

Key Points
Infusion of iHPs is safe and well tolerated in NHPs.
iHPs are hypoimmunogenic and can be administered with a low risk of alloimmunization.

Abstract
Administration of ex vivo expanded somatic myeloid progenitors has been explored as a way to facilitate a more rapid myeloid recovery and improve overall survival after myeloablation. Recent advances in induced pluripotent stem cell (iPSC) technologies have created alternative platforms for supplying off-the-shelf immunologically compatible myeloid progenitors, including cellular products derived from major histocompatibility complex (MHC) homozygous superdonors, potentially increasing the availability of MHC-matching cells and maximizing the utility of stem cell banking. However, the teratogenic and tumorigenic potential of iPSC-derived progenitor cells and whether they will induce alloreactive antibodies upon transfer remain unclear. We evaluated the safety and efficacy of using CD34+CD45+ hematopoietic progenitors derived from MHC homozygous iPSCs (iHPs) to treat cytopenia after myeloablative hematopoietic stem cell (HSC) transplantation in a Mauritian cynomolgus macaque (MCM) nonhuman primate (NHP) model. We demonstrated that infusion of iHPs was well tolerated and safe, observing no teratomas or tumors in the MCMs up to 1 year after HSC transplantation and iHP infusion. Importantly, the iHPs also did not induce significant levels of alloantibodies in MHC-matched or -mismatched immunocompetent MCMs, even after increasing MHC expression on iHPs with interferon-γ. These results support the feasibility of iHP use in the setting of myeloablation and suggest that iHP products pose a low risk of inducing alloreactive antibodies.
Introduction
Bone marrow (BM) suppression and associated neutropenia are major complications and limiting factors in radiotherapy and chemotherapy.1 The occurrence of neutropenia after chemotherapy or myeloablative conditioning significantly increases susceptibility to infection, resulting in increased health care costs and hospital resources.2 Despite the development of novel antifungal and antibacterial agents3,4 and supportive strategies, including administration of granulocyte colony-stimulating factor (G-CSF),5 infection remains a major cause of morbidity and mortality after chemotherapy. Since neutrophil recovery was identified as the most reliable factor in predicting favorable outcomes in patients with BM suppression,6 granulocyte transfusions have been used to treat neutropenia. However, the short half-life of granulocytes, the lack of consistency between granulocyte preparations, and the large number of donors required to obtain neutrophils for transient improvement limit the utility of this treatment strategy and may explain the only modest clinical effect reported in a recent clinical trial.7 To overcome these limitations, administration of ex vivo expanded myeloid progenitors has been explored as a way to facilitate a more rapid myeloid recovery and improve overall survival after myeloablation.8-15 Myeloid progenitors produced ex vivo from somatic hematopoietic stem/progenitor cells (HSPCs) have been shown to reduce BM aplasia and fungal and bacterial infections and improve overall survival after BM ablation in animal models.8 , -12,15 Recent studies have also demonstrated that HSPCs improve survival from sepsis in a mouse model.16 The benefits of a progenitor-based strategy are supported by clinical trials demonstrating the safety and feasibility of using non–HLA-matched ex vivo expanded cord blood progenitors as an alternative to granulocyte transfusion in patients with acute myeloid and other leukemias after intensive chemotherapy.17,18 This strategy is also effective at reducing the incidence of infection and antimicrobial use in neutropenic patients receiving myeloablative chemotherapy for acute myeloid leukemia after transfusion of common myeloid progenitors manufactured from G-CSF–mobilized peripheral blood HSPCs.19,20
Recent advances in reprogramming and induced pluripotent stem cell (iPSC) technologies have created alternative platforms for an off-the-shelf supply of immunologically compatible cellular products, including hematopoietic progenitors (HPs). iPSCs offer the possibility of generating autologous blood cells in unlimited numbers and avoiding HLA alloimmunization. However, the high costs of personalized stem cell therapy and its complexity make it currently impractical for broad application. Alternatively, creation of iPSC banks has been proposed to supply HLA-matched blood cells. However, iPSC banking using random donors is impractical because of the high variability of HLA. Generation and banking of iPSC lines from rare individuals who are homozygous at their HLA-A, -B, and -DR loci have been proposed as an alternative approach to circumvent these limitations.21-25 iPSC lines from these major histocompatibility complex (MHC) homozygous superdonors can significantly increase the degree of MHC matching between iPSC-derived cell-replacement products and recipients, thereby reducing alloimmunization and maximizing the utility of stem cell banks.
