

Key Points
Multi–TAA-Ts can be generated from patients who have failed prior chemo-immunotherapies for HL.
TAA-Ts are safe when given alone and persist long term in responders, and nivolumab increases persistence of TAA-Ts in vivo.

Abstract
Hodgkin lymphoma (HL) Reed Sternberg cells express tumor-associated antigens (TAA) that are potential targets for cellular therapies. We recently demonstrated that TAA-specific T cells (TAA-Ts) targeting WT1, PRAME, and Survivin were safe and associated with prolonged time to progression in solid tumors. Hence, we evaluated whether TAA-Ts when given alone or with nivolumab were safe and could elicit antitumor effects in vivo in patients with relapsed/refractory (r/r) HL. Ten patients were infused with TAA-Ts (8 autologous and 2 allogeneic) for active HL (n = 8) or as adjuvant therapy after hematopoietic stem cell transplant (n = 2). Six patients received nivolumab priming before TAA-Ts and continued until disease progression or unacceptable toxicity. All 10 products recognized 1 or more TAAs and were polyfunctional. Patients were monitored for safety for 6 weeks after the TAA-Ts and for response until disease progression. The infusions were safe with no clear dose-limiting toxicities. Patients receiving TAA-Ts as adjuvant therapy remain in continued remission at 3+ years. Of the 8 patients with active disease, 1 patient had a complete response and 7 had stable disease at 3 months, 3 of whom remain with stable disease at 1 year. Antigen spreading and long-term persistence of TAA-Ts in vivo were observed in responding patients. Nivolumab priming impacted TAA-T recognition and persistence. In conclusion, treatment of patients with r/r HL with TAA-Ts alone or in combination with nivolumab was safe and produced promising results. This trial was registered at www.clinicaltrials.gov as #NCT022039303 and #NCT03843294.
Introduction
Adoptive cellular therapy is promising for Hodgkin lymphoma (HL) as was previously demonstrated by the safety and efficacy of T-cell therapies targeting Epstein-Barr virus (EBV)-positive HL.1 However, only 30% to 40% of HLs express EBV-encoded antigens, precluding the broader application of this cell therapy in HL.1-3 Furthermore, the success of CD19 chimeric antigen receptor (CAR)-T in B-cell hematologic malignancies is not completely recapitulated in HL by the CD30-directed CARs.4,5 Despite lymphodepleting chemotherapy, the 1-year progression-free survival (PFS) for patients with active disease at the time of CAR-T infusion was only 36%.4-8 The unique inhibitory microenvironment of HL impairs the survival, activation, proliferation, and function of the infused T cells, and antigen modulation/loss is a well-known mechanism of resistance to CAR-T therapies.9 Similarly, checkpoint inhibitors (CPIs) targeting programmed-cell death protein 1 (PD-1) have been approved for relapsed HL; however; 30% to 40% of patients do not respond to CPIs with median PFS as little as 10 months.10-14 Therefore, development of strategies that can improve antigen recognition and simultaneously enhance T-cell function and persistence of tumor-specific T cells in vivo could enhance the potency of adoptive T-cell therapies in relapse/refractory (r/r) HL.
Targeting multiple non-EBV tumor-associated antigens (TAAs) presented through major histocompatibility complex to the native T-cell receptor offers advantage over single surface antigen targeting (eg, CAR-T) by providing clonal heterogeneity and reduced risk of antigen escape. In a first-in-human clinical trial, we recently showed that TAA-T cells targeting Wilms tumor gene-1 (WT1), Preferentially Expressed Antigen in Melanoma (PRAME), and Survivin were safe and induced disease stabilization in a variety of solid tumors.15 We hypothesized that TAA-Ts specific to these same antigens could be generated for patients with HL and that the addition of CPIs to the TAA-T infusions could provide a synergistic approach to optimize the rate, depth, and duration of clinical responses.
Here we present the safety of autologous and allogeneic TAA-T targeting of WT1, PRAME, and Survivin when given alone or in combination with the PD-1 inhibitor nivolumab to patients with r/r HL or as adjuvant therapy to patients considered high risk of relapse after autologous (ASCT) or allogeneic (HSCT) stem cell transplant. We characterized the TAA-T products for specificity and function, tracked the in vivo persistence of TAA-Ts over time, and assessed the impact of nivolumab on the function and persistence of the infused TAA-T cells.
Methods
Patients and treatment protocol
Patients with r/r HL were enrolled in 2 studies, Multi-institutional Prospective Research of Expanded Multi-antigen Specifically Oriented Lymphocytes for the Treatment of VEry High Risk Hematopoietic Malignancies (RESOLVE) (NCT022039303) and Phase I Study Utilizing Tumor Associated Antigen Specific T cells (TAA-T) with PD1 Inhibitor Nivolumab for Relapsed/Refractory Lymphoma (SUSTAIN)(NCT03843294), approved by the US Food and Drug Administration (IND 16135) and the Children’s National Hospital, The Johns Hopkins University, and University of Utah institutional review boards. Patients were eligible for TAA-T infusion if they had measurable disease (active arm) or were in remission after allogeneic or autologous HSCT but considered to have high risk of relapse after transplant (adjuvant arm). Details on the study treatment are provided in the supplemental Material and supplemental Appendix.
All patients were evaluated for toxicity for 6 weeks after the first TAA-T infusion or 28 days after additional infusions, which was considered the safety monitoring period using National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 (see supplemental Appendix). Patients were not allowed to receive any concurrent therapy during the safety monitoring period. For patients receiving nivolumab, immune-related adverse events were captured from the time of starting nivolumab until last follow-up.
Response was assessed according to the Lugano criteria.16 Outcome of patients at their last follow-up at the time of data cutoff (27 April 2021) was used for overall response and survival assessment.
Generation of TAA-Ts
Functional characterization of TAA-T cells
Flow cytometry for surface and intracellular markers and interferon γ (IFNγ) enzyme-linked Immunospot (ELISPOT) assay was performed as previously described and as described in the supplemental Methods.15,19
Tracking of TAA-Ts
Peripheral blood (PB) from recipients was drawn before starting nivolumab (if applicable), before TAA-T infusion, and at regular time points after infusion. Isolated PB mononuclear cells (PBMCs) were expanded ex vivo for 2 weeks after coculture with autologous dendritic cells and TAA-Ts characterized as above.
T-cell receptor β-chain immunosequencing
Immunosequencing of the CDR3 regions of human T-cell receptor (TCR) β chains was performed using the immunoSEQ assay (Adaptive Biotechnologies, Seattle, WA) as previously described.20-23
Statistical analysis
Descriptive statistics were used to summarize clinical characteristics, TAA-T products, exhaustion markers, and reactivity of TAA-T cells by anti-IFNγ assay using mean, standard deviation, standard error of mean (SEM), median, and range. Differences between antigen responses were calculated using univariate analysis of variance using GraphPad Prism. TCR sequences were analyzed using the Ilumina platform and are described in detail in the supplemental Material.
