

Key Points
Consistent with previous results, pola + BR has a tolerable safety profile.
The survival benefit of pola + BR vs BR persists with longer follow-up; efficacy in the pola + BR extension and randomized arms was similar.

Abstract
Polatuzumab vedotin plus bendamustine and rituximab (pola + BR) received regulatory approvals for relapsed/refractory diffuse large B-cell lymphoma (R/R DLBCL) based on primary results from the randomized arms of the GO29365 study. After the randomized phase, 106 additional patients received pola + BR in a single-arm extension cohort. We report updated results from the randomized arms and results of the extension cohort. In this phase 1b/2 study, patients with R/R DLBCL who were transplant ineligible received up to six 21-day cycles of pola + BR or BR. The primary end point of the randomized arms was the complete response (CR) rate at end of treatment. Primary objectives of the extension cohort were safety, pharmacokinetic profile, and efficacy of pola + BR. As of 7 July 2020, a total of 192 patients with R/R DLBCL were enrolled in the pola + BR cohort (n = 152 [safety run-in, n = 6; randomized, n = 40; extension cohort, n = 106]) or the BR cohort (n = 40). Significant survival benefit with pola + BR vs BR persisted in the randomized arms (median progression-free survival, 9.2 vs 3.7 months [hazard ratio, 0.39; 95% confidence interval, 0.23-0.66]; median overall survival, 12.4 vs 4.7 months [hazard ratio, 0.42; 95% confidence interval, 0.24-0.72]). In the extension cohort, the independent review committee–assessed objective response rate was 41.5%, and the CR rate was 38.7%; median independent review committee–assessed progression-free survival and overall survival were 6.6 months and 12.5 months, respectively. No new safety signals with pola + BR were identified. Pola + BR is an effective treatment option for patients with R/R DLBCL, with a well-characterized and manageable safety profile. This trial was registered at www.clinicaltrials.gov as #NCT02257567.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma. Although DLBCL is curable for the majority, ∼30% to 40% of patients are either refractory to first-line treatment or will relapse after an initial response.1 Approximately 50% of patients with relapsed or refractory (R/R) DLBCL are ineligible for standard second-line treatment with intensive salvage therapy and autologous stem cell transplantation (SCT), and prognoses for these patients are poor.1,2 Common treatment options in this setting include rituximab plus gemcitabine and oxaliplatin (R-GemOx), as well as bendamustine plus rituximab (BR). These have been evaluated in patients with transplantation-ineligible R/R DLBCL, with a median progression-free survival (PFS) of 3.6 to 6.7 months.3,4 However, there is no defined standard of care, and the treatment landscape is further evolving with the development of targeted therapies. Several treatments have now been approved in the second-line setting and beyond, including the antibody–drug conjugate polatuzumab vedotin in combination with BR (pola + BR)5,6 and the anti-CD19 monoclonal antibody tafasitamab in combination with lenalidomide.7 The CD19-directed chimeric antigen receptor (CAR) T-cell therapies axicabtagene ciloleucel, tisagenlecleucel, and lisocabtagene maraleucel have been approved in the third-line setting,8-10 as well as selinexor, a selective inhibitor of nuclear export.11
Polatuzumab vedotin is an antibody–drug conjugate targeting CD79b to deliver a microtubule polymerization inhibitor, monomethyl auristatin E.12-14 CD79b is a critical component of the B-cell receptor signaling pathway; it is expressed on all normal B cells and on most mature B-cell malignancies, including DLBCL.15,16 The phase 1b/2 GO29365 study (#NCT02257567) assessed the safety and efficacy of pola + BR in patients with R/R DLBCL. The study included a hard-to-treat patient population: 53% of patients in the randomized pola + BR arm were primary refractory, 75% were refractory to their last prior therapy, and 46% had received at least 3 previous lines of therapy. In the randomized cohort, after a median follow-up of 22.3 months, pola + BR significantly improved the survival of patients with R/R DLBCL compared with BR alone: median PFS, 9.5 vs 3.7 months (hazard ratio [HR], 0.36; 95% confidence interval [CI], 0.21-0.63; P = .001); and median overall survival (OS), 12.4 vs 4.7 months (HR, 0.42; 95% CI, 0.24-0.75; P = .002). The primary end point was met, with an independent review committee (IRC)-assessed complete response (CR) rate of 40.0% with pola + BR vs 17.5% with BR alone. The IRC-assessed best objective response (BOR) rate was 62.5% with pola + BR vs 25.0% with BR. Biomarker analysis suggested that patients derived benefit from pola + BR regardless of cell-of-origin or double-expressor lymphoma status.17 Based on these preliminary results, pola + BR received regulatory approvals in the United States and the European Union for patients with transplant-ineligible R/R DLBCL.5,6
Following the initial pola + BR vs BR randomized arms of the study, an additional 106 patients with R/R DLBCL were enrolled into a single-arm extension cohort and received pola + BR to further assess the safety, efficacy, and pharmacokinetic (PK) profile of this treatment combination. Here, we report updated results with a further 27 months of follow-up in the randomized pola + BR arm, as well as results of the single-arm extension cohort.
Methods
Study design and treatment
This phase 1b/2, open-label, multicenter, randomized study was designed to evaluate the safety, efficacy, and PK profile of pola + BR and pola + B + obinutuzumab in patients with R/R DLBCL or follicular lymphoma. Here, data are reported from the arms of the study onto which patients with R/R DLBCL were enrolled and assigned treatment with pola + BR or BR; this includes an initial safety run-in phase (phase 1b), followed by randomized cohorts and a single-arm extension cohort (phase 2) (Figure 1). PK results in the randomized DLBCL cohorts have been reported previously.18
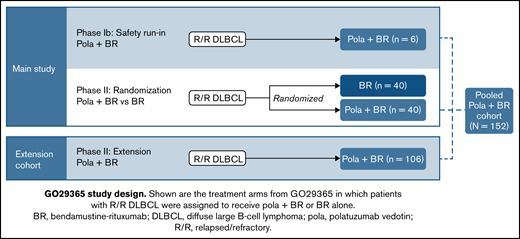
GO29365 study design. Shown are the treatment arms from GO29365 in which patients with R/R DLBCL were assigned to receive pola + BR or BR alone.
GO29365 study design. Shown are the treatment arms from GO29365 in which patients with R/R DLBCL were assigned to receive pola + BR or BR alone.
The study protocol was approved by the institutional review boards or ethics committees at participating institutions in accordance with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines, including Good Clinical Practice, and the ethical principles originating from the Declaration of Helsinki.19,20 Informed consent was given by all patients. An internal monitoring committee reviewed data regularly during the conduct of the study.
