receive either treatment of their initial infusion followed by the alternative treatment after a washout of 18 ± 10 days. PK of rVWF alone was evaluated at a dose of 80 IU/kg body weight VWF:RCo and repeated after 6 months, with an overall study participation of 12 months. PK analyses were performed on plasma samples obtained 1 hour before infusion and at 11 time points up to 96 hours after infusion. The presence of VWF multimers was determined semiquantitatively prior to infusion, at 6 and 12 hours after infusion.
Treatment of bleeds
Bleeding episodes were to be treated with an initial infusion of 40 to 60 IU/kg VWF:RCo rVWF for minor to moderate bleeds such as epistaxis, oral bleeding, or menorrhagia and up to 80 IU/kg VWF:RCo for major bleeds, which included bleeds such as severe or refractory epistaxis or menorrhagia, gastrointestinal bleeding, central nervous system trauma, hemarthrosis, or posttraumatic hemorrhage. To ensure an immediate hemostatic level of FVIII:C, the initial dose of rVWF was to be administered together with rFVIII at a ratio of 1.3:1 ± 0.2 VWF:RCo/FVIII:C and subsequently without rFVIII as long as therapeutic FVIII:C levels were maintained.
In major bleeding episodes, subsequent doses were to be administered every 8 to 12 hours for 3 days to maintain the trough level of VWF:RCo > 50 IU/dL and then as deemed necessary by the investigator for up to 7 days. If the VWF:RCo level was >150 or 200 IU/dL, a planned treatment was to be delayed by ≥12 or 24 hours, respectively. The use of antifibrinolytic agents during the study was at the discretion of the physician in charge.
Treatment of each bleed was rated according to a predefined 4-point scale (Table 1) based on the actual number of infusions administered to control the bleed vs the treating physician’s estimate of the number of infusions required.24 The primary end point was the extent of control of the bleeding episodes, which was defined as the number of subjects with a mean hemostatic efficacy rating score of <2.5. Secondary efficacy outcomes included the number of treated bleeding episodes with a hemostatic efficacy rating of excellent or good and the number of infusions and units of rVWF/rFVIII or rVWF required to control a bleed.
Safety
Safety was evaluated by clinical assessments of adverse events, hematology panels, coagulation panels (including soluble P-selectin and D-dimers), serum chemistry, urinalysis, viral serology, and immunologic assessments. Nijmegen-modified Bethesda assays were used to determine neutralizing antibodies against VWF:RCo, VWF:collagen binding activity (VWF:CB), FVIII binding activity (VWF:FVIIIB), and FVIII.26 The presence of total binding anti-VWF antibodies, antihost cell (CHO) proteins, murine immunoglobulin (Ig)G, and anti-human Furin (total Ig) was determined using a proprietary enzyme-linked immunosorbent assay employing polyclonal anti-human Ig antibodies.27
Test products
rVWF is a purified glycoprotein synthesized in a genetically engineered CHO cell line.28 The manufacturing process of rVWF is based on a cell culture system that expresses both rFVIII (ADVATE) and rVWF; no exogenously added raw materials of human or animal origin are used in the cell culture and purification processes.28 Physiologic saline was used as a placebo for the crossover PK assessment.
Hemostatic and multimer assay methodology
A modified VWF:RCo assay based on the Dade Behring coagulation analyzer (BCS) method was used to determine VWF:RCo activity.23,29 VWF:Ag and VWF:CB levels were quantitated using standard sandwich enzyme-linked immunosorbent assays using polyclonal rabbit anti-human VWF antibodies (DAKO) or collagen type III (Biozol), respectively. FVIII activity (FVIII:C) was measured with a 1-stage clotting assay (Siemens Coagulation Analyzer CA-7000, Sysmex; APPT Actin FS, Siemens). VWF multimer patterns were assessed using sodium dodecyl sulfate-agarose gel electrophoresis and using western blot with luminescence video imaging.