Results
Patient characteristics
Blood for TAA-T manufacture was procured from 14 patients (9 men and 5 women) with r/r HL from May 2015 to June 2020. In total, 12 products were manufactured from 10 patients receiving TAA-T infusions with or without nivolumab (Table 1). Two of the 10 patients received TAA-Ts generated from matched heathy donors (allogeneic), and the remaining 8 received patient-derived TAA-T infusions (autologous). Of the 4 enrolled patients who did not receive TAA-Ts, 1 patient was diagnosed with myelodysplastic syndrome before starting protocol therapy and hence came off protocol, and TAA-T manufacture failed to meet the dose needed for the other 3 patients. Three of the 10 infused patients (all with active disease at time of procurement) required repeat procurement before their first infusion because of insufficient cell dose, and 1 of these 3 patients received only 1 TAA-T infusion instead of the planned 2 infusions.
Patient characteristics
| Patient ID | Diagnosis | Age/sex | Previous therapies | Donor source for TAA-Ts | Dose of each TAA-T infusion | No. of TAA-T infusions | Treatment arm |
|---|---|---|---|---|---|---|---|
| Patient 1 | HL, NS | 54/M | Upfront: ABVDX6, XRT, Relapse: R-ICEx4, auto-HSCT, BvX7, Gem-VinoX2, Allo-HSCT | Allo | 0.5107/m2 | 1 | Active |
| Patient 2 | HL, NS | 36/M | Upfront: ABVDX6, XRT, Relapse: ICEX4, Auto-HSCT, Relapse#2 EverolimusX3, BvX3, NivoX11 | Auto | 0.5107/m2 | 3 | Active |
| Patient 3 | HL, NLP | 23/M | Upfront: ABVE-PCX4, Relapse: DA-EPOCH-R, R-ICEX4, XRT, Allo-HSCT | Allo | 4107/m2 | 1 | Adjuvant (TAA-T infused 6 mo after HSCT) |
| Patient 4 | HL, NS | 16/F | Upfront: ABVE-PC X 5, Bv-Gem, Ifos-Vino-Bortezomib,Nivo, Nivo+Bv, Auto-HSCT | Auto | 0.5e6/m2 | 1 | Adjuvant (TAA-T infused 2 mo after HSCT) |
| Patient 5 | HL, gray zone | 47/M | Upfront: ABVDX6, IF XRT, for refractory disease: ICE + BvX2, Auto-HSCT, Bv maintenance, XRT + rituximab, Nivolumab | Auto | 2107/m2 | 2 | Active |
| Patient 6 | HL, NS | 36/M | Upfront: ABVDX3, XRT, Relapse: ICEX2, BvX3, NivoX4 | Auto | 2107/m2 | 2 | Active |
| Patient 7 | HL, NS | 33/F | Upfront: ABVDX4, relapse: ICEX3, auto-HSCT, BvX2, Lenalidomide, Bv+Benda, PembroX4, anti-CD25 Mab,Vinblastine+Prednisone | Auto | 2107/m2 | 2 | Active |
| Patient 8 | HL, NS | 36/F | Upfront: ABVDX4, ICEX2, Gem+Oxaliplatin, Bv, acalabrutinib+pembro, anti-CD25 Mab, PembroX5 | Auto | 2107/m2 | 2 | Active |
| Patient 9 | HL, NS | 39/M | Upfront: ABVDX6, Relapse: Gem-Cisp-DexX3; Bv; ICEX2, auto-HSCT, NivoX28 doses, ESHAPX4 | Auto | 2107/m2 | 2 | Active |
| Patient 10 | HL, NS | 31/F | Upfront: ABVDX6, ICEX2, auto-HSCT, BvX3 | Auto | 2107/m2 | 1 | Active |
| Patient ID | Diagnosis | Age/sex | Previous therapies | Donor source for TAA-Ts | Dose of each TAA-T infusion | No. of TAA-T infusions | Treatment arm |
|---|---|---|---|---|---|---|---|
| Patient 1 | HL, NS | 54/M | Upfront: ABVDX6, XRT, Relapse: R-ICEx4, auto-HSCT, BvX7, Gem-VinoX2, Allo-HSCT | Allo | 0.5107/m2 | 1 | Active |
| Patient 2 | HL, NS | 36/M | Upfront: ABVDX6, XRT, Relapse: ICEX4, Auto-HSCT, Relapse#2 EverolimusX3, BvX3, NivoX11 | Auto | 0.5107/m2 | 3 | Active |
| Patient 3 | HL, NLP | 23/M | Upfront: ABVE-PCX4, Relapse: DA-EPOCH-R, R-ICEX4, XRT, Allo-HSCT | Allo | 4107/m2 | 1 | Adjuvant (TAA-T infused 6 mo after HSCT) |
| Patient 4 | HL, NS | 16/F | Upfront: ABVE-PC X 5, Bv-Gem, Ifos-Vino-Bortezomib,Nivo, Nivo+Bv, Auto-HSCT | Auto | 0.5e6/m2 | 1 | Adjuvant (TAA-T infused 2 mo after HSCT) |
| Patient 5 | HL, gray zone | 47/M | Upfront: ABVDX6, IF XRT, for refractory disease: ICE + BvX2, Auto-HSCT, Bv maintenance, XRT + rituximab, Nivolumab | Auto | 2107/m2 | 2 | Active |
| Patient 6 | HL, NS | 36/M | Upfront: ABVDX3, XRT, Relapse: ICEX2, BvX3, NivoX4 | Auto | 2107/m2 | 2 | Active |
| Patient 7 | HL, NS | 33/F | Upfront: ABVDX4, relapse: ICEX3, auto-HSCT, BvX2, Lenalidomide, Bv+Benda, PembroX4, anti-CD25 Mab,Vinblastine+Prednisone | Auto | 2107/m2 | 2 | Active |
| Patient 8 | HL, NS | 36/F | Upfront: ABVDX4, ICEX2, Gem+Oxaliplatin, Bv, acalabrutinib+pembro, anti-CD25 Mab, PembroX5 | Auto | 2107/m2 | 2 | Active |
| Patient 9 | HL, NS | 39/M | Upfront: ABVDX6, Relapse: Gem-Cisp-DexX3; Bv; ICEX2, auto-HSCT, NivoX28 doses, ESHAPX4 | Auto | 2107/m2 | 2 | Active |
| Patient 10 | HL, NS | 31/F | Upfront: ABVDX6, ICEX2, auto-HSCT, BvX3 | Auto | 2107/m2 | 1 | Active |
Doses: ≥18 years: 240 mg every 2 weeks or 480 mg every 4 weeks; <18 years: 3 mg/kg, max 240 mg every 2 weeks. ABVD, doxorubicin, bleomycin, vincristine, dacarbazine; ABVE-PC, doxorubicin, bleomycin, vincristine, etoposide, prednisone, and cyclophosphamide; Allo-HSCT, allogeneic hematopoietic stem cell transplantation; Auto-HSCT, autologous hematopoietic stem cell transplantation; Benda, bendamustine; Bv, brentuximab; CCR, continued complete remission; Cisp, cisplatin; DA-EPOCH-R, dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab; Dex, dexamethasone; ESHAP, etoposide, steroids, ara-C, and cisplatin; Gem, gemcitabine; ICE, ifosphamide, carboplatin, etoposide; Ifos, ifosphamide; Mab, monoclonal antibody; Nivo, nivolumab; NLP, modular lymphocyte predominant; NS, nodular sclerosing; PD, progressive disease; Pembro, pembrolizumab; PMR, partial metabolic response; SD, stable disease nivolumab dose; Vino, vinorelbine; XRT, involved field radiation.
Active disease.