In the present study, we evaluated the feasibility and safety of using iPSC-derived CD34+ HPs (iHPs) for the treatment of cytopenia after myeloablative HSPC transplantation and assessed the immunogenicity of MHC homozygous iHPs in a preclinical nonhuman primate (NHP) model. This model employs Mauritian cynomolgus macaques (MCMs), which are descended from a small founder population, limiting their MHC diversity to only 7 common haplotypes (M1-M7),26-28 making it possible to rapidly select MHC identical, homozygous, and heterozygous MCMs by genetic screening. Evaluating iPSC-derived blood products in NHPs provides the proof of concept to initiate clinical trials and counterbalances the potential risks to individuals enrolled in clinical trials.
Materials and methods
Experimental animals
In this study, we used a total of 22 female MCMs, housed at the Wisconsin National Primate Research Center (WNPRC). None of the animals was previously pregnant or exposed to allogeneic tissue. All animal experiments were approved by the University of Wisconsin Institutional Animal Care and Use Committee.
MHC and blood group typing
MHC genotyping was performed by Genetics Services at WNPRC, as previously described.29 Molecular genetic blood group typing was performed by VRL Laboratories (San Antonio, TX).
Reprogramming NHP fibroblast cells
We collected skin punch biopsies to harvest skin fibroblasts from homozygous MCMs (M2/M2, M3/M3, and M6/M6) and reprogrammed them with combinations of oriP/EBNA1-based episomal vectors, as previously described.30,31 iPSC colonies that appeared within 3 to 4 weeks were seeded onto mouse embryonic fibroblasts (MEFs) for expansion and characterization. MHC genotypes of generated iPSCs are listed in supplemental Table 1.
Donor CD34+ cell collection
To collect CD34+ cells, we subcutaneously injected the MCMs with G-CSF (filgrastim) at a dose of 100 μg/kg once per day for 3 days. We collected BM aspirates 2 to 11 hours after the last G-CSF injection, as described previously.32 We isolated mononuclear cells from the aspirates by density gradient centrifugation, performed magnetic-activated cell sorting enriched for CD34+ cells using human CD34 antibody clone 563 (BD Biosciences), and cryopreserved the CD34+ cells until further use. CD34+ HSPCs were thawed 2 days before transplantation and cultured overnight in serum-free expansion medium (Stem Cell Technologies) containing 100 ng/mL of human stem cell factor (Peprotech), 100 ng/mL of human FMS-related tyrosine kinase 3 ligand (Peprotech), and 50 ng/mL of human thrombopoietin (Peprotech). The next day, we transduced the CD34+ HSPCs with enhanced green fluorescent protein (eGFP) or tdTomato chimeric lentivirus with SIV capsid33 at a multiplicity of infection of 10 in the presence of 5 μg/mL of rapamycin (Reagents Direct) for 24 hours.34 The packaging plasmid encoding SIV capsid proteins to escape host-mediated restrictions of TRIM5α was provided by Theodora Hatziioannou (Aaron Diamond AIDS Research Center). On the day of transplantation, the cells were washed 3 times with phosphate-buffered saline and resuspended in plasmalyte containing 2% autologous serum and 5 U/mL of heparin.
iPSC tagging with fluorescent markers and differentiation in OP9 coculture
All iPSC lines were maintained in an undifferentiated state in primate embryonic stem cell medium (ReproCell, Yokohama, Japan) by passaging on MEFs every 3 days. iPSCs were tagged with either eGFP or tdTomato using chimeric lentiviruses with SIV capsid.33 eGFP+ or tdTomato+ iPSCs were fluorescence-activated cell sorted and cultured on MEFs. Differentiation of NHP iPSCs to iHPs was performed as described previously.30 Floating blood cells at days 8 to 10 of coculture were collected, evaluated by flow cytometry with antibodies to CD34, CD45, and CD90 (supplemental Table 2), and frozen until use. On day 3 after HSPC transplantation, eGFP- or tdTomato-tagged iHPs were thawed and transduced once again with eGFP or tdTomato lentivirus at a multiplicity of infection of 10 overnight to ensure maximum tagging of cells and avoid transgene silencing after differentiation. On day 4 of transplantation, the cells were washed 3 times with phosphate-buffered saline and resuspended in Plasma-Lyte containing 2% autologous serum and 5 U/mL of heparin. Aliquot of cells used for infusion was resuspended in Methocult GF+H4435 complete colony-forming unit medium (Stem Cell Technologies). Colonies were counted on days 12 to 14 of culture.