Statistical methods
For the primary efficacy analysis of the rate of subjects with treatment success, which was defined as a mean efficacy rating score <2.5 (Table 1), the null hypothesis of a rate of ≤0.65 was tested on a 5% 1-sided level implicitly, by computing the Clopper-Pearson interval of proportions at the 90% confidence level. Similarly, for the secondary efficacy analysis, the rate of all treated bleeds with excellent or good treatment outcome was tested against the null hypothesis of ≤0.60 on a 5% 1-sided level implicitly, by computing the Clopper-Pearson 90% confidence interval (CI) of the proportions.
PK parameters of VWF:RCo, VWF:Ag, and VWF:CB including area under the plasma concentration/time curve from time 0 to infinity per dose (AUC0-∞/Dose), elimination phase half-life (T1/2); mean residence time (MRT), clearance (CL), incremental recovery (IR), and volume of distribution at steady state (Vss) were summarized by mean (standard deviation) and 2-sided 95% CIs for the mean, median, and interquartile ranges (IQR). The AUC0-∞/Dose was also calculated for FVIII activity (FVIII:C). For crossover PK analysis, AUC0-∞/Dose of VWF:RCo of rVWF alone and rVWF:rFVIII were compared by calculating the 90% 2-sided CI for the difference of the mean natural logarithms of AUC0-∞/Dose between rVWF alone and rVWF:rFVIII. The error variance used to calculate these CIs was obtained from an analysis of variance model that consisted of fixed effect terms that model the subject effect, the sequence effect, the period effect, and the drug effect. Safety (adverse events) was evaluated descriptively.
Results
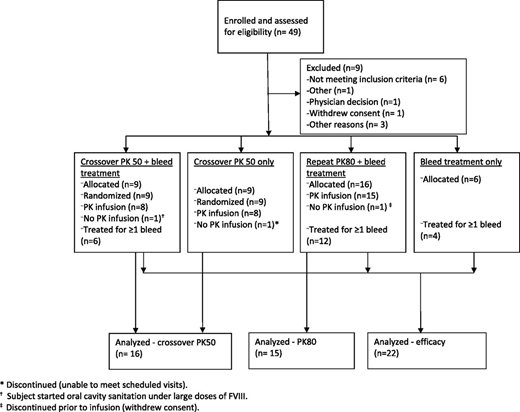
Patient disposition and demographics
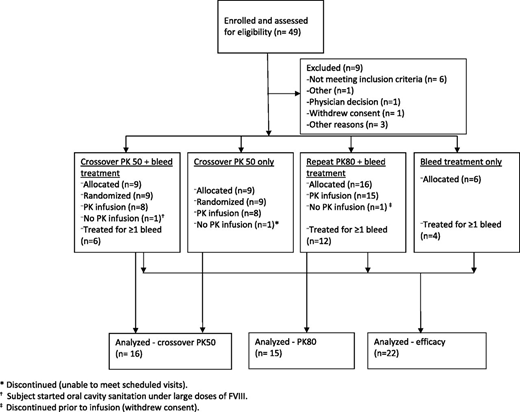
Hemostatic efficacy
Twenty-two subjects (17 type 3, 4 type 2A, and 1 type 2N VWD) experienced ≥1 bleeding episode that was treated with rVWF, and 9 of 31 subjects allocated for bleed treatment did not experience a bleed during the study. A total of 192 bleeding episodes (122 minor, 61 moderate, 7 major/severe, and 2 unknown severity) treated with rVWF were assessed for hemostatic efficacy according to a predefined 4-point scale (Table 1). Most bleeding episodes (several of which occurred in multiple locations) were mucosal (epistaxis, menorrhagia, mouth/oral cavity; n = 106, 55.2%), followed by 59 (30.7%) joint bleeds, 37 (19.3%) bleeding episodes in other sites including soft tissue and superficial bleeds, and 6 (3.1%) gastrointestinal bleeds. An antifibrinolytic agent, such as tranexamic acid, was administered during 45 of 192 (23.4%) treated bleeding episodes in 7 of 22 (31.8%) subjects. All bleeds were treated successfully, with an overall treatment success rate on a subject level of 100% (n = 22; Clopper-Pearson exact 90% CI: 87.3-100.0).