Eight patients with r/r HL received 1 to 3 TAA-T infusions with doses ranging from 0.5 to 4 × 107/m2 following a median of 6 lines of previous therapies (Table 1). Of these, 2 patients received TAA-T alone (patient 1 received a single infusion at a dose of 0.5 × 107/m2 and patient 2 received 3 infusions at a dose of 0.5 × 107/m2 per infusion), and 6 patients received 2 TAA-T infusions at a dose of 2 × 107/m2 per infusion with nivolumab before and after their first TAA-T infusion and continued nivolumab per protocol (patients 5-10).
Adjuvant arm.
Two patients (patients 3 and 4; Table 1) received a single TAA-T infusion (1 allogeneic, 1 autologous) while in complete remission (CR). Patient 3 received 3 lines of prior therapy to achieve a second and then received haploidentical allogeneic HSCT. He received a single infusion of donor-derived TAA-Ts at a dose of 4 × 107/m2 approximately 6 months after HSCT. Patient 4 with primary refractory HL received 5 lines of therapy before ASCT and received a single infusion of autologous TAA-T at 0.5 × 107/m2 2 months after ASCT and at 8 weeks after TAA-T received a 5-month course of brentuximab and nivolumab.
TAA-T product characterization
Median time from collection to clinical freeze was 28 days (range, 22-31 days), which is a mean ± SEM of 6.9 ± 1.64-fold expansion at the time of clinical freeze (Table 2; Figure 1A). Products were polyclonal with CD3+T (mean ± SEM, 92.4 ± 2.5%), CD4+ helper T (10.8 ± 0.1%), CD8+ cytotoxic T (61.3 ± 6.5%), TCRαβ (69 ± 8.4%), TCRʏδ (31.7 ± 8.6%) with minimal CD3-CD56+ natural killer cells (3.9 ± 2%), and CD3+CD56+ (26.7 ± 2%) as detected by immunophenotype analysis and TCRVβ sequencing (Figure 1B-C). There was <10% lysis (release criteria for TAA-T infusion) of nonpulsed autologous Phytohaemagglutinin blasts at an effector: target ratio of 40:1 (mean ± SEM, 3.2 ± 0.99%), suggesting there was no auto- or allo-reactivity of the TAA-T products (Figure 1D).
Product characterization
| Patient ID | Percentage | SFC/1 × 105 T cells | Fold expansion | Total no. of cells frozen | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B cells | T cells | CD4+ | CD8+ | Natural Killer T-cells | Natural killer cells | CD3+ TCRab | CD3+ TCRgd | Mono | DCs | Actin | WT1 | Prame | Survivin | TAA* | |||
| Patient 1 | 0 | 99.5 | 10.9 | 29.3 | 42 | 0.92 | 16.3 | 73.5 | 0.3 | 0 | 7 | 16 | 179.5 | 2 | 88 | 23.2 | 1.39e8 |
| Patient 2 | 1.58 | 84.9 | 10.2 | 74.6 | 52.3 | 0.52 | 80.3 | 4.1 | 1.08 | 0.16 | 1 | 2.5 | 5 | 0 | 5.5 | 12.3 | 2.45107 |
| Patient 3 | 0 | 84.5 | 20 | 39 | 25 | 0.3 | 66 | 23.1 | 0 | 0 | 154.5 | 139 | 335 | 132.5 | 445 | 5 | 1.36e8 |
| Patient 4 | 0.2 | 99.1 | 16.9 | 81.6 | 2.2 | 14.8 | 91.8 | 69 | 0.2 | 0 | 5.5 | 10.5 | 72.5 | 5 | 48 | 5.91 | 1e8 |
| Patient 5 | 0.05 | 83.3 | 1.74 | 42.7 | 23.7 | 16.8 | 45.3 | 42.2 | 0.01 | 0.04 | 3.5 | 5 | 4 | 1 | 25.5 | 4.92 | 9.35107 |
| Patient 6 | 0.1 | 80.9 | 4.8 | 69.8 | 27.9 | 4.1 | 82.7 | 12.3 | 0 | 0 | 2.5 | 16 | 234 | 2 | 182.5 | 3.86 | 1.39108 |
| Patient 6_2 | 0 | 97.3 | 7.1 | 77 | 58.3 | 2.6 | 71.6 | 26 | 0 | 0 | 7.5 | 15 | 12 | 5 | 27 | 4.1 | 4.95 × 107 |
| Patient 7 | 0.07 | 96 | 20 | 75.7 | 45.1 | 1.1 | 87.4 | 11.5 | 0 | 0.02 | 18.5 | 24.5 | 15.5 | 30 | 20.5 | 5.57 | 1.3e8 |
| Patient 7_2 | 0 | 99.3 | 10.4 | 85.4 | 1.44 | 0.17 | 94..1 | 4 | 0 | 0 | 5.5 | 1 | 3 | 6 | 9 | 1.83 | 1.7 × 107 |
| Patient 8 | 0.3 | 98 | 1.73 | 43.3 | 33.7 | 0.6 | 39.5 | 58 | 0.1 | 0 | 7.5 | 37 | 67 | 18 | 22 | 5.97 | 1.88e8 |
| Patient 9 | 0.05 | 98.3 | 9.5 | 87.5 | 11.2 | 0 | 97.5 | 1.9 | 0 | 0.02 | 9.5 | 11 | 16 | 9 | 11.5 | 6.52 | 1.37e8 |
| Patient 10 | 0.09 | 99.5 | 11.8 | 69.5 | 3.59 | 0 | 81.4 | 16.1 | 0 | 0 | 1.5 | 15.5 | 1 | 14 | 10.5 | 2.31 | 4.38107 |
| Patient ID | Percentage | SFC/1 × 105 T cells | Fold expansion | Total no. of cells frozen | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B cells | T cells | CD4+ | CD8+ | Natural Killer T-cells | Natural killer cells | CD3+ TCRab | CD3+ TCRgd | Mono | DCs | Actin | WT1 | Prame | Survivin | TAA* | |||
| Patient 1 | 0 | 99.5 | 10.9 | 29.3 | 42 | 0.92 | 16.3 | 73.5 | 0.3 | 0 | 7 | 16 | 179.5 | 2 | 88 | 23.2 | 1.39e8 |
| Patient 2 | 1.58 | 84.9 | 10.2 | 74.6 | 52.3 | 0.52 | 80.3 | 4.1 | 1.08 | 0.16 | 1 | 2.5 | 5 | 0 | 5.5 | 12.3 | 2.45107 |
| Patient 3 | 0 | 84.5 | 20 | 39 | 25 | 0.3 | 66 | 23.1 | 0 | 0 | 154.5 | 139 | 335 | 132.5 | 445 | 5 | 1.36e8 |
| Patient 4 | 0.2 | 99.1 | 16.9 | 81.6 | 2.2 | 14.8 | 91.8 | 69 | 0.2 | 0 | 5.5 | 10.5 | 72.5 | 5 | 48 | 5.91 | 1e8 |
| Patient 5 | 0.05 | 83.3 | 1.74 | 42.7 | 23.7 | 16.8 | 45.3 | 42.2 | 0.01 | 0.04 | 3.5 | 5 | 4 | 1 | 25.5 | 4.92 | 9.35107 |
| Patient 6 | 0.1 | 80.9 | 4.8 | 69.8 | 27.9 | 4.1 | 82.7 | 12.3 | 0 | 0 | 2.5 | 16 | 234 | 2 | 182.5 | 3.86 | 1.39108 |
| Patient 6_2 | 0 | 97.3 | 7.1 | 77 | 58.3 | 2.6 | 71.6 | 26 | 0 | 0 | 7.5 | 15 | 12 | 5 | 27 | 4.1 | 4.95 × 107 |
| Patient 7 | 0.07 | 96 | 20 | 75.7 | 45.1 | 1.1 | 87.4 | 11.5 | 0 | 0.02 | 18.5 | 24.5 | 15.5 | 30 | 20.5 | 5.57 | 1.3e8 |
| Patient 7_2 | 0 | 99.3 | 10.4 | 85.4 | 1.44 | 0.17 | 94..1 | 4 | 0 | 0 | 5.5 | 1 | 3 | 6 | 9 | 1.83 | 1.7 × 107 |
| Patient 8 | 0.3 | 98 | 1.73 | 43.3 | 33.7 | 0.6 | 39.5 | 58 | 0.1 | 0 | 7.5 | 37 | 67 | 18 | 22 | 5.97 | 1.88e8 |
| Patient 9 | 0.05 | 98.3 | 9.5 | 87.5 | 11.2 | 0 | 97.5 | 1.9 | 0 | 0.02 | 9.5 | 11 | 16 | 9 | 11.5 | 6.52 | 1.37e8 |
| Patient 10 | 0.09 | 99.5 | 11.8 | 69.5 | 3.59 | 0 | 81.4 | 16.1 | 0 | 0 | 1.5 | 15.5 | 1 | 14 | 10.5 | 2.31 | 4.38107 |
TAA represents spot-forming cells (SFC) per 1 × 105 T cells in response to a mixture of WT1, PRAME, and Survivn added to the same experimental well.