Pretransplantation conditioning regimen, monitoring of engraftment, and posttransplantation care
On day −2, the recipient macaques from all 3 groups underwent total-body irradiation at 500 cGy for 2 days, sparing the lungs and eyes, as described previously.32 On the day of transplantation, autologous HSPCs tagged with either eGFP or tdTomato or left unmarked were transplanted into the recipient macaques from all 3 groups IV at a dose of 3 × 106 cells per kg body weight. Ceftriaxone was started 4 days after transplantation and continued until the absolute neutrophil count was >500 cells per μL. Where indicated, we infused iHPs IV at a dose of 30 × 106 per kg body weight on day 4 posttransplantation. After transplantation, peripheral blood was drawn from the recipient macaques twice per week for the first month after iHP infusion, weekly for the second and third months, and monthly for the remainder of the study to monitor blood counts and engraftment. Blood counts were monitored using an SXS-1000i automated hematology analyzer (Sysmex Corporation, Kobe, Japan) with confirmatory blood smear evaluations. BM aspirates were collected from the macaques in groups 2 and 3 at 2, 4, and 8 months post-iHP infusion. Colon and lymph node biopsies were collected at 2 and 8 months post-iHP infusion. Presence of iHPs in peripheral blood and biopsied tissues was monitored by flow cytometry and genomic DNA polymerase chain reaction (PCR) to track eGFP+ or tdTomato+ cells using specific primers for eGFP (forward, 5′-AGCTGTTCACCGGGGTGGT-3′; reverse, 5′-GTCTTGTAGTTGCCGTCGT-3′) and tdTomato (forward, 5′-GGTCATCAAAGAGTTCATGCG-3′; reverse, 5′-GGGAAGGACAGCTTCTTGTA-3′). Primers specific to β-actin (forward, 5′-GCAGGAGATGGCCACGGCGCC-3′; reverse, 5′-TCTCCTTCTGCATCCTGTCGGC-3′) were used as an internal control. Antibodies used for flow cytometry are listed in supplemental Table 2. Recipient macaques were closely monitored for signs of pain or stress. Euthanasia was used in the event of an untreatable infection, in consultation with the WNPRC veterinarians.
Assessment of alloimmune response toward homozygous iHPs
For an assessment of MHC alloimmunization, immunocompetent MCMs were divided into 4 groups and infused with iHPs at 10 × 106/kg derived from wild-type nonmodified iPSCs once per week for 3 weeks (supplemental Table 3). Serum samples were collected 1 week after each immunization and then 2 and 5 months after final immunization. We tested for MHC alloantibodies by flow cytometry using a panel of herpesvirus papio immortalized B lymphoid cells (BLCLs) from MCMs with genotype matching donor MHC.35 Serial dilutions of serum were incubated with BLCLs for 1 hour at 37°C and 5% carbon dioxide. After washing twice with magnetic-activated cell sorting buffer, we detected bound alloantibodies by incubating cells with the rhesus recombinant antirhesus immunoglobulin G1/3 [1B3]-FITC monoclonal antibody, engineered and produced by the National Institutes of Health Nonhuman Primate Reagent Resource (catalog #PR-1233, RRID:AB_ AB_2819288), and Ghost Violet 540 cell viability dye (TONBO Biosciences) for 30 minutes in the dark. Plates were washed twice and then analyzed by flow cytometry using the MACSQuant Analyzer 10 (Miltenyi Biotec) and FlowJo software. Median fluorescence intensity (MFI) was determined for all samples, and normalized MFI (nMFI) was calculated by dividing the MFI of the serum sample postimmunization by the MFI of the serum sample before immunization.35,36 To generate positive control serum, MCMs received PHA-P–stimulated MHC-mismatched PBMCs at 1.85 × 106/kg IV and 5.5 × 106/kg intradermally. A cutoff nMFI of ≥2 was considered positive.35
Results
Derivation of MCM iPSCs and de novo generation of CD34+ iHPs
MCMs with desired MHC homozygous and heterozygous genotypes were selected after genetic MHC and blood type screening. Transgene-free MCM iPSCs were generated by reprogramming skin fibroblasts with EBNA/oriP-based episomal plasmids30,31 (supplemental Table 1). For in vivo tracking, we tagged iPSCs with eGFP or tdTomato via lentiviral transfection. iHPs were generated from MCM iPSCs in coculture with OP9 in the presence of CHIR.30 Multiple rounds of differentiation were performed to collect and freeze CD34+ iHPs until obtaining the desired numbers of cells (Figure 1A). iHPs produced in these cultures have CD34+CD45+CD90+ multipotential HP phenotype and are highly enriched in granulocyte-macrophage, granulocyte, and macrophage colony-forming cells (Figure 1B-D). Additionally, the iHPs expressed MHC class I weakly and were mostly MHC class II− (Figure 1E).