All 192 bleed treatments (100%; Clopper-Pearson 95% CI: 98.1-100.0%) were rated as either excellent (96.9%, 186 of 192) or good (3.1%, 6 of 192; Table 3). One infusion was adequate to treat 157 bleeds (81.8%; median, 1; range, 1-4 infusions). For 10 of these treatments in 3 subjects, the first infusion of rVWF was inadvertently administered without rFVIII, all with excellent efficacy. Four infusions, which was the maximum number of infusions administered during the trial, were required for 1 mucosal bleed (in the genital tract and oral cavity simultaneously). The median dose of rVWF and rFVIII administered per bleed was 46.5 IU/kg VWF:RCo and 33.6 IU/kg, respectively.
Excellent treatment efficacy ratings were given to 97.5% of minor bleeds (119 of 122) and 96.7% of moderate bleeds (59 of 61). Treatment of 6 of 7 major/severe bleeds (85.7%, 3 joint bleeds and 3 gastrointestinal tract bleeds) was rated as excellent, and treatment of 1 major bleed in the nasopharyngeal tract was rated good. A median of 2 infusions (range, 1-3) was required to control major bleeds. As expected, the amount of rVWF administered per bleed was generally higher for bleeds of greater severity; however, 1 major/severe gastrointestinal tract bleed was controlled after a single infusion of rVWF:rFVIII at a dose of 57.5 IU/kg VWF:RCo and 41.5 IU/kg rFVIII.
In a subanalysis of mucosal bleeds (menorrhagia, nasopharyngeal, or oral cavity; N = 100), efficacy for 98% of bleed treatments was rated excellent (Table 4). The cause of bleed appeared not to have an impact on efficacy, with “excellent” hemostatic efficacy ratings in 97.5% (160 of 165) of spontaneous and 100% (26 of 26) of traumatic bleeds.
PK
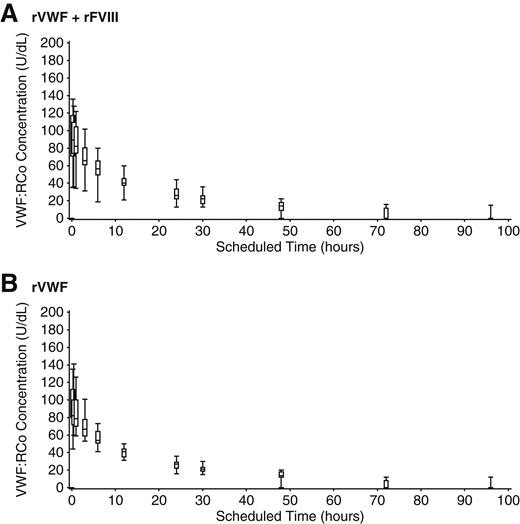
Plasma concentrations of VWF:RCo increased rapidly in VWD patients after a single infusion of rVWF at a dose of 50IU/kg VWF:RCo, with a consistent PK profile when administered alone or together with rFVIII (Table 5; Figure 3). The ratio between the geometric mean AUC0-∞/Dose of VWF:RCo, estimated within the analysis of variance model, showed close agreement for rVWF administered alone vs with rFVIII (1.02; 90% CI, 0.93-1.11). The mean VWF:RCo CL was 0.03 dL/kg/h, and IR was 1.9 [(U/dL)/(U VWF:RCo/kg)] with or without rFVIII; mean (95% CI) terminal T1/2 was 22.5 hours (17.4-27.6 hours) for rVWF:rFVIII and 22.6 hours (19.5-25.7 hours) for rVWF alone. All PK parameters for VWF:Ag and VWF:CB were consistent with VWF:RCo, with no influence of rFVIII on rVWF PK parameters.