Patients received bendamustine 90 mg/m2 intravenously (IV) on days 2 and 3 of cycle 1, and days 1 and 2 of subsequent cycles, plus rituximab IV (375 mg/m2 on day 1 of each cycle). Patients treated with polatuzumab vedotin received 1.8 mg/kg IV on day 2 of cycle 1, and day 1 of subsequent cycles. Patients received treatment of up to six 21-day cycles. Prophylaxis with granulocyte colony-stimulating factor for neutropenia was at the discretion of clinicians in the randomized phase 2 component and was required in each cycle of therapy for patients treated in the extension cohort.
Patient population
Patients aged ≥18 years were eligible if they had histologically confirmed R/R DLBCL (excluding transformed follicular lymphoma), received ≥1 prior line of therapy, had an Eastern Cooperative Oncology Group performance status of 0 to 2, and were considered transplant ineligible by the treating physician or experienced treatment failure with prior autologous SCT. Patients with current grade >1 peripheral neuropathy (PN) were excluded. Additional inclusion and exclusion criteria have been described previously.17
Study assessments and end points
The primary end point of the randomized arms was to determine the CR rate by positron emission tomography/computed tomography (PET-CT) imaging at the end of treatment (EOT; 6-8 weeks after cycle 6 day 1, or last dose of study treatment) as determined centrally by an IRC using the modified Lugano criteria (supplemental Material).17,21 Secondary objectives included safety, tolerability, and efficacy, including objective response rate (ORR; achievement of CR or partial response), BOR (defined as a CR or partial response while on study based on PET-CT or CT imaging only, as determined by the investigator or IRC),15 duration of response (DOR), PFS, and OS. The primary objectives of the extension cohort included assessments of safety, PK profile, and efficacy of pola + BR.
Adverse events (AEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Scans were assessed by the investigators and the IRC, and response was determined by using the modified Lugano criteria.21
Data analysis and statistical methods
The primary safety population included all patients who received at least 1 dose of any study medication in any cohort. Efficacy was evaluated in the intention-to-treat population. Under the assumption of 40% PET-CT CR rate at EOT, a sample size of 100 patients in the extension cohort was selected to provide a precise 95% CI (Clopper–Pearson exact CI of 30%-50%) for the CR rate. Furthermore, this 95% CI completely excludes the 95% CI (17.5% [11% to 26%]) of the CR rate observed in the randomized BR arm. Response rates were reported as percentages with associated 95% Clopper–Pearson CIs. Time-to-event end points, including DOR, PFS, and OS, were summarized as median survival time estimated using Kaplan–Meier methodology with 95% Brookmeyer and Crowley CIs. Differences in time-to-event end points between the pola + BR and BR arms were compared for exploratory purposes and reported as hazard ratios (HRs) using stratified Cox regression. Exploratory subgroup analyses (ie, refractory status, number of previous lines of therapy) were conducted on BOR, PFS, and OS. All reported P values are two-sided and not α-controlled.
Results
Patient population
Between 15 October 2014 and 9 July 2019, a total of 192 patients with R/R DLBCL from 46 sites across 12 countries in Asia, Australia, Europe, and North America were enrolled into the pola + BR or BR cohorts. Patients were enrolled into different arms of the trial as follows: phase 1b safety run-in (pola + BR; n = 6) and phase 2 randomized arms (pola + BR and BR; n = 40 per arm). A total of 106 patients were later enrolled into the extension cohort and received pola + BR. As of the clinical cutoff date (7 July 2020), median follow-up was 48.9 and 48.3 months for the randomized arms (pola + BR and BR, respectively) and 15.2 months for the extension cohort.
Demographic and baseline characteristics are described in Table 1. Baseline characteristics in the extension cohort were generally similar to those of the randomized arms. Patients in the extension cohort were heavily pretreated, and a large proportion were refractory; 39.6% of patients had received ≥3 prior lines of therapy, 68.9% were primary refractory, and 76.4% were refractory to their last prior therapy.
Patient demographic and baseline clinical characteristics
| ITT | Randomized | Extension cohort, pola + BR (n = 106) | Pooled, pola + BR (N = 152) | |
|---|---|---|---|---|
| BR (n = 40) | pola + BR (n = 40) | |||
| Median (range) age, y | 71 (30-84) | 67 (33-86) | 70 (24-94) | 69 (24-94) |
| Age ≥65 y | 26 (65) | 23 (58) | 77 (73) | 103 (68) |