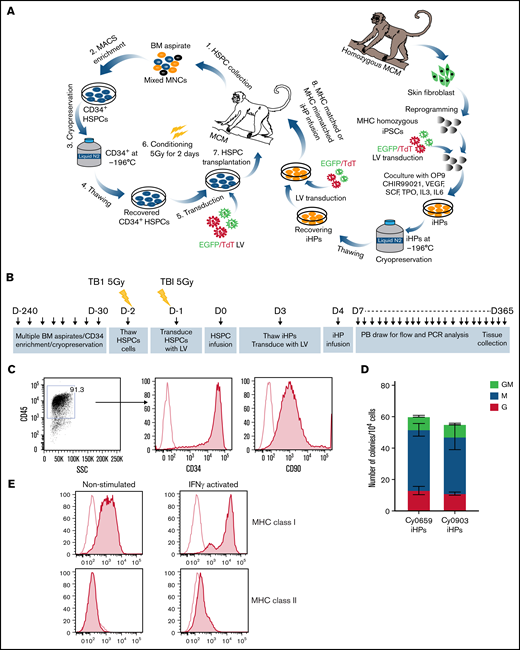
Characterization of iHPs. (A) Schematic diagram of HSPC transplantation and iHP infusion experiments. (B) Timeline for experiments. (C) Representative flow cytometric dot plot and histograms showing expression of CD34 and CD90 (filled histogram) vs unstained control (empty histogram in iHPs; cells are gated on live CD45+ cells). (D) Colony-forming cell (CFC) potential of iHPs. Results are mean ± standard deviation for 3 independent experiments. (E) Representative flow cytometric experiment showing MHC class I and class II expression (filled histograms) relative to unstained controls (empty histogram) in iHPs before and after activation with interferon-γ (IFN-γ). Cells are gated on live CD45+ cells. LV, lentivirus; PB, peripheral blood.
Characterization of iHPs. (A) Schematic diagram of HSPC transplantation and iHP infusion experiments. (B) Timeline for experiments. (C) Representative flow cytometric dot plot and histograms showing expression of CD34 and CD90 (filled histogram) vs unstained control (empty histogram in iHPs; cells are gated on live CD45+ cells). (D) Colony-forming cell (CFC) potential of iHPs. Results are mean ± standard deviation for 3 independent experiments. (E) Representative flow cytometric experiment showing MHC class I and class II expression (filled histograms) relative to unstained controls (empty histogram) in iHPs before and after activation with interferon-γ (IFN-γ). Cells are gated on live CD45+ cells. LV, lentivirus; PB, peripheral blood.
Infusion of iHPs in the setting of autologous myeloablative HSPC transplantation
A schematic diagram of the experimental design is shown in Figure 1A. Three groups of MCMs underwent myeloablative total-body irradiation followed by transplantation of cryopreserved BM autologous CD34+ HSPCs, some of which were tagged with eGFP or tdTomato (Table 1). Control group 1 included animals undergoing autologous HSPC transplantation. Animals in groups 2 and 3 received a single dose of cryopreserved iHPs tagged with eGFP or tdTomato from homozygous MHC-matched iPSCs (group 2) or homozygous MHC-mismatched iPSCs (group 3) 4 days after autologous HSPC transplantation. The iHP infusion dose was 30 × 106/kg, with colony-forming unit numbers within the 2.1 to 2.4 × 105/kg range. As part of standard posttransplantation supportive care, peripheral blood transfusions were administered to animals with platelet counts <20 000/mL. One animal in control group 1 developed thrombocytopenia refractory to transfusions, and 1 animal in group 3 developed diarrhea and thrombocytopenia and was unresponsive to treatment; termination of study participation was necessary on days 43 and 29 after HSPC transplantation, respectively. All other animals were followed for at least 1 year posttransplantation.