Phenotype of TAA-T products. (A) Fold expansion (n = 12). (B) Immunophenotype of the TAA-T products (n = 10). (C) Polyclonality of TAA-T as assessed by TCR-Vβ deep sequencing (n = 6). (D) Lack of cytotoxicity of TAA-T (effectors) against non–antigen-pulsed PHA blasts (targets) at an various effector to target ratios (n = 10). (E) Exhaustion markers on TAA-T products (n = 6). PHA, phytohaemagglutinin
Phenotype of TAA-T products. (A) Fold expansion (n = 12). (B) Immunophenotype of the TAA-T products (n = 10). (C) Polyclonality of TAA-T as assessed by TCR-Vβ deep sequencing (n = 6). (D) Lack of cytotoxicity of TAA-T (effectors) against non–antigen-pulsed PHA blasts (targets) at an various effector to target ratios (n = 10). (E) Exhaustion markers on TAA-T products (n = 6). PHA, phytohaemagglutinin
Expression of exhaustion markers PD1, TIM3, and LAG3 on CD3+ TAA-T (percentage ± SEM) was 29 ± 0.277%, 6.4 ± 0.24%, and 3.99 ± 0.1%, respectively, and not different between products (Figure 1E). Next, we evaluated whether the PBMCs used for manufacturing of TAA-Ts affected the success or failure of the product. A product was considered successful if TAA-Ts were generated to meet the cell dose. CD4+PD1+ T cells were significantly higher in the PBMC material that did not result in successful product (n = 4) compared with successful products (n = 8; 27% vs 0.2%; P = .0039; supplemental Figure 1A). There was no difference in expression of TIM3 and LAG3 on CD4+ T cells or PD1, TIM3, and LAG3 on CD3+ or CD8+ subsets. Similarly, we did not find a significant difference between monocytic and granulocytic myeloid-derived suppressor cells between failed and successful products (P = .57; supplemental Figure 1B-C).
TAA-T products are polyclonal and polyfunctional and recognize multiple TAAs
Antigen specificity was evaluated using the IFNγ ELISPOT assay (Figure 2A). All products demonstrated a response to the staphylococcal enterotoxin B–positive control, with a median of 485 (range, 228-809) IFNγ spot-forming cells (SFCs)/2.5 × 105 TAA-T (values not included in the figure). Nonspecific activity to actin showed a median of 5.25 (range, 1-154.5) IFNγ SFCs/2.5 × 105. Response to specific antigens showed the following: WT1, median of 15.75 (range, 2.5-139) IFNγ SFCs/2.5 × 105; PRAME, median of 41.5 (range, 4-335) IFNγ SFCs/2.5 × 105; Survivin, median of 7 (range, 0-132.5) IFNγ SFCs/2.5 × 105 (Table 2). Eight products recognized at least 1 antigen, 5 recognized 2 antigens, and 1 recognized all 3 antigens. Two products did not recognize any TAA. There was, however, no correlation between antigen specificity of the TAA-T product and clinical response (supplemental Table 1). On restimulation with TAAs, the TAA-T products were polyfunctional, secreting IFNγ and tumor necrosis factor α (TNFα) from both CD4+ and CD8+ T-cell compartments (Figure 2B-C).

Functional characterization of TAA-T products. (A) Tumor antigen specificity as measured by IFNγ ELISPOT assay of the 12 infused products after overnight restimulation with overlapping 15mer pepmixes of actin (irrelevant control antigen), WT1, PRAME, and Survivin. (B) Polyfunctionality as assessed by the release of IFNγ and TNFα by CD3+, CD4+, and CD8+ TAA-Ts in response to irrelevant antigen. Actin and TAA (PRAME) shown in a representative dot plot of TAA-T product and summarized for 6 TAA-T products (C). * and ** denote P < .01 for difference between actin and PRAME.
Functional characterization of TAA-T products. (A) Tumor antigen specificity as measured by IFNγ ELISPOT assay of the 12 infused products after overnight restimulation with overlapping 15mer pepmixes of actin (irrelevant control antigen), WT1, PRAME, and Survivin. (B) Polyfunctionality as assessed by the release of IFNγ and TNFα by CD3+, CD4+, and CD8+ TAA-Ts in response to irrelevant antigen. Actin and TAA (PRAME) shown in a representative dot plot of TAA-T product and summarized for 6 TAA-T products (C). * and ** denote P < .01 for difference between actin and PRAME.
TAA-Ts are safe when given alone or in combination with nivolumab
To distinguish the effects of infusing TAA-T with associated therapies, patients were not allowed to receive any therapy during the 6-week safety monitoring period after the first TAA-T infusion. There were no dose-limiting toxicities and no TAA-T infusion-related adverse events within the safety monitoring period (Table 3). One patient developed a grade 1 cytokine release syndrome presenting with fever, fatigue, and elevation in C-reactive protein that resolved spontaneously without requiring any anticytokine therapy. No patients developed neurotoxicity related to the TAA-Ts, although 1 patient (patient 5) developed autoimmune encephalitis presenting with grade 3 seizures 2 months after the second TAA-T while receiving nivolumab. It was not possible to determine a definitive causal relationship to either nivolumab or TAA-Ts. Adverse events that were possibly related to TAA-T included grade 1 fatigue (n = 2) and myalgia (n = 1), all of which resolved spontaneously. Two patients developed abnormalities in thyroid function tests before the first TAA-T attributed to nivolumab and 1 had clinical hypothyroidism diagnosed 4 weeks after the TAA-T. All values corrected with thyroid supplementation and remained stable. One patient (patient 4) developed grade 1 myositis and discontinued nivolumab after 5 months (approximately 6 months after TAA-T).