| Male sex | 25 (63) | 28 (70) | 52 (49) | 84 (55) |
| ECOG PS score | ||||
| 0 | 17 (43) | 12 (30) | 30 (28) | 44 (29) |
| 1 | 14 (35) | 21 (53) | 62 (59) | 87 (57) |
| 2 | 8 (20) | 6 (15) | 14 (13) | 20 (13) |
| Ann Arbor stage III/IV | 36 (90) | 34 (85) | 84 (79) | 122 (80) |
| Bulky disease | 14 (35.0) | 10 (25) | 28 (26) | 39 (26) |
| IPI score 3-5 at enrollment | 29 (73) | 22 (55) | 70 (66) | 94 (62) |
| Median no. of prior therapies (range) | 2 (1-5) | 2 (1-7) | 2 (1-7) | 2 (1-7) |
| 1 line | 12 (30) | 11 (28) | 37 (35) | 50 (33) |
| 2 lines | 9 (23) | 11 (28) | 27 (26) | 42 (28) |
| ≥3 lines | 19 (48) | 18 (45) | 42 (40) | 60 (39) |
| WHO 2016 classification (central pathology review)* | 40 | 40 | 104 | 150 |
| DLBCL NOS | 40 (100) | 38 (95) | 98 (94) | 142 (95) |
| ABC | 20 (50) | 19 (48) | 50 (48) | 73 (49) |
| GCB | 17 (43) | 15 (38) | 42 (40) | 58 (39) |
| Follicular lymphoma | 0 | 1 (3) | 0 | 1 (1) |
| Burkitt lymphoma | 0 | 1 (3) | 0 | 1 (1) |
| T-cell/histiocyte-rich large B-cell lymphoma | 0 | 0 | 1 (1) | 1 (1) |
| High-grade B-cell lymphoma with MYC and BCL2, and/or BCL6 rearrangements (DLBCL morphology) | 0 | 0 | 5 (5) | 5 (3) |
| Prior SCT | 6 (15) | 10 (25) | 17 (16) | 27 (18) |
| Prior CAR T-cell therapy | 0 | 0 | 1 (1) | 1 (1) |
| DOR of last treatment ≤12 mo | 34 (85) | 32 (80) | 92 (87) | 129 (85) |
| Median (range) time from last treatment, mo | 2.7 (1-97) | 4.3 (1-386) | 3.2 (1-232) | 3.4 (1-386) |
| Primary refractory† | 28 (70) | 21 (53) | 73 (69) | 97 (64) |
| Refractory to last prior therapy‡ | 33 (83) | 30 (75) | 81 (76) | 116 (76) |
| ITT | Randomized | Extension cohort, pola + BR (n = 106) | Pooled, pola + BR (N = 152) | |
|---|---|---|---|---|
| BR (n = 40) | pola + BR (n = 40) | |||
| Median (range) age, y | 71 (30-84) | 67 (33-86) | 70 (24-94) | 69 (24-94) |
| Age ≥65 y | 26 (65) | 23 (58) | 77 (73) | 103 (68) |
| Male sex | 25 (63) | 28 (70) | 52 (49) | 84 (55) |
| ECOG PS score | ||||
| 0 | 17 (43) | 12 (30) | 30 (28) | 44 (29) |
| 1 | 14 (35) | 21 (53) | 62 (59) | 87 (57) |
| 2 | 8 (20) | 6 (15) | 14 (13) | 20 (13) |
| Ann Arbor stage III/IV | 36 (90) | 34 (85) | 84 (79) | 122 (80) |
| Bulky disease | 14 (35.0) | 10 (25) | 28 (26) | 39 (26) |
| IPI score 3-5 at enrollment | 29 (73) | 22 (55) | 70 (66) | 94 (62) |
| Median no. of prior therapies (range) | 2 (1-5) | 2 (1-7) | 2 (1-7) | 2 (1-7) |
| 1 line | 12 (30) | 11 (28) | 37 (35) | 50 (33) |
| 2 lines | 9 (23) | 11 (28) | 27 (26) | 42 (28) |
| ≥3 lines | 19 (48) | 18 (45) | 42 (40) | 60 (39) |
| WHO 2016 classification (central pathology review)* | 40 | 40 | 104 | 150 |
| DLBCL NOS | 40 (100) | 38 (95) | 98 (94) | 142 (95) |
| ABC | 20 (50) | 19 (48) | 50 (48) | 73 (49) |
| GCB | 17 (43) | 15 (38) | 42 (40) | 58 (39) |
| Follicular lymphoma | 0 | 1 (3) | 0 | 1 (1) |
| Burkitt lymphoma | 0 | 1 (3) | 0 | 1 (1) |
| T-cell/histiocyte-rich large B-cell lymphoma | 0 | 0 | 1 (1) | 1 (1) |
| High-grade B-cell lymphoma with MYC and BCL2, and/or BCL6 rearrangements (DLBCL morphology) | 0 | 0 | 5 (5) | 5 (3) |
| Prior SCT | 6 (15) | 10 (25) | 17 (16) | 27 (18) |
| Prior CAR T-cell therapy | 0 | 0 | 1 (1) | 1 (1) |
| DOR of last treatment ≤12 mo | 34 (85) | 32 (80) | 92 (87) | 129 (85) |
| Median (range) time from last treatment, mo | 2.7 (1-97) | 4.3 (1-386) | 3.2 (1-232) | 3.4 (1-386) |
| Primary refractory† | 28 (70) | 21 (53) | 73 (69) | 97 (64) |
| Refractory to last prior therapy‡ | 33 (83) | 30 (75) | 81 (76) | 116 (76) |
Data are presented as n (%) unless otherwise specified. Pooled pola + BR cohort includes patients from the phase 1b safety run-in (n = 6), phase 2 randomized arm (n = 40), and phase 2 extension cohort (n = 106). ABC, activated B-cell; ECOG PS, Eastern Cooperative Oncology Group performance status; GCB, germinal center B-cell; IPI, International Prognostic Index; ITT, intention-to-treat; NOS, not otherwise specified; WHO, World Health Organization.
Initial diagnosis unknown for some patients.
Defined as no response or progression or relapse within 6 months of first antilymphoma therapy end date.
Defined as no response or progression or relapse within 6 months of last antilymphoma therapy end date.
Efficacy
Randomized cohorts.
Updated efficacy data from the randomized cohort (pola + BR vs BR) are shown in Table 2. With an additional 27 months of follow-up in the randomized pola + BR arm was 62.5% vs 25.0%; best CR rate was 52.5% vs 22.5%, respectively. The median IRC-assessed DOR (95% CI) was 10.9 months (5.7-40.7) with pola + BR vs 10.6 months (4.0-19.6) with BR (HR, 0.60; 95% CI, 0.25-1.43; P = .25); the median investigator-assessed DOR was 12.7 vs 4.1 months (HR, 0.42; 95% CI, 0.19-0.91; P = .02) with pola + BR vs BR.