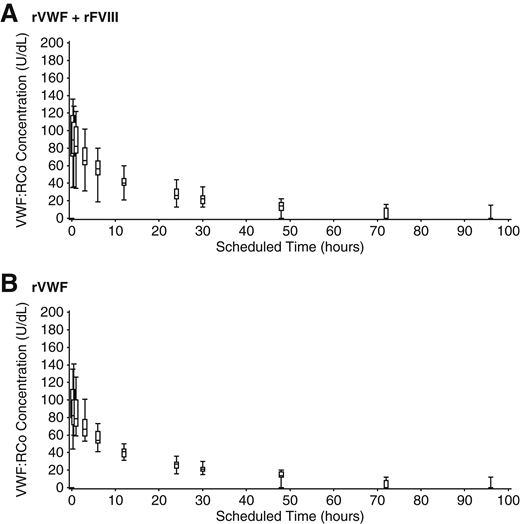
VWF:RCo of rVWF. Median, IQR, and range of VWF:RCo after infusion of 50 IU VWF:RCo/kg rVWF alone and together with rFVIII.
VWF:RCo of rVWF. Median, IQR, and range of VWF:RCo after infusion of 50 IU VWF:RCo/kg rVWF alone and together with rFVIII.
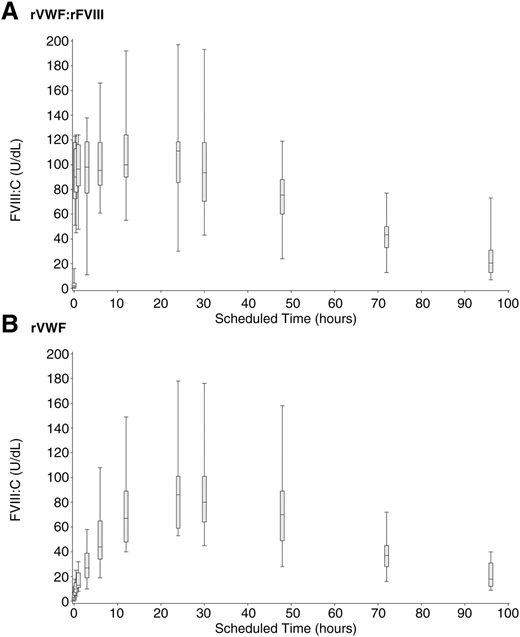
rVWF alone (ie, without rFVIII) induced a substantial stabilization of endogenous FVIII:C, as shown by the progression of median FVIII:C over 96 hours (Figure 4), rising above 40% by 6 hours after infusion and increasing monotonically to a peak of 86.0 U/dL at 24 hours after infusion. Immediate hemostatic FVIII:C levels after infusion with rVWF:rFVIII were also observed, as expected, followed by sustained stabilization of FVIII:C.
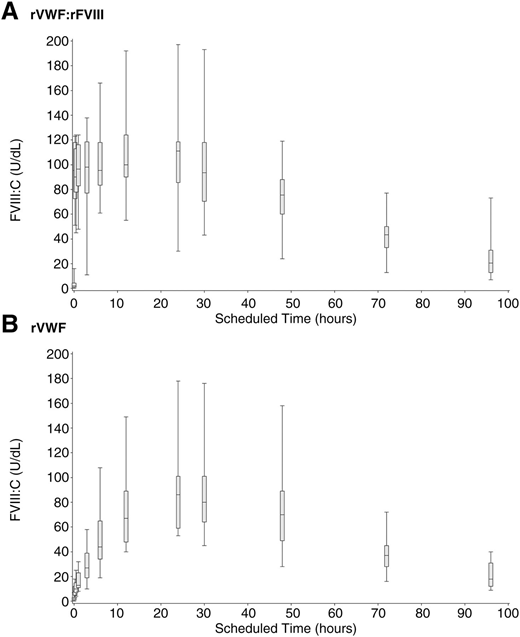
FVIII:C PK (median, IQR and range) in severe VWD patients after rVWF infusion. Median, IQR, and range of FVIII:C (U/dL) over 96 hours in severe VWD patients (N = 16) in a nonbleeding state. (A) rVWF (50 IU VWF:RCo/kg) administered together with rFVIII (38.5 IU/kg). (B) rVWF alone (50 IU VWF:RCo/kg).