Safety and outcomes of patients receiving TAA-Ts ± nivolumab
| Patient ID | Disease status before TAA-Ts (DS/SPD/cm2) | Disease status at 6 wk after first TAA-Ts (DS/SPD/cm2) | Therapy after TAA-Ts | Outcome at last follow-up | Survival at last follow-up | GVHD/CRS | Safety attributed to TAA-T cells | IrAE |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | DS = 5/7.08 | PD (DS = 5/15.15) | None* | PD (32 d)* | Dead (6 mo) | None* | None | N/A |
| Patient 2 | DS = 4/3.5 | PMR (DS = 3/3.5) | None | PD (6 mo) | Unknown | None | None | N/A |
| Patient 3 | DS = 2/<1 | CCR DS = 2/<1 | None | CCR (42 mo) | Alive (42 mo) | None | None | N/A |
| Patient 4 | DS = 3/13.28 | CCR DS = 3 | Nivolumab +Bv started 2 mo from TAA-T | CCR (40 mo) | Alive (40 mo) | None | None | Myositis 6 mo after TAA-T |
| Patient 5 | DS = 4 | DS = 3 | Nivolumab resumed after first TAA-T | SD (20 mo) | Alive (20 mo) | None | None | Autoimmune encephalitis 6 mo after TAA-T |
| Patient 6 | DS = 5/9 | PMR (DS = 4/5.4) | Nivolumab resumed 6 wk after first TAA-T† | CR (20 mo) | Alive (20 mo) | None | None | Hypothyroidism before TAA-T |
| Patient 7 | DS = 5/23.5 | SD (DS = 5/26.3) | Nivolumab resumed 6 wk after first TAA-T | PD (6 mo) | Alive (12 mo) | Grade 1 skin autoreactivity | None | None |
| Patient 8 | DS = 5/37.9 | SD (DS = 5/50.9) | Nivolumab resumed 6 wk after first TAA-T | PD (6 mo) | Alive (15 mo) | Grade 1 CRS | None | None |
| Patient 9 | DS = 5/29.7 | SD (DS = 5/43.5) | Nivolumab resumed 6 wk after first TAA-T; ongoing | SD (12 mo) | Alive (12 mo) | None | None | Hypothyroidism before TAA-T |
| Patient 10 | DS = 5/6.57 | SD (DS = 5/6.57) | Nivolumab resumed 6 wk after first TAA-T; ongoing | SD (12 mo) | Alive (12 mo) | None | None | Hypothyroidism before TAA-T |
| Patient ID | Disease status before TAA-Ts (DS/SPD/cm2) | Disease status at 6 wk after first TAA-Ts (DS/SPD/cm2) | Therapy after TAA-Ts | Outcome at last follow-up | Survival at last follow-up | GVHD/CRS | Safety attributed to TAA-T cells | IrAE |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | DS = 5/7.08 | PD (DS = 5/15.15) | None* | PD (32 d)* | Dead (6 mo) | None* | None | N/A |
| Patient 2 | DS = 4/3.5 | PMR (DS = 3/3.5) | None | PD (6 mo) | Unknown | None | None | N/A |
| Patient 3 | DS = 2/<1 | CCR DS = 2/<1 | None | CCR (42 mo) | Alive (42 mo) | None | None | N/A |
| Patient 4 | DS = 3/13.28 | CCR DS = 3 | Nivolumab +Bv started 2 mo from TAA-T | CCR (40 mo) | Alive (40 mo) | None | None | Myositis 6 mo after TAA-T |
| Patient 5 | DS = 4 | DS = 3 | Nivolumab resumed after first TAA-T | SD (20 mo) | Alive (20 mo) | None | None | Autoimmune encephalitis 6 mo after TAA-T |
| Patient 6 | DS = 5/9 | PMR (DS = 4/5.4) | Nivolumab resumed 6 wk after first TAA-T† | CR (20 mo) | Alive (20 mo) | None | None | Hypothyroidism before TAA-T |
| Patient 7 | DS = 5/23.5 | SD (DS = 5/26.3) | Nivolumab resumed 6 wk after first TAA-T | PD (6 mo) | Alive (12 mo) | Grade 1 skin autoreactivity | None | None |
| Patient 8 | DS = 5/37.9 | SD (DS = 5/50.9) | Nivolumab resumed 6 wk after first TAA-T | PD (6 mo) | Alive (15 mo) | Grade 1 CRS | None | None |
| Patient 9 | DS = 5/29.7 | SD (DS = 5/43.5) | Nivolumab resumed 6 wk after first TAA-T; ongoing | SD (12 mo) | Alive (12 mo) | None | None | Hypothyroidism before TAA-T |
| Patient 10 | DS = 5/6.57 | SD (DS = 5/6.57) | Nivolumab resumed 6 wk after first TAA-T; ongoing | SD (12 mo) | Alive (12 mo) | None | None | Hypothyroidism before TAA-T |
Data cutoff date was 27 April 2021.
Allo-HSCT, allogeneic hematopoietic stem cell transplant; Bv brentuximab vedotin; CCR, continued complete remission; CR, complete response; CRS, cytokine release syndrome; DS, Deauville Score: SPD sum of the product of the diameters; IrAE, immune-related adverse events; PD, progressive disease; PMR, partial metabolic response; SD, stable disease; XRT, radiation; N/A, not applicable.
Patient came off study before 6-wk safety monitoring because of clinical progression, received nivolumab as standard of care per treating physician, and developed grade 4 GVHD attributed to nivolumab.
Received nivolumab for 4 cycles followed by XRT to residual mediastinal mass and went into CR and proceeded to allogeneic HSCT at 6 months from TAA-Ts.
Clinical outcomes
Adjuvant treatment.
At a median follow-up of 41 months (range, 40-42 months) the 2 patients in the adjuvant arm have maintained a continued complete remission (Figure 3A).

Clinical outcomes. Swimmers plot showing the outcome of patients after receiving TAA-Ts with or without nivolumab. (A) Patients in remission at the time of TAA-T infusion. (B) Patients with measurable disease at the time of TAA-T infusion. The timing of nivolumab is indicated by the blue bar in relation to the TAA-T infusion, which is indicated by an asterisk.
Clinical outcomes. Swimmers plot showing the outcome of patients after receiving TAA-Ts with or without nivolumab. (A) Patients in remission at the time of TAA-T infusion. (B) Patients with measurable disease at the time of TAA-T infusion. The timing of nivolumab is indicated by the blue bar in relation to the TAA-T infusion, which is indicated by an asterisk.
Active disease.
Median time to last follow-up for the 8 patients with active disease was 12.6 months (range, 3.7-21 months; Figure 3B). Patient 1 with relapsed disease after allogeneic HSCT received donor-derived TAA-Ts and developed progressive disease at 6 weeks. He then received nivolumab off protocol and achieved a CR but developed grade 4 graft-versus-host disease (GVHD) and died of complications related to GVHD. Patient 2 initially had symptom relief and an observed positron emission tomography (PET) response of their bony lesions after first TAA-T dose and received 2 more TAA-T infusions to maintain this response, but subsequently developed disease progression at 6 months from the first infusion.