Animal groups used in HSPC transplantation studies
| Recipient ID | MHC/ABO | BM CD34+ cells | iPSC donor, MHC/ABO | iHPs | iHPs, CFCs per kg | |||
|---|---|---|---|---|---|---|---|---|
| Dose per kg | Total dose | Tag | Total dose (30 × 106/kg) | Tag | ||||
| Group 1: control, autologous HSPCs only | ||||||||
| cy0679 | M1/M3 AB | 5 × 106 | 11 × 106 | eGFP | — | — | — | |
| cy0694 | M3/M3 B | 3 × 106 | 12 × 106 | eGFP | — | — | — | |
| cy0659 | M3/M3 AB | 3 × 106 | 20 × 106 | eGFP | ||||
| Group 2: autologous HSPCs + MHC-matched homozygous iHPs | ||||||||
| cy0668 | M3/M4 B | 3 × 106 | 19 × 106 | eGFP | M3/M3 AB | 180 × 106 | tdTomato | 2.1 × 105 |
| cy0898 | M2/M3 AB | 3 × 106 | 9 × 106 | tdTomato | M3/M3 AB | 100 × 106 | eGFP | 2.4 × 105 |
| cy0959 | M1/M3 AB | 3 × 106 | 12 × 106 | No tag | M3/M3 AB | 120 × 106 | eGFP | 2.4 × 105 |
| Group 3: autologous HSPCs + MHC-mismatched homozygous iHPs | ||||||||
| cy0936 | M5/M6 AB | 3 × 106 | 12 × 106 | eGFP | M3/M3 AB | 120 × 106 | tdTomato | 2.2 × 105 |
| cy0932 | M1/M4 AB | 3 × 106 | 11 × 106 | No tag | M3/M3 AB | 110 × 106 | eGFP | 2.3 × 105 |
| cy0925 | M1/M3 B | 3 × 106 | 12 × 106 | No tag | M2/M2 B | 115 × 106 | eGFP | 2.2 × 105 |
| Recipient ID | MHC/ABO | BM CD34+ cells | iPSC donor, MHC/ABO | iHPs | iHPs, CFCs per kg | |||
|---|---|---|---|---|---|---|---|---|
| Dose per kg | Total dose | Tag | Total dose (30 × 106/kg) | Tag | ||||
| Group 1: control, autologous HSPCs only | ||||||||
| cy0679 | M1/M3 AB | 5 × 106 | 11 × 106 | eGFP | — | — | — | |
| cy0694 | M3/M3 B | 3 × 106 | 12 × 106 | eGFP | — | — | — | |
| cy0659 | M3/M3 AB | 3 × 106 | 20 × 106 | eGFP | ||||
| Group 2: autologous HSPCs + MHC-matched homozygous iHPs | ||||||||
| cy0668 | M3/M4 B | 3 × 106 | 19 × 106 | eGFP | M3/M3 AB | 180 × 106 | tdTomato | 2.1 × 105 |
| cy0898 | M2/M3 AB | 3 × 106 | 9 × 106 | tdTomato | M3/M3 AB | 100 × 106 | eGFP | 2.4 × 105 |
| cy0959 | M1/M3 AB | 3 × 106 | 12 × 106 | No tag | M3/M3 AB | 120 × 106 | eGFP | 2.4 × 105 |
| Group 3: autologous HSPCs + MHC-mismatched homozygous iHPs | ||||||||
| cy0936 | M5/M6 AB | 3 × 106 | 12 × 106 | eGFP | M3/M3 AB | 120 × 106 | tdTomato | 2.2 × 105 |
| cy0932 | M1/M4 AB | 3 × 106 | 11 × 106 | No tag | M3/M3 AB | 110 × 106 | eGFP | 2.3 × 105 |
| cy0925 | M1/M3 B | 3 × 106 | 12 × 106 | No tag | M2/M2 B | 115 × 106 | eGFP | 2.2 × 105 |
CFC, colony-forming cell.