FVIII:C PK (median, IQR and range) in severe VWD patients after rVWF infusion. Median, IQR, and range of FVIII:C (U/dL) over 96 hours in severe VWD patients (N = 16) in a nonbleeding state. (A) rVWF (50 IU VWF:RCo/kg) administered together with rFVIII (38.5 IU/kg). (B) rVWF alone (50 IU VWF:RCo/kg).
Multimer analysis
The presence and in vivo degradation of ULM was established by an observed increase in the median percentage from zero before infusion to 30% 15 minutes after infusion of rVWF, followed by a substantial decline between 12 and 24 hours. Near-baseline levels were observed in almost all subjects by 96 hours after infusion (Figure 5).

VWF multimer activity. (A) Proportion of large VWF multimers. (B) VWF multimers and degradation products pre- and postinfusion of rVWF in a subject with type 3 VWD.
VWF multimer activity. (A) Proportion of large VWF multimers. (B) VWF multimers and degradation products pre- and postinfusion of rVWF in a subject with type 3 VWD.
Safety
Eight of a total of 125 adverse events (AEs; 6.4%) that were observed during the trial were considered to have a causal relationship to rVWF (Table 6). Six of these AEs in 4 subjects were not serious. One subject experienced mild infusion site paresthesia, moderate dysgeusia, and moderate tachycardia, 1 subject showed a mild ECG T-wave inversion, 1 subject experienced mild generalized pruritus, and 1 subject had a mild hot flush. One subject experienced 2 simultaneous serious AEs (chest discomfort and increased heart rate). These symptoms improved after 10 minutes of oxygen treatment, the subject fully recovered within 3 hours, and no clinical cardiac symptomatology was observed. This AE was assessed as serious due to hospitalization for the purpose of observation. The subject had a history of allergic responses to cryoprecipitate and a pdVWF concentrate.
No subject developed anti-VWF neutralizing or binding antibodies, FVIII neutralizing antibodies, or antibodies against rFurin, CHO host cell proteins, or murine IgG. There were no clinical signs of a thrombotic event in any subject, and no clinically significant abnormal d-dimer values after treatment were observed. No clinical laboratory values (hematology, serology, clinical chemistry, or urinalysis) were reported as AEs.
Discussion
As the first rVWF formulation, vonicog alfa offers minimization of any potential risk for transmission of adventitious agents and other blood-borne pathogens. In an initial phase 1 trial in patients with severe VWD,23 rVWF was shown to be safe with a similar PK profile, a tendency toward a longer VWF:RCo terminal half-life, and sustained stabilization of FVIII:C compared with a plasma-derived VWF:FVIII. In the present phase 3 clinical trial, we further investigated the safety of rVWF and evaluated the PK and efficacy using different dosing regimens with and without rFVIII for the treatment of bleeding episodes in subjects with severe type 1, 2, or 3 VWD.
The PK profile of rVWF in subjects with VWD was evaluated in a nonbleeding state, by assessment of VWF:RCo (a surrogate for the platelet-dependent function of rVWF), VWF:Ag; VWF:CB (a surrogate for adhesion of rVWF to subendothelial collagen); and FVIII activity. There was no apparent difference in VWF PK between the 2 doses of rVWF (50 vs 80 IU/kg VWF:RCo) or between rVWF and rVWF:rFVIII. The observed mean VWF:RCo T1/2 (22.6 hours for rVWF alone and 22.5 hours for rVWF administered together with rFVIII) is considerably longer than that of pdVWF products, which has been reported to range between 12.8 and 15.8 hours.30 The observed IR of 1.9 U/dL VWF: RCo/kg is consistent with that reported in the previously published phase 1 trial23 and similar to the IR reported for commercial pdVWF/pdFVIII concentrates.13,24,31 Compared with pdVWF/FVIII concentrates, the pharmacokinetic profile for rVWF shows a tendency toward a longer VWF:RCo T1/2. VWF:Ag and VWF:CB parameters were consistent with VWF:RCo and, as predicted, no influence of rFVIII was observed. Stabilization of endogenous FVIII was very rapid after administration of rVWF alone, reaching hemostatic levels (>40 IU/dL) within 6 hours (median) and sustained on a high level >70 IU/dL through 48 hours followed by gradual decline in FVIII:C over the 96-hour observation period, but with continued hemostatic levels (>40 IU/dL) up to 72 hours after infusion.