The remaining 6 patients with active disease received nivolumab before and after TAA-T infusion. Patient 5 had a positron emission tomography (PET) avid mediastinal mass that has remained stable at last follow-up (21 months). Patient 6 achieved a complete metabolic response at 6 months after 2 doses of TAA-Ts and nivolumab for 4 months and then proceeded to allogeneic HSCT. He remains in CR at 12 months after HSCT (20.4 months after first TAA-Ts). Patients 9 and 10 remain on nivolumab with stable disease (SD) at 12 months from study entry. Two patients (patients 7 and 8) developed progressive disease (PD) at 6 months and came off protocol. Interestingly, both patients went on to achieve CR after receiving standard chemotherapy despite previously being considered chemo-refractory.
Long-term TAA-T cell persistence and evidence of antigen spreading are observed in responding patients
The 2 patients (patients 3 and 4) who received TAA-Ts as adjuvant treatment showed long-term (up to 2 years) persistence of TAA-T cells in the PB as detected by IFNγ ELISPOT assay (Figure 4A-B). The T cells were specific to 1 or more TAA antigens and showed presence of low-frequency T cells to nontargeted TAAs not included in the product, consistent with evidence of antigen spreading as has previously been described with adoptively transferred EBV/latent membrane protein(LMP)-specific T-cell therapy for HL1 (supplemental Table 2). The detected TAA-specific T cells were polyfunctional, with CD4+- and CD8+-specific T cells producing IFNγ and TNFα on restimulation, suggesting a functional in vivo tumor antigen-specific response (supplemental Figures S2 and S3). In contrast, patients who received TAA-T alone for active disease (patients 1 and 2) had very low frequencies of TAA-Ts detected in PB after infusion (Figure 4C-D). Specifically, patient 2 had detectable T cells specific for TAAs by week 6 after infusion that coincided with improvement in bone pain and a minor metabolic response. He received 2 additional TAA-T infusions, and TAA-specific T cells were detected in the PB before the third TAA-T infusion. However, this clinical benefit did not persist, and the patient ultimately developed disease progression at a time when TAA-specific T cells were no longer detectable in vivo (Figure 4C).
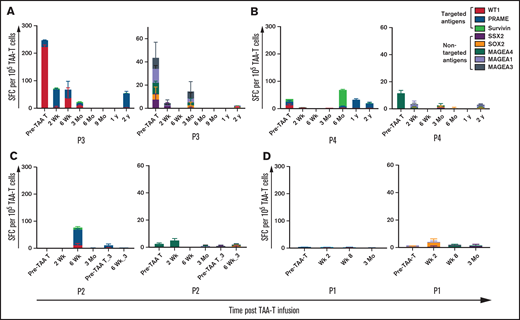
Recognition of tumor specific antigens over time. Recovery of antigen-specific T cells and antigen spreading as detected by IFNγ ELISPOT assay in patients receiving TAA-T alone in responders (A-B) and nonresponders (C-D). For each figure, reactivity to the targeted antigens WT1, PRAME, and Survivin is shown in panel i and nontargeted antigens MAGE family, SSX2, and SOX2 is in panel ii.
Recognition of tumor specific antigens over time. Recovery of antigen-specific T cells and antigen spreading as detected by IFNγ ELISPOT assay in patients receiving TAA-T alone in responders (A-B) and nonresponders (C-D). For each figure, reactivity to the targeted antigens WT1, PRAME, and Survivin is shown in panel i and nontargeted antigens MAGE family, SSX2, and SOX2 is in panel ii.
Priming with nivolumab enhances TAA-T–specific response and persistence
All 6 patients with active lymphoma who received TAA-T with nivolumab had detectable TAA-Ts as detected by IFNγ ELISPOT assay at 2 to 6 weeks, and 4 had detectable TAA-Ts at 3 months (Figure 5A-F). Interestingly, only responding patients (ie, patients who achieved SD, PR, or CR to nivolumab at 3 months from the start of the drug) demonstrated reactivity to targeted TAAs before TAA-T infusion after receiving nivolumab priming (median, 9.5 IFNγ SFCs/1 × 105 cells; range, 4-288) and also at 3 months (median IFNγ SFCs/1 × 105 cells of 56; range, 4.5-185; Figure 6A). In contrast, the 2 nonresponders who had disease progression to ongoing nivolumab after TAA-Ts had minimal detectable TAA-specific T cells in vivo before TAA-Ts (median IFNγ SFCs/1 × 107 5 of 6; range, 0-6) and at 3 months (median IFNγ SFCs/1 × 107 5 of 3; range, 0-5.5). The differences of the TAA-T reactivity, however, were not statistically significant (P = .38).

Recognition of tumor specific antigens over time. Recovery of antigen-specific T cells and antigen spreading as detected by IFNγ ELISPOT assay in patients receiving TAA-T and nivolumab in responders (A-D) and nonresponders (E-F). For each figure, reactivity to the targeted antigens WT1, PRAME, and Survivin is shown in panel i and nontargeted antigens MAGE family, SSX2, and SOX2 in panel ii.
Recognition of tumor specific antigens over time. Recovery of antigen-specific T cells and antigen spreading as detected by IFNγ ELISPOT assay in patients receiving TAA-T and nivolumab in responders (A-D) and nonresponders (E-F). For each figure, reactivity to the targeted antigens WT1, PRAME, and Survivin is shown in panel i and nontargeted antigens MAGE family, SSX2, and SOX2 in panel ii.

Impact of nivolumab on persistence of functional TAA-T cells. (A) Comparison of T-cell reactivity to the targeted antigens as detected by IFNγ ELISPOT assay after nivolumab priming and before TAA-T infusion and at 3 months between responding (R) and nonresponding patients (NR). (B) Persistence of polyfunctional TAA-T cells over time in responders. (C) Persistence of polyfunctional TAA-T cells over time in nonresponders. (D) Representative plot of patient demonstrating recovery of in vivo polyfunctional CD4+ and CD8+ TAA-T cells secreting both IFNγ and TNFα was detected after brief ex vivo expansion after restimulation with antigens at several follow-up time points.
Impact of nivolumab on persistence of functional TAA-T cells. (A) Comparison of T-cell reactivity to the targeted antigens as detected by IFNγ ELISPOT assay after nivolumab priming and before TAA-T infusion and at 3 months between responding (R) and nonresponding patients (NR). (B) Persistence of polyfunctional TAA-T cells over time in responders. (C) Persistence of polyfunctional TAA-T cells over time in nonresponders. (D) Representative plot of patient demonstrating recovery of in vivo polyfunctional CD4+ and CD8+ TAA-T cells secreting both IFNγ and TNFα was detected after brief ex vivo expansion after restimulation with antigens at several follow-up time points.
The TAA-T responses were polyfunctional (secreting both IFNγ and TNFα in response to antigen restimulation), largely restricted to the CD4+ compartment (representative patient sample shown in Figure 6D), only observed in responders, and decreased over time for nonresponders (Figure 6B-C).
TAA-relevant T-cell clones are detected in PB and persist over time
To explore longitudinal effects of treatment on the peripheral TCRβ repertoire, we sequenced the CDR3 region of the TAA-T product and PBMCs obtained before and after TAA-T infusion. PBMCs were isolated at study entry (for those receiving nivolumab priming), baseline before TAA-T infusion (for all patients), and 2 and 6 weeks and 3 months after TAA-T. Immunosequencing of the TCRβ locus showed that posttreatment peripheral repertoires were similarly diverse as baseline (supplemental Figure 4A-B).