Animals were monitored posttreatment by complete blood count with differential and platelet counts and followed up clinically for any postinfusion toxicities (Figure 2). Evaluation of nadir and blood count recovery revealed no significant differences between groups in nadir duration or time after transplantation at which white blood cells became self-sustaining >500/μL. On average, animals infused with iHPs required slightly fewer blood transfusions (Table 2), but differences in the number of administered transfusions were not significant between groups. None of the animals showed clinical signs of toxicity after iHP infusion. Although all animals developed diarrhea after HSPC transplantation, routine fecal cultures on days 30 to 35 posttransplantation revealed Candida species in 2 control group animals, whereas routine fecal cultures performed in 5 animals infused with iHPs were negative for Candida species.
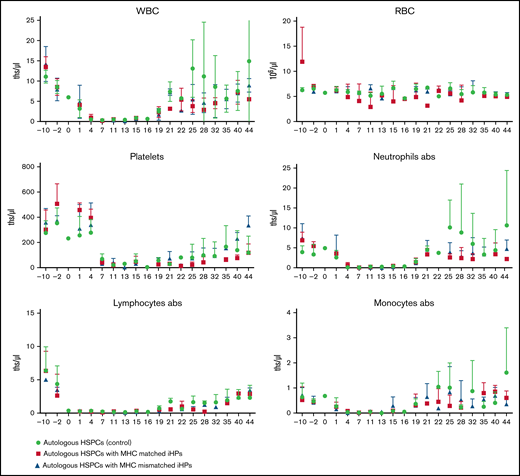
Complete blood counts to monitor hematopoietic reconstitution. Results are mean ± standard deviation of 3 different animals in each group. abs, absolute counts; ths/μL, thousands/μL.
Complete blood counts to monitor hematopoietic reconstitution. Results are mean ± standard deviation of 3 different animals in each group. abs, absolute counts; ths/μL, thousands/μL.
Clinical parameters
| Parameter | Group 1 | Group 2 | Group 3 |
|---|---|---|---|
| Blood transfusions | 2.3 ± 0.6 | 2 ± 0.5 | 1.7 ± 0.3 |
| WBCs | |||
| Days <500 | 2.3 ± 1.3 | 2 ± 0.5 | 2.6 ± 0.3 |
| Nadir, n/μL | 286 ± 101 | 196 ± 23 | 256 ± 58 |
| Days of nadir | 8 ± 0.5 | 7 ± 0 | 9 ± 2 |
| Days to >500 | 13 ± 1.5 | 12 ± 1.3 | 14.6 ± 1.7 |
| Days to >1 000 | 17 ± 1.2 | 17 ± 1 | 19 ± 1 |
| ANC | |||
| Days <500 | 3.6 ± 1.2 | 3 ± 0.5 | 4 ± 0 |
| Nadir, n/μL | 70 ± 50 | 63 ± 14.5 | 43 ± 23 |
| Days of nadir | 7 ± 1.1 | 6 ± 1 | 8 ± 1.3 |
| Days to >500 | 15 ± 1.7 | 15 ± 2.3 | 18 ± 0 |
| Days to >1 000 | 19.6 ± 0.6 | 19.6 ± 1.2 | 19 ± 1 |
| Platelets | |||
| Nadir, n/μL | 9 000 ± 7 549 | 7 000 ± 6 027 | 12 666 ± 5 487 |
| Days of nadir | 18 ± 5.5 | 20 ± 2.9 | 15 ± 2.8 |
| Days to >20 000 | 20 ± 6 | 29.5 ± 4.5 | 18 ± 3.7 |
| Parameter | Group 1 | Group 2 | Group 3 |
|---|---|---|---|
| Blood transfusions | 2.3 ± 0.6 | 2 ± 0.5 | 1.7 ± 0.3 |
| WBCs | |||
| Days <500 | 2.3 ± 1.3 | 2 ± 0.5 | 2.6 ± 0.3 |
| Nadir, n/μL | 286 ± 101 | 196 ± 23 | 256 ± 58 |
| Days of nadir | 8 ± 0.5 | 7 ± 0 | 9 ± 2 |
| Days to >500 | 13 ± 1.5 | 12 ± 1.3 | 14.6 ± 1.7 |
| Days to >1 000 | 17 ± 1.2 | 17 ± 1 | 19 ± 1 |
| ANC | |||
| Days <500 | 3.6 ± 1.2 | 3 ± 0.5 | 4 ± 0 |
| Nadir, n/μL | 70 ± 50 | 63 ± 14.5 | 43 ± 23 |
| Days of nadir | 7 ± 1.1 | 6 ± 1 | 8 ± 1.3 |
| Days to >500 | 15 ± 1.7 | 15 ± 2.3 | 18 ± 0 |
| Days to >1 000 | 19.6 ± 0.6 | 19.6 ± 1.2 | 19 ± 1 |
| Platelets | |||
| Nadir, n/μL | 9 000 ± 7 549 | 7 000 ± 6 027 | 12 666 ± 5 487 |
| Days of nadir | 18 ± 5.5 | 20 ± 2.9 | 15 ± 2.8 |
| Days to >20 000 | 20 ± 6 | 29.5 ± 4.5 | 18 ± 3.7 |
Data are presented as mean ± standard error.