The lower limit of the 90% CI of the rate of subjects with successful treatment (100%; 90% CI: 87.3-100.0%) was well above the predefined limit of 65%. A hemostatic efficacy rating of excellent or good was achieved for 100% of treated bleeding events (Clopper-Pearson exact 90% CI: 87.3-100.0), which again is significantly higher than the predefined 60% limit (a conservative estimate determined for this prospective clinical trial) and is consistent with the reported response rate of 97% with commercial pdVWF:pdFVIII.32 There were no hemostatic ratings of poor/none during the trial, which compares favorably with results previously reported in a prospective clinical trial of a pdVWF/FVIII concentrate. In that study, for 10 of 208 bleeds (ranging from mild to major at various sites of bleeding) in VWD, type 3 patients received a poor or none efficacy score using a slightly different rating scale.13
Most bleeding events resolved after 1 infusion of rVWF:rFVIII or rVWF. Only 1 bleed (in the genital tract and oral cavity concomitantly, in a type 3 VWD patient) required 4 infusions, which was the maximum number of infusions administered to treat a bleed during the trial. Furthermore, although rFVIII was to be administered together with rVWF for the initial infusion to ensure an initial rise to hemostatic levels of rFVIII:C, in the 10 treatments where subjects were inadvertently adminstered rVWF alone for the initial infusion, efficacy was rated as excellent. A potential limitation of the present study is the low number of subjects with severe non-type 3 VWD (type 1 [N = 2] and type 2 [N = 6]). However, as documented previous bleed treatment was prerequisite for inclusion in the study and most bleeds occur in subjects with type 3 VWD, the majority of subjects enrolled either had type 3 VWD or did not present with a bleed suitable for rVWF treatment during the study. The presence of ULM in rVWF may result in improved platelet and collagen binding and therefore provide more effective treatment outcomes. It has been well established that the size of the highly polymerized VWF multimers directly correlates with their hemostatic activity.32-34 More recently, in vitro experiments showed that, although highly multimerized rVWF (which have a higher number of collagen and platelet binding sites per molecule) effectively promoted platelet binding and thus have a stronger hemostatic potential, VWF multimers of lower molecular weight showed decreased platelet adhesion properties.35 In comparison, plasma-derived VWF deficient in ULM was less effective in mediating platelet adhesion to collagen, as reflected by delayed platelet binding kinetics. This advantage may predict efficacy in the very challenging situation of bleeding from gastrointestinal angiodysplasia related to VWD, which is historically difficult to manage with VWF replacement therapy.36
ADAMTS13 degrades the ULM of VWF that is normally synthesized and released by vascular endothelial cells in healthy individuals. In the present trial, the initial increase in median percentage of ULM to 30% after 15 minutes (the earliest time point measured) after rVWF:rFVIII or rVWF treatment was followed by a substantial decline (between 12 and 24 hours) and ULM reduction to baseline levels by 96 hours, which supports the notion that ADAMTS13 effectively functions to cleave the infused ULM in rVWF. This outcome confirms that the susceptibility of rVWF to ADAMTS13 cleavage allows adequate physiologic processing of rVWF on administration and results in a molecule with low thrombogenicity.37 However, the enhanced rVWF and FVIII PK profiles suggest that the highly multimerized rVWF molecules may be eliminated more slowly from the circulation and may be more effective in stabilizing FVIII, without resulting in excessively high FVIII:C levels.