Using a binomial model with Benjamini-Horschberg correction for multiple comparisons (controlling false discovery rate at 0.01), we identified T-cell clones enriched at higher frequency in the product and not present in the pretreatment sample as TAA-T relevant clones. On average, there were 246 TAA-T relevant clones (range, 162-348). This patient-specific set of TAA-T relevant clones was longitudinally tracked in the periphery, and the cumulative frequency (number of TAA-T relevant T cells/total T-cell count) was compared across peripheral samples (Figure 7A). Relative to pretreatment (frequency = 0), there was a significantly higher frequency of newly detected enriched product clones in the PB in patients receiving TAA-T alone or with nivolumab priming for all posttreatment samples (Plmm < .005 for 2 and 6 weeks and 3 months after TAA-T relative to pretreatment; Figure 7B). Hence, despite not using prescribed lymphodepletion, TAA-T relevant clones were trackable in PB samples, albeit at low frequency (maximum, 0.2%). To assess the effect of nivolumab on the infused TAA-Ts, we tracked 400 enriched TCR clones in patient 6 (responder); 278 of these were newly detected and not present before TAA-T, of which 48 were expanded in the product and detected in at least 1 other follow-up sample. Twenty-six of the 48 clones were not detected before TAA-Ts as shown in Figure 7C but were present in samples obtained from at least 2 other time points. An apparent increase of these clones was observed on day 90 when the patient was in a complete remission, suggesting a potential impact of nivolumab that resumed after TAA-Ts.
![Persistence of new TAA-T clones expanded in the product that were not present at baseline. (A) Using TCRVβ deep sequencing, new clones detected in the TAA-T product were identified and tracked in PB over time. (B) Longitudinal tracking of new clones expanded in product over time for patients receiving TAA-T alone. Multi-institutional Prospective Research of Expanded Multi-antigen Specifically Oriented Lymphocytes for the Treatment of VEry High Risk Hematopoietic Malignancies (RESOLVE) and TAA-T with nivolumab [TAA-T with nivolumab on Phase I Study Utilizing Tumor Associated Antigen Specific T cells (TAA-T) with PD1 Inhibitor Nivolumab for Relapsed/Refractory Lymphoma (SUSTAIN) trial]. (C) Tracking of clones enriched in TAA-T product and not present at baseline in patient 6.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/2/10.1182_bloodadvances.2021005343/3/m_advancesadv2021005343f7.png?Expires=1767697976&Signature=Wt2XRQbJDOHrxWxn6FENrD-fV-UTZt1j0SxZE3xlUtQJzGWFVCkJKbw6d0aAqxPbXZ1ZP6U-WAwBu-mA~x587WHjSaPBrswU5KDea2YldiL~jp8ug~pimP2MZ46jiLC5SzHMfWMg7lfYBtzLMbEL~b04hHDakLLek1hV8OfbkDBoK7xY3AngUBLZF4joQfBrmoiL3fhGt~JaDAfx9ZA6ta-7-c650WDZmaTNzwQBRjUGkprHDrnT1pq~XKcz6mEY9h7vY8rZ-~RD3pjqapsv8y4KAwClZD02s~skE1VRWzNpyTTSShkxtArXfeur6R1up~3zpiDNUZVtGwVa0zgNMg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Persistence of new TAA-T clones expanded in the product that were not present at baseline. (A) Using TCRVβ deep sequencing, new clones detected in the TAA-T product were identified and tracked in PB over time. (B) Longitudinal tracking of new clones expanded in product over time for patients receiving TAA-T alone. Multi-institutional Prospective Research of Expanded Multi-antigen Specifically Oriented Lymphocytes for the Treatment of VEry High Risk Hematopoietic Malignancies (RESOLVE) and TAA-T with nivolumab [TAA-T with nivolumab on Phase I Study Utilizing Tumor Associated Antigen Specific T cells (TAA-T) with PD1 Inhibitor Nivolumab for Relapsed/Refractory Lymphoma (SUSTAIN) trial]. (C) Tracking of clones enriched in TAA-T product and not present at baseline in patient 6.
Persistence of new TAA-T clones expanded in the product that were not present at baseline. (A) Using TCRVβ deep sequencing, new clones detected in the TAA-T product were identified and tracked in PB over time. (B) Longitudinal tracking of new clones expanded in product over time for patients receiving TAA-T alone. Multi-institutional Prospective Research of Expanded Multi-antigen Specifically Oriented Lymphocytes for the Treatment of VEry High Risk Hematopoietic Malignancies (RESOLVE) and TAA-T with nivolumab [TAA-T with nivolumab on Phase I Study Utilizing Tumor Associated Antigen Specific T cells (TAA-T) with PD1 Inhibitor Nivolumab for Relapsed/Refractory Lymphoma (SUSTAIN) trial]. (C) Tracking of clones enriched in TAA-T product and not present at baseline in patient 6.
Exhausted T cells in the tumor microenvironment affect response to TAA-Ts
To determine the mechanism of TAA-T resistance we further studied patient 7 who had initially experienced dramatic symptom relief with resolution of her B symptoms 6 weeks after the first TAA-T negligible expansion of TAA-specific T cells in the PB as shown in Figure 5E. On imaging, this patient had a mixed response at 3 months, but at 6 months had PD. Biopsy of an involved lymph node at the time of disease progression showed increased CD3+ and CD4+ T cells and T-regulatory cells (CD4+CD25highCD127low; supplemental Figure 5A). Further evaluation of these tumor infiltrating lymphocytes (TILs) demonstrated high expression of exhaustion markers PD1, TIM3, and LAG3 on both CD4+ and CD8+ T-cell compartments (supplemental Figure 5B). Ex vivo expansion of the TILs with the TAA overlapping 15mers representing full length proteins WT1, Prame and Survivin (pepmixes) with the Th1 cytokine milieu resulted in a product with decreased T-regulatory cells and exhaustion marker phenotype (supplemental Figure 5B); however, the TILs did not demonstrate any TAA specificity (supplemental Figure 5C). PBMCs isolated at the time of disease progression (at month 6) and ex vivo expanded similar to the TILs demonstrated the same exhausted phenotype with high expression of LAG3 and PD1 compared with PB samples obtained before PD (supplemental Figure 5B). However, unlike the TILs, these PB-derived T cells demonstrated TAA-specific reactivity to PRAME (supplemental Figure 5C). Evaluation of patient 8 who developed disease progression after TAA-Ts but subsequently responded to conventional chemotherapy showed a decrease in exhaustion markers (PD1, LAG3, and TIM3) with a small concurrent increase in T cells reactive to TAA antigens at the time of remission compared with the time of relapse (supplemental Figure 5D).
Discussion
We report the safety, efficacy, and immune biology correlates of infusing multiantigen-specific non–gene-engineered tumor-associated antigen-specific T cells given in combination with nivolumab or alone in 10 patients with r/r HL. The 2 patients who received TAA-Ts as adjuvant therapy after transplant (allogeneic and autologous) have maintained sustained remissions for more than 3 years despite having chemo-refractory disease before transplant. Although both these patients had preexisting responses to 1 of the targeted antigens, they demonstrated antigen spreading at long-term follow-up. Although efficacy was not the primary end point of this study, 7 of 8 patients with active disease achieved SD at 6 weeks after the TAA-T infusion. One patient achieved CR and proceeded to allogeneic HSCT, whereas 3 remain with SD at a median follow-up of 12.6 months (range, 3.4-21 months). The 2 patients with PD went on to achieve CR after receiving chemotherapy despite being previously chemo-refractory, suggesting that CPI can rebalance the immunostimulatory and immunosuppressive immune composition of the HL microenvironment, leading to restored chemosensitivity (supplemental Figure 5D). Although numbers are small, clinical responses in patients receiving combination TAA-T and nivolumab were achieved without increased immune-related adverse events compared with those seen in clinical trials with single agent nivolumab,10,11 Given all patients received a washout of 6 weeks with no other systemic therapy during the safety monitoring period after TAA-Ts, our results demonstrate that the combinatorial approach is safe and warrants evaluation in phase 2 studies.