Summary of efficacy outcomes
| Outcome | Randomized cohorts | Extension cohort, pola + BR (n = 106) | |
|---|---|---|---|
| BR (n = 40) | pola + BR (n = 40) | ||
| EOT response, n (%) | |||
| IRC-assessed ORR | 7 (17.5) | 17 (42.5) | 44 (41.5) |
| CR | 7 (17.5) | 17 (42.5) | 41 (38.7) |
| PR | 0 | 0 | 3 (2.8) |
| SD | 1 (2.5) | 6 (15.0) | 4 (3.8) |
| PD | 6 (15.0) | 8 (20.0) | 19 (17.9) |
| Missing/NE | 26 (65.0) | 9 (22.5) | 39 (36.8) |
| INV-assessed ORR | 7 (17.5) | 19 (47.5) | 45 (42.5) |
| CR | 6 (15.0) | 17 (42.5) | 39 (36.8) |
| PR | 1 (2.5) | 2 (5.0) | 6 (5.7) |
| SD | 0 | 1 (2.5) | 1 (0.9) |
| PD | 27 (67.5) | 13 (32.5) | 40 (37.7) |
| Missing/NE | 6 (15.0) | 7 (17.5) | 20 (18.9) |
| Best responses, n (%) | |||
| ORR (IRC) | 10 (25.0) | 25 (62.5) | 60 (56.6) |
| CR | 9 (22.5) | 21 (52.5) | 56 (52.8) |
| PR | 1 (2.5) | 4 (10.0) | 4 (3.8) |
| SD | 9 (22.5) | 5 (12.5) | 17 (16.0) |
| PD | 8 (20.0) | 6 (15.0) | 17 (16.0) |
| Missing/NE | 13 (32.5) | 4 (10.0) | 12 (11.3) |
| ORR (INV) | 13 (32.5) | 28 (70.0) | 66 (62.3) |
| CR | 8 (20.0) | 23 (57.5) | 53 (50.0) |
| PR | 5 (12.5) | 5 (12.5) | 13 (12.3) |
| SD | 2 (5.0) | 1 (2.5) | 7 (6.6) |
| PD | 22 (55.0) | 7 (17.5) | 29 (27.4) |
| Missing/NE | 3 (7.5) | 4 (10.0) | 4 (3.8) |
| Median DOR, mo (95% CI) | |||
| IRC-assessed | 10.6 (4.0-19.6) | 10.9 (5.7-40.7) | 9.5 (7.9-12.1) |
| INV-assessed | 4.1 (2.6-12.7) | 12.7 (5.8-27.9) | 8.7 (5.9-12.1) |
| Median PFS, mo (95% CI) | |||
| IRC-assessed | 3.7 (2.1-4.5) | 9.2 (6.0-13.9) | 6.6 (5.1-9.2) |
| INV-assessed | 2.0 (1.5-3.7) | 7.5 (4.9-17.0) | 5.9 (4.8-7.5) |
| 24-mo PFS probability, % (95% CI) | |||
| IRC-assessed | 9.1 (0.0-18.9) | 28.4 (13.8-43.0) | — |
| Median OS, months (95% CI) | 4.7 (3.7-8.3) | 12.4 (9.0-32.0) | 12.5 (8.3-23.1) |
| Outcome | Randomized cohorts | Extension cohort, pola + BR (n = 106) | |
|---|---|---|---|
| BR (n = 40) | pola + BR (n = 40) | ||
| EOT response, n (%) | |||
| IRC-assessed ORR | 7 (17.5) | 17 (42.5) | 44 (41.5) |
| CR | 7 (17.5) | 17 (42.5) | 41 (38.7) |
| PR | 0 | 0 | 3 (2.8) |
| SD | 1 (2.5) | 6 (15.0) | 4 (3.8) |
| PD | 6 (15.0) | 8 (20.0) | 19 (17.9) |
| Missing/NE | 26 (65.0) | 9 (22.5) | 39 (36.8) |
| INV-assessed ORR | 7 (17.5) | 19 (47.5) | 45 (42.5) |
| CR | 6 (15.0) | 17 (42.5) | 39 (36.8) |
| PR | 1 (2.5) | 2 (5.0) | 6 (5.7) |
| SD | 0 | 1 (2.5) | 1 (0.9) |
| PD | 27 (67.5) | 13 (32.5) | 40 (37.7) |
| Missing/NE | 6 (15.0) | 7 (17.5) | 20 (18.9) |
| Best responses, n (%) | |||
| ORR (IRC) | 10 (25.0) | 25 (62.5) | 60 (56.6) |
| CR | 9 (22.5) | 21 (52.5) | 56 (52.8) |
| PR | 1 (2.5) | 4 (10.0) | 4 (3.8) |
| SD | 9 (22.5) | 5 (12.5) | 17 (16.0) |
| PD | 8 (20.0) | 6 (15.0) | 17 (16.0) |
| Missing/NE | 13 (32.5) | 4 (10.0) | 12 (11.3) |
| ORR (INV) | 13 (32.5) | 28 (70.0) | 66 (62.3) |
| CR | 8 (20.0) | 23 (57.5) | 53 (50.0) |
| PR | 5 (12.5) | 5 (12.5) | 13 (12.3) |
| SD | 2 (5.0) | 1 (2.5) | 7 (6.6) |
| PD | 22 (55.0) | 7 (17.5) | 29 (27.4) |
| Missing/NE | 3 (7.5) | 4 (10.0) | 4 (3.8) |
| Median DOR, mo (95% CI) | |||
| IRC-assessed | 10.6 (4.0-19.6) | 10.9 (5.7-40.7) | 9.5 (7.9-12.1) |
| INV-assessed | 4.1 (2.6-12.7) | 12.7 (5.8-27.9) | 8.7 (5.9-12.1) |
| Median PFS, mo (95% CI) | |||
| IRC-assessed | 3.7 (2.1-4.5) | 9.2 (6.0-13.9) | 6.6 (5.1-9.2) |
| INV-assessed | 2.0 (1.5-3.7) | 7.5 (4.9-17.0) | 5.9 (4.8-7.5) |
| 24-mo PFS probability, % (95% CI) | |||
| IRC-assessed | 9.1 (0.0-18.9) | 28.4 (13.8-43.0) | — |
| Median OS, months (95% CI) | 4.7 (3.7-8.3) | 12.4 (9.0-32.0) | 12.5 (8.3-23.1) |
BR, bendamustine-rituximab; CI, confidence interval; CR, complete response; DOR, duration of response; EOT, end of treatment; INV, investigator; IRC, Independent Review Committee; NE, not evaluable; ORR, objective response rate; OS, overall survival; PD, progressive disease; PFS, progression-free survival; pola, polatuzumab vedotin; PR, partial response; SD, stable disease.
The median IRC-assessed PFS (95% CI) was 9.2 months (6.0-13.9) with pola + BR vs 3.7 months (2.1-4.5) with BR (HR, 0.39; 95% CI, 0.23-0.66; P < .0003) (Figure 2); median investigator-assessed PFS was 7.5 vs 2.0 months (HR, 0.33; 95% CI, 0.20-0.56; P < .0001) with pola + BR and BR, respectively (supplemental Figure 1). Median OS (95% CI) was 12.4 months (9.0-32.0) vs 4.7 months (3.7-8.3) with pola + BR vs BR (HR, 0.42; 95% CI, 0.24-0.72; P = .001). The 24-month OS probability (95% CI) was 38% (22.5-53.9) with pola + BR vs 17.0% (3.6-30.4) with BR. The 24-month PFS probability (95% CI) was 28.4% (13.9-43.0) with pola + BR vs 9.1% (0-18.9) with BR. Event-free survival in the randomized cohort is reported in supplemental Table 1.