ANC, absolute neutrophil count; WBC, white blood cell.
Flow cytometric analysis of peripheral blood revealed presence of iPSC-derived cells within granulocyte and monocyte gates, which peaked between 7 and 15 days posttransplantation (3-11 days after iHP infusion; Figure 3A-B). The presence of iPSC derivatives in peripheral blood was confirmed by genomic PCR (Figure 3C). Phenotypic analysis of GFP+ or tdTomato+ populations within granulocyte and monocyte gates (Figure 3B) reveled that they were low/negative for CD11b and CD14 lineage markers in all animals. This can be explained by the relatively low expression of CD11b by iPSC-derived granulocytes, as shown in our prior in vitro studies,37 and by the CD14− phenotype of immature cells of monocyte/macrophage lineage. It is well recognized that CD14 appears relatively late in monocytic differentiation.38 To evaluate possible migration and retention of iHPs or iHP progeny within tissues, we performed BM, colon, and lymph node biopsies at 2, 4, and 8 months posttransplantation and performed genomic PCR to detect eGFP or tdTomato. Although we detected no positive signals in BM cells, footprints of infused cells were detected in lymph node biopsy of cy0668 2 months after MHC-matched iHP infusion and in colon and lymph node biopsies of cy0925 8 months after MHC-mismatched iHP infusion (Figure 4). Interestingly, cy0925 showed a longer persistence of eGFP signal in peripheral blood after iHP infusion (Figure 3C).
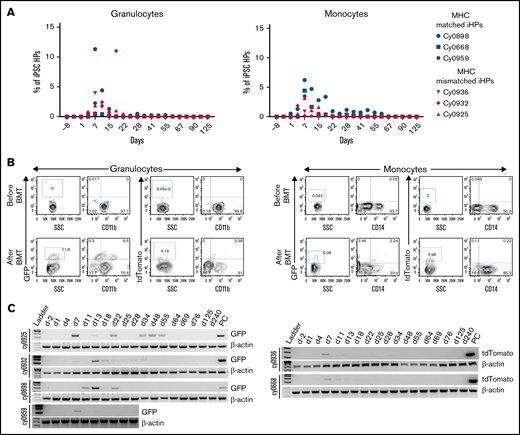
Detection of iPSC-derived cells in peripheral blood. (A) Percentage of eGFP+ or tdTomato+ cells within granulocyte or monocyte forward scatter (FSC) vs side scatter (SSC) gate after gating on live single cells in individual animals. (B) Representative dot plots showing eGFP or tdTomato vs CD11b or CD14 expression by cells within granulocyte or monocyte FSC vs SSC gate. (C) Genomic PCR analysis using primers specific for eGFP or tdTomato to detect iPSC-derived cells in the peripheral blood. Peripheral blood mononuclear cells from animal undergoing transplantation with eGFP-transduced HSPCs were used as positive control for animals that received eGFP-tagged iHPs. iHPs expressing tdTomato were used as positive control for animals that received iHPs tagged with tdTomato. Peripheral blood collected before transplantation was used as negative control.
Detection of iPSC-derived cells in peripheral blood. (A) Percentage of eGFP+ or tdTomato+ cells within granulocyte or monocyte forward scatter (FSC) vs side scatter (SSC) gate after gating on live single cells in individual animals. (B) Representative dot plots showing eGFP or tdTomato vs CD11b or CD14 expression by cells within granulocyte or monocyte FSC vs SSC gate. (C) Genomic PCR analysis using primers specific for eGFP or tdTomato to detect iPSC-derived cells in the peripheral blood. Peripheral blood mononuclear cells from animal undergoing transplantation with eGFP-transduced HSPCs were used as positive control for animals that received eGFP-tagged iHPs. iHPs expressing tdTomato were used as positive control for animals that received iHPs tagged with tdTomato. Peripheral blood collected before transplantation was used as negative control.