Similar to our original observations after the administration of EBV-specific T cells to patients with EBV+ HL,1 we were able to demonstrate that TAA-T cells given alone or in combination with CPIs elicited antigen spreading responses in responding patients. These data are also consistent with observations from recent clinical trials administering T cells targeting alternative tumor-associated antigen combinations given without CPI for the treatment of hematologic malignancies.24-26 Unlike nonresponding patients, responders to ongoing nivolumab showed persistence of polyfunctional CD4+ TAA-T cells at 3 to 6 months in the PB without any additional TAA-T infusions, which suggests that nivolumab may influence the reactivity and persistence of TAA-Ts in vivo, supporting our hypothesis that nivolumab aids in T-cell persistence and is consistent with prior immune CPI trials.27-29 The impact of lymphodepleting chemotherapy before TAA-T infusion could further facilitate the homing of the TAA-T cells to tumor sites and will be explored in our subsequent study.
In our previous study that demonstrated the safety and efficacy of TAA-T for solid tumors, maximum clinical benefit was seen in patients receiving multiple TAA-T infusions.15 The number of TAA-Ts manufactured from PB limited our ability to administer multiple doses to these patients with heavily pretreated HL, thus suggesting that apheresis collections for TAA-T manufacture may be preferable for this population. Our patients had received a median of 6 lines of prior therapy and were extremely lymphopenic, with increased evidence of exhaustion and myeloid-derived suppressor cells that are known to impair T-cell expansion and function.30-32 The 2 patients who were recollected after exposure to nivolumab had successful products generated, which suggests that nivolumab priming before collection could be used as a future strategy to generate TAA-T cells for high-risk patients receiving CPIs.33 Importantly, TAA-T cell therapy offers a potential option for patients who do not experience a durable remission to a CPI alone, especially when used as consolidation in high-risk patients after autologous stem cell transplant or those who have failed prior CPIs.34,35 Assessment of product attributes in our study provides opportunity to increase potency of the TAA-T product for future studies. The high frequency of γ-δ T cells seen in our TAA-T products will be compared with efficacy in larger cohorts, because these unique immune T cells with properties of both innate and adaptive immune responses to tumor- and viral-associated antigens are known to contribute to antitumor immunity. Although the frequency of antigen-specific T cells was relatively low in our products, any relationship to efficacy is limited by the small sample size. Moreover, we did not identify a correlation between antigen specificity and efficacy in our prior solid tumor TAA-T cell study15 ; however, this question will be explored in a larger cohort of HL patients in the future.
As illustrated by patient 7, the inability of TILs to recognize TAAs despite an expansion of TAA-Ts in the PB is reflective of the intensely inhibitory tumor microenvironment in HL.36 The presence of TILs expressing other inhibitory checkpoints such as LAG3 and TIM3 with a corresponding decrease in these markers after ex vivo expansion in a Th1-promoting cytokine milieu confirms that developing other strategies, in addition to PD1 blockade, are essential to overcome tumor-induced immunosuppression in vivo.37,38 We previously showed that adoptively transferred EBV-specific T cells engineered to express a dominant negative transforming growth factor β (TGFβ) receptor II can overcome the inhibitory and antiproliferative effects of TGFβ in the tumor microenvironment.1,2,39 Future studies will therefore explore broadening the portfolio of targeted antigens by the TAA-T product, correlating exhaustion markers with product efficacy, and rendering the products resistant to TGFβ in combination with CPIs.
Limitations of our study include its small sample size and heterogenous patient population. Although patients receiving TAA-Ts for adjuvant therapy could have experienced benefit from the immune reconstitution from their transplant, the recovery of polyfunctional TAA-Ts in the PB even 2 years after TAA-Ts is suggestive of protective TAA-specific antitumor immune responses in vivo. The persistence of polyfunctional TAA-T in responders to nivolumab and the emergence of new product clones, although in smaller frequencies over time, are suggestive of a synergistic role of nivolumab, and therefore, long-term follow-up with immune correlates in this population is required. Future studies evaluating TCR clonotypes to the epitopes of targeted and nontargeted antigens will provide further insight into the TAA-T–specific response.
In summary, autologous multi-TAA-T cells can be generated from patients who have failed multiple prior immunotherapies for HL and are well tolerated when given alone or with checkpoint inhibitors. The expansion and persistence of infused TAA-T cells was associated with clinical benefit to patients including durable remissions.
Acknowledgments
The authors thank Anne Angiolillo for thoughtful feedback in reviewing the data and the manuscript and Matthew Knight and Kathryn G Boland (Adaptive Biotechnologies, Seattle, WA) for help with TCRVβ sequencing.
This work was supported in part by National Institutes of Health grant P01-CA-015396 (R.A., C.M.B., and P.J.H.) and Ben’s Run Foundation (C.M.B.). This work was also supported by grants from the American Society of Hematology Scholar Award (H.D.), the Leukemia & Lymphoma Society Translational Research Program (H.D. and C.M.B.), Children’s Cancer Foundation, Inc. (H.D.), and The Safeway Foundation (H.D.).
Authorship
Contribution: H.D., P.J.H., and C.M.B. conceived and designed the study. P.M. and F.H., provided administrative and regulatory support; H.D., R.S., M.G., B.H. R.A., and C.M.B. provided patient care and trial recruitment; H.D., M.T., M.M., K.T., M.G., M.S., P.M., C.L., A.S., S.A.B collected and compiled the data; H.D., P.J.H., M.G., C.L., and C.M.B. analyzed and interpreted the data; H.D. and C.M.B. wrote the manuscript; and all authors provided final approval of the manuscript and are accountable for all aspects of work.
Conflict-of-interest disclosure: H.D. has served on advisory board for Pfizer and on the scientific advisory board of Children’s Cancer Foundation, Inc. P.J.H. is a cofounder, consultant, and on the board of directors of Mana Therapeutics; on the scientific advisory board and a consultant for Cellevolve; on a scientific advisory board of Maxcyte; and has intellectual property on TAA-specific T cells and virus-specific T cells. C.M.B. owns stock or other ownership interests in Mana Therapeutics, NexImmune, Torque, and Cabaletta Bio; has a consulting or advisory role for Repertoire Immune Medicines, NexImmune, Cellectis, and Cabaletta Bio; and has received patents or royalties for TAA-specific T cells and HIV-specific T cells. B.H. has received research funding from CRISPR Therapeutics, Celgene, Roche, and Miragen. S.A.B. have a financial interest in Adaptive Biotechnologies. All remaining authors declare no competing financial interests.
Correspondence: Catherine M. Bollard, Center for Cancer and Immunology Research, Children’s National Hospital, 111 Michigan Ave NW, 20010 Washington, DC; e-mail: cbollard@childrensnational.org.