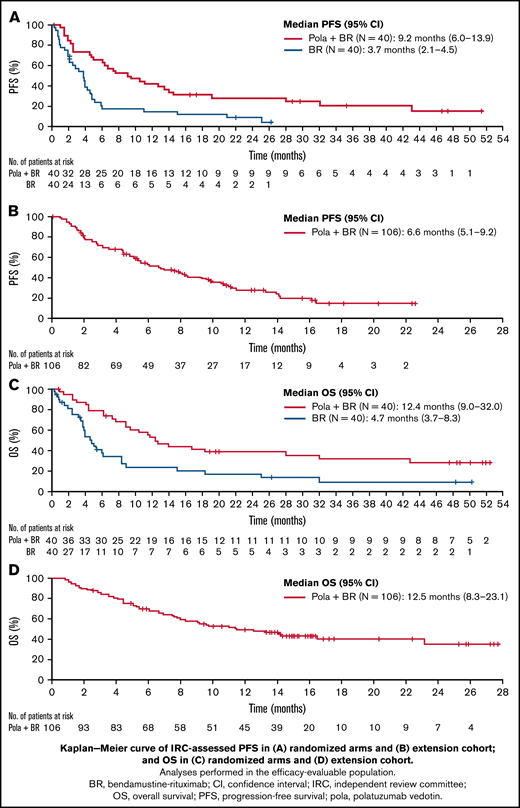
Kaplan-Meier curves of survival. IRC-assessed PFS in randomized arms (A), IRC-assessed PFS in the extension cohort (B), OS in randomized arms (C), OS in the extension cohort (D). Analyses performed in the efficacy-evaluable population.
Kaplan-Meier curves of survival. IRC-assessed PFS in randomized arms (A), IRC-assessed PFS in the extension cohort (B), OS in randomized arms (C), OS in the extension cohort (D). Analyses performed in the efficacy-evaluable population.
Extension cohort.
Following enrollment of the randomized trial, 106 patients with R/R DLBCL were enrolled into the extension cohort and received pola + BR. The IRC-assessed ORR at EOT was 41.5% and the IRC-assessed PET-CT CR rate at EOT was 38.7% (n = 41; 95% CI, 29.4-48.6) (Table 2). The BOR rate was 56.6%, and the best CR rate was 52.8%. The median (95% CI) DOR was 9.5 months (7.9-12.1) by IRC assessment and 8.7 months (5.9-12.1) by investigator assessment. The median (95% CI) IRC-assessed PFS was 6.6 months (5.1-9.2); PFS by investigator assessment was similar (Table 2; supplemental Figure 1). Median OS was 12.5 months (95% CI, 8.2-23.1) (Figure 2); the 12-month OS probability was 50.2% (95% CI, 40.4-60.1).
Subgroup analysis.
An exploratory analysis of BOR rates and survival of patients in the pooled pola + BR cohorts (all patients enrolled to receive pola + BR in the safety run-in, randomized, and extension cohorts [N = 152]) according to line of therapy and refractory status is shown in Figure 3 and supplemental Table 2. Patients derived benefit from pola + BR, regardless of the number of previous lines of therapy received or refractory status. Higher response rates, and longer median PFS and OS, were observed in patients who received pola + BR in earlier lines of treatment compared with later lines, and in patients who were not refractory to prior therapies compared with those who were refractory. Strong efficacy benefit was seen in patients without primary refractory disease and in patients receiving pola + BR as second-line therapy, with a median OS of 32.0 months and 18.4 months, respectively. Aside from these subgroups, there were no trends in baseline characteristics to identify patients who would likely achieve longer survival.
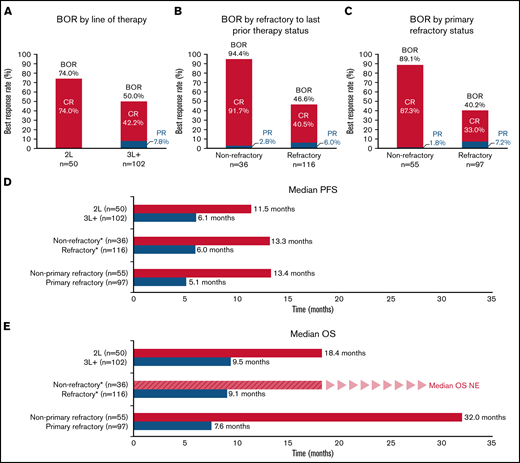
Subgroup analyses. IRC-assessed BOR (A-C), IRC-assessed PFS (D), OS (E). *Refractory to last prior treatment. BOR, best objective response; CR, complete response; NE, not evaluable; OS, overall survival; PFS, progression-free survival; PR, partial response; 2L, patients had received one prior line of therapy before treatment with Pola + BR; 3L+ patients had received two or more prior lines of therapy before treatment with Pola + BR.
Subgroup analyses. IRC-assessed BOR (A-C), IRC-assessed PFS (D), OS (E). *Refractory to last prior treatment. BOR, best objective response; CR, complete response; NE, not evaluable; OS, overall survival; PFS, progression-free survival; PR, partial response; 2L, patients had received one prior line of therapy before treatment with Pola + BR; 3L+ patients had received two or more prior lines of therapy before treatment with Pola + BR.
Safety
Safety results comparing the randomized arms (pola + BR vs BR in patients with R/R DLBCL) were previously reported.17 Briefly, in the randomized pola + BR arm, the most frequent all-grade AEs were anemia (53.8%), neutropenia (53.8%), and thrombocytopenia (48.7%); the most common grade 3 to 4 AEs were neutropenia (46.2%), thrombocytopenia (41.0%), and anemia (28.2%). No new safety signals emerged with longer follow-up.
Here, we report safety in all evaluable patients from the pooled pola + BR cohort (ie, those who received pola + BR in the safety run-in, randomized, and extension cohorts [N = 151]). The median duration of treatment exposure was 3.2 months (range, 0-7 months; 25th-75th percentile range, 1.4-3.7); the median number of treatment cycles received was 5. The median dose intensity for bendamustine (adjusted for dose modifications and delays) was 95.5% (range, 7-107) in the pooled pola + BR arm (n = 151); in the randomized phase 2 cohort, median dose intensity for bendamustine in the BR arm (n = 39) was 95.6% (range, 64-103). Overall, in the pooled pola + BR cohort, 80 (53.0%) patients discontinued treatment with polatuzumab vedotin early; the most common reason for treatment discontinuation was progressive disease (PD; n = 40 [26.5%]). Bendamustine was discontinued in 54.3% of patients, and rituximab was discontinued in 53.0% of patients in the pooled pola + BR cohort; PD was also the most common reason for bendamustine (26.5%) and rituximab (26.5%) treatment discontinuation.