rVWF:rFVIII was well tolerated in this trial. AEs were generally mild and transient and occurred at rates comparable to those observed previously with rVWF-rFVIII or with pdVWF-pdFVIII concentrates.12,23,25 The rate of events considered to be related to treatment was low (6.4%), comprising 6 nonserious and 2 serious AEs. The 2 simultaneous serious events (chest discomfort and increased heart rate), which occurred in a single patient with a history of allergic reactions to cryoprecipitate and a pdVWF concentrate, are known potential risks of factor replacement therapies. No anaphylactic or severe allergic reactions were reported during the trial. As the ULM were shown to be highly effective in hemostasis, thrombogenic risk was carefully assessed in the clinical development of this product. No thrombotic events observed following treatment with rVWF:rFVIII, either during the present trial or in the previous phase 1 trial.23 In rare cases, alloantibody development against VWF has been observed in patients with type 3 VWD.38 In the present study, none of the patients developed anti-VWF binding or neutralizing antibodies to VWF. Furthermore, as the initial and repeated rVWF PK results are in close agreement in this cohort of subjects with VWD, it can be concluded that there is no evidence of any inhibitory allo-antibodies that would influence the T1/2 or other PK parameters of rVWF.
In summary, results of this clinical trial provide evidence that rVWF:rFVIII is safe and hemostatically effective in severe VWD patients in a variety of clinical bleeding presentations. The finding that a single infusion was required for the majority of bleeds may be a reflection of the high ULM content of VWF and the long VWF:RCo T1/2 relative to plasma-derived VWF/FVIII concentrates, which may predict greater efficacy than the plasma-derived concentrates in high risk bleeding situations such as surgery or gastrointestinal bleeding. Finally, the sustained stabilization of endogenous FVIII has the potential to obviate the need for rFVIII after the first infusion when additional infusions are required.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Amy Shapiro, Patrick Fogarty, Tadeusz Robak, Andrzej Mital, Tatiana Andreeva, Farida Perina, Elena Vinogradove, Toshko Lissitchov, Cédric Hermans, Ingrid Pabinger-Fasching, Inge Scharrer, Thynn Thynn Yee, John Pasi, Maria Mazzucconi, Francesco Demartis, Victor Jiménez Yuste, Anna Olsson, Charles Hay, Tadashi Matsushita, Katsuyuki Fukutake, Vijay Ramanan, Ross Baker, Simon J. McRae, and Hideji Hanabusa for providing patients and data, Reinhard Schneppenheim and Florian Oyen for VWF gene mutation analysis, Ulrich Budde for VWF multimer analyses, Barbara Valenta-Singer, Judit Koranyi, Siddhesh Darne, Jeanette Fait, Lorraine Ampaw, Bettina Ploder, Wolfgang Draxler, Bernadette Keohane, Paz Carrasco, Britany Wilcher, Vanessa Nguyen, Nadia Agopyan, Frank Horling, Herbert Gritsch, and Claudia Apostol for operational support.
This study was supported by research funding from Baxalta.
Authorship
Contribution: B.E. and I.P. were responsible for conception and design; J.C.G., G.C., J.W., P.K., M.R., and F.W.G.L. provided patients and performed data collection; O.O.-S., M.C., S.F., B.G.P., I.P., and B.E. performed data analysis and interpretation; M.C. and B.G.P. wrote the manuscript; and all authors critically revised and approved the final manuscript.
Conflict-of-interest disclosure: J.C.G. has served on advisory boards for Baxalta, Bayer, and CSL Behring. G.C. has served on advisory boards for Novo Nordisk, Bayer, Baxalta, CSL Behring, and Kedrion. J.W. has acted as a consultant and participated in expert groups for Bayer, Baxalta, and NovoNordisk and given lectures for Bayer, Baxalta, CSL Behring, GSK, NovoNordisk, Octapharma, and Pfizer. P.K. has received honoraria for serving on an advisory board for CSL Behring. M.R. has received research funding from Biogen, Bayer, CSL, Novo Nordisk, OPKO, Pfizer, SPARKS, and Baxalta; has served on advisory boards for Biogen, Baxalta, and CSL; and has acted as a consultant for Biogen and Tacere. F.W.G.L. has served on advisory boards for, and received unrestricted research grants from, CSL Behring and Baxalta. O.O.-S., M.C., S.F., B.G.P., I.P., and B.E are employees of Baxalta.
Correspondence: Bruce Ewenstein, Baxalta US Inc, 650 East Kendall St, Cambridge, MA 02142; e-mail: bruce.ewenstein@baxalta.com.