In the pooled pola + BR cohort, at least one AE of any grade was reported in 150 (99.3%) patients; 122 (80.8%) patients had at least one grade 3 to 4 AE. Compared with AEs previously reported for the randomized pola + BR arm, similar frequencies were observed in the pooled cohort of patients treated with pola + BR, in which the most common any-grade AEs were neutropenia (n = 56 [37.1%]), diarrhea (n = 54 [35.8%]), nausea (n = 50 [33.1%]), anemia (n = 49 [32.5%]), and thrombocytopenia (n = 40 [26.5%]); the most common grade 3 to 4 AEs were neutropenia (n = 49 [32.5%]), infections and infestations (n = 33 [21.9%]), thrombocytopenia (n = 31 [20.5%]), and anemia (n = 19 [12.6%]) (Table 3). Study treatment was discontinued in 31 (20.5%) patients in the pooled pola + BR group due to AEs, with the most common reasons including thrombocytopenia, neutropenia, PN, and sepsis. In the randomized BR arm, study treatment was discontinued in 5 (12.8%) patients, with discontinuations mainly due to neutropenia and thrombocytopenia.
AEs according to treatment arm and in all patients treated with pola + BR
| AE | Randomized, BR (n = 39) | Pooled, pola + BR* (N = 151) | ||
|---|---|---|---|---|
| All grade | Grade 3-4 | All grade | Grade 3-4 | |
| Neutropenia | 15 (38.5) | 13 (33.3) | 56 (37.1) | 49 (32.5) |
| Thrombocytopenia | 12 (30.8) | 9 (23.1) | 49 (32.5) | 31 (20.5) |
| Anemia | 10 (25.6) | 7 (17.9) | 49 (32.5) | 19 (12.6) |
| Infections and infestations† | 20 (51.3) | 8 (20.5) | 74 (49.0) | 33 (21.9) |
| Diarrhea | 11 (28.2) | 2 (5.1) | 54 (35.8) | 6 (4.0) |
| Nausea | 16 (41.0) | 0 | 50 (33.1) | 1 (0.7) |
| Pyrexia | 9 (23.1) | 0 | 44 (29.1) | 2 (1.3) |
| Fatigue | 14 (35.9) | 1 (2.6) | 40 (26.5) | 3 (2.0) |
| Decreased appetite | 8 (20.5) | 0 | 39 (25.8) | 4 (2.6) |
| PN‡ | 3 (7.7) | 0 | 47 (31.1) | 3 (2.0) |
| AE | Randomized, BR (n = 39) | Pooled, pola + BR* (N = 151) | ||
|---|---|---|---|---|
| All grade | Grade 3-4 | All grade | Grade 3-4 | |
| Neutropenia | 15 (38.5) | 13 (33.3) | 56 (37.1) | 49 (32.5) |
| Thrombocytopenia | 12 (30.8) | 9 (23.1) | 49 (32.5) | 31 (20.5) |
| Anemia | 10 (25.6) | 7 (17.9) | 49 (32.5) | 19 (12.6) |
| Infections and infestations† | 20 (51.3) | 8 (20.5) | 74 (49.0) | 33 (21.9) |
| Diarrhea | 11 (28.2) | 2 (5.1) | 54 (35.8) | 6 (4.0) |
| Nausea | 16 (41.0) | 0 | 50 (33.1) | 1 (0.7) |
| Pyrexia | 9 (23.1) | 0 | 44 (29.1) | 2 (1.3) |
| Fatigue | 14 (35.9) | 1 (2.6) | 40 (26.5) | 3 (2.0) |
| Decreased appetite | 8 (20.5) | 0 | 39 (25.8) | 4 (2.6) |
| PN‡ | 3 (7.7) | 0 | 47 (31.1) | 3 (2.0) |
Data are presented as n (%). Shown are all-grade AEs occurring in ≥20% of patients and grade 3 to 4 AEs in ≥10% of patients (safety-evaluable population).
Includes all patients with DLBCL who received at least 1 dose of pola + BR.
System organ class grouped term.
Includes PN, peripheral sensory neuropathy, muscular weakness, paresthesia, muscle atrophy, hypoesthesia, gait disturbance, decreased vibratory sense, hypotonia, and neuralgia.
In the pooled pola + BR cohort, serious AEs were reported in 86 (57.0%) patients. The most common serious AEs (≥5% of patients) were febrile neutropenia (9.9%), sepsis (9.9%), pneumonia (9.3%), and pyrexia (8.6%). Fatal AEs occurred in 17 (11.3%) of 151 patients in the pooled pola + BR cohort and in 10 (25.6%) of 39 patients in the randomized BR arm (supplemental Material).
PN events of any grade occurred in 47 (31.1%) patients in the pooled pola + BR cohort and were reversible in the majority of cases. Three patients experienced grade 3 PN, two of which were reported as muscular weakness and resolved within 2 weeks; there was 1 death due to PD before PN resolution. Secondary malignancies were reported in 4 patients and included large granular lymphocytic leukemia, malignant melanoma, and prostate cancer; 1 patient had both squamous cell carcinoma and myelodysplastic syndrome.
Subsequent antilymphoma therapies
Of all patients treated with pola + BR in the study (including the extension cohort), 4 patients proceeded to receive consolidative SCT (autologous [n = 1] or allogeneic [n = 3]). Nine patients received CAR T-cell therapy after pola + BR, including 1 patient who discontinued pola + BR after 3 cycles to bridge to CAR T-cell therapy. For patients treated with CAR T-cell therapy after pola + BR, OS after treatment with pola + BR ranged from 11.5 to 28.0 months; 4 patients are alive and remain in follow-up.
Discussion
R/R DLBCL continues to be an area of high unmet clinical need, despite recent advances and approved therapies. More effective, less toxic, and broadly available therapies for R/R DLBCL are of paramount importance, particularly for transplant-ineligible patients. In the initial results of the GO29365 study, pola + BR showed a significant survival benefit vs BR in the randomized phase 2 comparison, leading to regulatory approvals.5,6 Here, we report long-term results of this randomized cohort with >4 years of follow-up, as well as new data from a single-arm extension cohort of patients receiving pola + BR (n = 106). Response rates observed in the single-arm extension cohort were highly consistent with the response data reported for the randomized cohort, confirming the efficacy of the pola + BR combination in a larger population of patients with R/R DLBCL, the majority of whom were refractory. Overall, these results reinforce the clinical benefit of pola + BR, indicating durable disease control in a substantial proportion of patients. In keeping with the original report,17 this combination has a manageable safety profile, with no new safety signals identified.
With a median of 48 months of follow-up, the significant improvement in PFS and OS persists for patients randomized to receive pola + BR vs BR alone. Ten patients (25%) from the randomized pola + BR cohort had an ongoing DOR of >2 years (range, 26-49 months) and a PFS of >2 years (range, 28-51 months). Eleven (28%) of the 40 patients randomized to receive pola + BR experienced OS beyond 2 years (range, 28-53 months); of these, 2 received consolidative therapy with either an allogenic SCT (n = 1) or CAR T-cell therapy (n = 1). The Kaplan–Meier curves of PFS and OS tended to plateau beyond 2 years; however, this observation should be interpreted with caution as there were relatively few patients at the tail end of the curve. There were no clear trends in baseline patient characteristics for the long-term survivors in the randomized cohort; patients ranged in age from 56 to 86 years, and patients who were refractory to previous treatments were also included, confirming that various types of patients can benefit from the combination of pola + BR.
Overall, the baseline characteristics in the extension cohort were similar to those of the randomized pola + BR cohort, with some exceptions. Although the median age was similar between the 2 groups, the proportion of patients aged ≥65 years was 73% in the extension cohort compared with 58% in the randomized pola + BR cohort. Although the majority of patients in the randomized arm and extension cohort had lymphoma that was refractory to the last treatment, the extension cohort included more patients with primary refractory lymphomas (69%) compared with the pola + BR randomized group (53%). One limitation of the study was that patients with transformed follicular lymphoma were excluded, although in the immunochemotherapy era, outcomes have been shown to be similar for these patients.22 The ongoing POLARGO (Randomized Phase III Study of Polatuzumab Vedotin Plus Rituximab, Gemcitabine, and Oxaliplatin in Relapsed/Refractory Diffuse Large B-Cell Lymphoma; #NCT04182204) study of pola-R-GemOx vs R-GemOx in R/R DLBCL does not exclude patients with transformed follicular lymphoma, and it may therefore show the utility of polatuzumab vedotin in this setting.
Response rates in the extension cohort were highly consistent with those observed in the randomized pola + BR arm. It is noteworthy that most patients whose lymphomas responded to pola + BR achieved a CR (extension cohort ORR, 42%; CR rate, 39%). The median DORs in the extension cohort and randomized pola + BR arm were comparable (9.5 and 10.9 months, respectively). Differences between investigator- and IRC-assessed DOR in the randomized cohort were noted and considered to be due to the small number of responders in the BR arm and expected variations in the IRC assessments. Median PFS of the extension cohort (6.6 months [95% CI, 5.1-9.2]) appeared shorter than in the randomized pola + BR arm (9.2 months [95% CI, 6.0-13.9]) but may reflect differences in patient population, along with the shorter duration of follow-up in the extension cohort. Importantly, median OS in the extension cohort (12.5 months [95% CI, 8.3-23.1]) was comparable to that observed in the randomized pola + BR arm (12.4 months [95% CI, 9.0-32.0]).
A pooled cohort of all patients assigned to treatment with pola + BR in GO29365 (N = 152) enabled further subgroup analyses exploring predictors of outcome. All subgroups examined appeared to derive benefit from polatuzumab vedotin; however, those with the best survival outcomes had received fewer lines of prior treatment and were not considered refractory to their last line of treatment, supporting the idea that baseline characteristics can have an impact on the clinical outcome for some patient subgroups. The greatest benefit was observed in patients who received pola + BR as second-line treatment, those who were not primary refractory, and those who were not refractory to their last prior therapy; best CR rates were 74%, 89%, and 92% in these subgroups, respectively.
Pola + BR has a well-characterized and tolerable safety profile. No new safety signals were identified with longer follow-up of patients in the randomized cohort, or in this full safety analysis of all patients treated with pola + BR in the GO29365 study (N = 151). The most common grade 3 to 4 AEs in the pooled pola + BR cohort were neutropenia (33%), infections and infestations (22%), and thrombocytopenia (21%). PN was experienced by 31% of patients, with most events being grade 1 to 2; very few patients (n = 3) experienced grade 3 PN, and this was mostly improved or resolved over time. Pola + BR is a time-limited therapy; most patients in this high-risk group who have an enduring unmet medical need (eg, patients ineligible for consolidative SCT due to age, comorbidity, or heavy pretreatment) were able to tolerate this regimen.
Other approved treatments have shown efficacy in R/R DLBCL, including CAR T-cell therapies, which are available in the third-line setting and beyond. However, not all patients are suitable for treatment with CAR T-cell therapy, due in part to a lack of effective bridging therapy; in addition, some data suggest that most patients who receive CAR T-cell therapy will eventually experience PD.23,24 CAR T-cell therapy is associated with distinct toxic effects and requires specialized care, whereas the combination of pola + BR is readily available and can be delivered to a wide population of patients, and it may therefore be a suitable option for patients unable to receive CAR T-cell therapy. Nine patients in this trial successfully received CAR T-cell treatment after pola + BR, highlighting this possible option in the sequence of treatment regimens for R/R DLBCL. Polatuzumab vedotin may also be an effective treatment to use as a bridge to CAR T-cell therapy; however, this area is evolving, and the use of bendamustine before apheresis needs to be further assessed due to the risk of lymphodepletion.
The novel treatment combination tafasitamab-lenalidomide has also been recently approved for transplant-ineligible patients with R/R DLBCL.7,25 An observed CR rate of 40% and a median DOR and PFS of 43.9 and 11.6 months, respectively, were reported in the single-arm L-MIND (Tafasitamab Plus Lenalidomide in Relapsed or Refractory Diffuse Large B-Cell Lymphoma) study, with >35 months of follow-up.26 Again, it is not possible to directly compare these results vs those with pola + BR, as patients who had received >3 prior therapies were excluded from L-MIND, and the study largely excluded patients who were primary refractory. In addition, the tafasitamab-lenalidomide regimen is administered until disease progression, rather than for a fixed period. Median OS was similar between the non-primary refractory subgroup of patients in the pooled pola + BR cohort of GO29365 (32.0 months) and patients treated with tafasitamab-lenalidomide in L-MIND (33.5 months).27
In conclusion, longer follow-up of the randomized cohorts and new data from an additional 106 patients treated with pola + BR further support the initial results from the GO29365 study. Patient-level pooled retrospective analyses from the SCHOLAR-1 (Retrospective Non-Hodgkin Lymphoma Research) study revealed that outcomes for patients with refractory retrospective DLBCL research were consistently poor, with a CR rate of 7% and a median OS of 6.3 months.24 Given the encouraging CR rate and significant durable disease control reported here in hard-to-treat patients with R/R DLBCL, in which the vast majority are primary refractory or refractory to their last prior therapy, pola + BR is an effective treatment option with a well-characterized and manageable safety profile. As the treatment landscape for patients with R/R DLBCL continues to evolve, pola + BR has emerged as a desirable therapeutic option that may have the potential to serve as a second-line treatment as a bridge to autologous SCT or CAR T-cell therapy. Ongoing development includes clinical trials of polatuzumab vedotin in the first-line setting in combination with chemoimmunotherapy (POLARIX [A Study Comparing the Efficacy and Safety of Polatuzumab Vedotin With Rituximab-Cyclophosphamide, Doxorubicin, and Prednisone (R-CHP) Versus Rituximab-Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone (R-CHOP) in Patients with Previously Untreated DLBCL], phase 3, #NCT03274492); in the R/R setting in combination with R-GemOx (POLARGO, phase 3, #NCT04182204); and with rituximab, ifosfamide, carboplatin, and etoposide (Pola-R-ICE [A Study of Polatuzumab Vedotin With Rituximab, Ifosfamide, Carboplatin, and Etoposide in patients with relapsed/refractory DLBCL], phase 2, #NCT04665765).
Acknowledgments
The authors thank the participating patients and their families, and the research nurses, study coordinators, and operations staff. Third-party medical writing assistance, under the direction of the authors, was provided by Lucinda Sinclair of Ashfield MedComms, an Ashfield Health company, and was funded by F. Hoffmann–La Roche Ltd.
The study was designed with input from investigators and was sponsored by Genentech, Inc. and F. Hoffmann–La Roche Ltd.
Authorship
Contribution: J.H., L.M., G.K., and L.H.S. designed the clinical study and interpreted clinical data; Y.M.C. contributed to the statistical analysis; and L.M. and L.H.S. wrote the manuscript with input and approval of the final version from all coauthors; and all authors collected and analyzed the data, reviewed the data, provided critical review of the manuscript, confirmed the completeness and accuracy of the results and the trial’s fidelity to the protocol, and agreed on its submission for publication.
Conflict-of-interest disclosure: L.H.S. reports honoraria/consulting fees from AbbVie, Acerta, Amgen, Apobiologix, AstraZeneca, Celgene, Chugai, Gilead, Incyte, Janssen, Kite, Karyopharm, Lundbeck, Merck Sharp & Dohme, MorphoSys, F. Hoffmann–La Roche Ltd./Genentech, Inc., Sandoz, Seattle Genetics, Servier, Teva, Takeda, TG Therapeutics, and Verastem; and research funding from F. Hoffmann–La Roche Ltd./Genentech Inc. and Teva. M.H. reports honoraria/consulting fees from Bristol Myers Squibb, F. Hoffmann–La Roche Ltd., Gilead, Merck Sharp & Dohme, and Takeda. S.O. reports honoraria and consultancy/advisory fees from AbbVie, AstraZeneca, F. Hoffmann–La Roche Ltd., Gilead, Janssen, Merck Sharp & Dohme, and Novartis; and research funding from Amgen, AstraZeneca, BeiGene, Epizyme, F. Hoffmann–La Roche Ltd., Janssen, and Merck Sharp & Dohme. A.F.H. reports consulting fees from AstraZeneca, Bristol Myers Squibb, Genentech, Inc., Karyopharm, Merck Sharp & Dohme, Seattle Genetics, Takeda, and Tubulis; and research funding from ADC Therapeutics, AstraZeneca, Bristol Myers Squibb, Genentech, Inc., Gilead, Merck Sharp & Dohme, and Seattle Genetics. S.A. reports consulting fees from Bristol Myers Squibb, F. Hoffmann–La Roche Ltd., Novartis, and Pfizer. C.R.F. reports consulting fees from AbbVie, Bayer, BeiGene, Celgene, Denovo Biopharma, F. Hoffmann–La Roche Ltd./Genentech, Inc., Gilead, Karyopharm, OptumRx, Pharmacyclics/Janssen, and Spectrum; and research funding from AbbVie, Acerta, Burroughs Wellcome Fund, Celgene, Eastern Cooperative Oncology Group, F. Hoffmann–La Roche Ltd./Genentech, Inc., Gilead, Janssen, Millennium/Takeda, National Cancer Institute, Pharmacyclics, TG Therapeutics, and V Foundation. T.M.K. reports consulting fees from AstraZeneca, Boryung, F. Hoffmann–La Roche Ltd./Genentech, Inc., Hanmi, Novartis, Sanofi, and Takeda; and research funding from AstraZeneca-KHIDI outside this work. A.M. reports honoraria, speakers bureau, and travel expenses from Celgene and F. Hoffmann–La Roche Ltd.; and research funding from Pfizer. M.O. reports honoraria from Takeda; research funding from AbbVie, Archigen, Bayer, Celgene, F. Hoffmann–La Roche Ltd., Janssen, Merck Sharp & Dohme, and Takeda; and travel support from Abdi İbrahim, F. Hoffmann–La Roche Ltd., JAZZ, and Takeda. V.S. reports honoraria from F. Hoffmann–La Roche Ltd.; and consulting fees from Novartis. G.S. reports advisory boards/consulting fees from AbbVie, BeiGene, Bristol Myers Squibb/Celgene, Debiopharm, Epizyme, F. Hoffmann–La Roche Ltd./Genentech, Inc., Genmab, Incyte, Janssen, Kite/Gilead, Miltenyi, MorphoSys, Novartis, Regeneron, and VelosBio. G.K., J.H., and L.M. are employees of F. Hoffmann–La Roche Ltd./Genentech, Inc.; and report ownership interests in F. Hoffmann–La Roche Ltd./Genentech, Inc. Y.M.C. is an employee of and reports ownership interests in F. Hoffmann–La Roche Ltd. M.J.M. reports honoraria from Bayer, F. Hoffmann-La Roche Ltd./Genentech, Inc., GlaxoSmithKline, Immunovaccine Technologies, Janssen, Pharmacyclics, Seattle Genetics, and Takeda; advisory role/consulting fees from Bayer, Daiichi Sankyo, F. Hoffmann–La Roche Ltd., Genentech, Inc., Juno Therapeutics, Merck Sharp & Dohme, Rocket Medical, Seattle Genetics, Takeda, and Teva; research funding from Bayer, F. Hoffmann–La Roche Ltd./Genentech, Inc., GlaxoSmithKline, IGM Biosciences, Immunovaccine Technologies, Janssen, Pharmacyclics, Rocket Medical, and Seattle Genetics; and travel, accommodations, and expenses from Bayer, F. Hoffmann–La Roche Ltd./Genentech, Inc., and Seattle Genetics.
Correspondence: Laurie H. Sehn, BC Cancer Centre for Lymphoid Cancer, 675 West 10th Ave, Vancouver, BC V5Z 1L3, Canada; e-mail: Lsehn@bccancer.bc.ca.