Detection of eGFP- or tdTomato-tagged iPSC-derived cells by genomic PCR in biopsy specimens collected from BM, colon, and lymph nodes. BM, lymph node, and colon biopsies collected from animal that did not receive any eGFP or tdTomato cells were used as negative control. Peripheral blood mononuclear cells from animal undergoing transplantation with eGFP-transduced HSPCs were used as positive control for animals that received eGFP-tagged iHPs. Day-10 iHPs expressing tdTomato were used as positive control for animals that received iHPs tagged with tdTomato.
Detection of eGFP- or tdTomato-tagged iPSC-derived cells by genomic PCR in biopsy specimens collected from BM, colon, and lymph nodes. BM, lymph node, and colon biopsies collected from animal that did not receive any eGFP or tdTomato cells were used as negative control. Peripheral blood mononuclear cells from animal undergoing transplantation with eGFP-transduced HSPCs were used as positive control for animals that received eGFP-tagged iHPs. Day-10 iHPs expressing tdTomato were used as positive control for animals that received iHPs tagged with tdTomato.
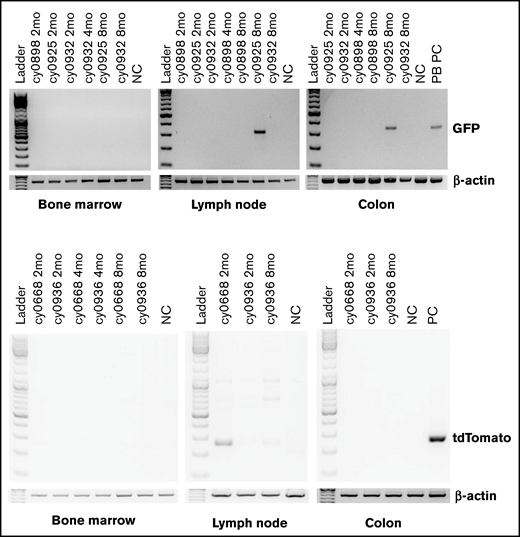
To assess safety, complete necropsies were performed by WNPRC American College of Veterinary Pathologist Board–certified veterinary pathologists, with detailed gross and microscopic examination of all organs, on all MCMs that underwent HSPC transplantation and received iHP infusion. No teratomas or other neoplastic lesions were identified in these animals, suggesting the safety of administration. No residual iPSC-derived cells were detected in tissues of any MCMs analyzed 1 year after iHP infusion. However, we detected eGFP by PCR signal in the heart, pancreas, colon, lymph node, lung, and brain specimens obtained at 25 days after MHC-mismatched iHP infusion in cy0959 (Figure 5). These findings, along with the detection of fluorescent tags in lymph node and colon biopsies of 2 other MCMs, suggest that iHPs/iHP progeny after infusion can migrate and persist for a limited time in peripheral tissues.
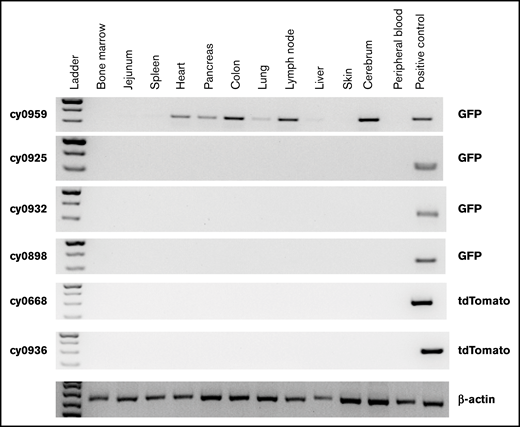
Detection of eGFP- or tdTomato-tagged iPSC-derived cells by genomic PCR in collected postmortem tissues. Peripheral blood mononuclear cells from animal undergoing transplantation with eGFP-transduced HSPCs were used as positive control for animals that received eGFP-tagged iHPs. Tissues were collected 25 days (cy0959) or 1 year (remaining animals) after iHP infusion. Day-10 iHPs expressing tdTomato were used as positive control for animals that received iHPs tagged with tdTomato.
Detection of eGFP- or tdTomato-tagged iPSC-derived cells by genomic PCR in collected postmortem tissues. Peripheral blood mononuclear cells from animal undergoing transplantation with eGFP-transduced HSPCs were used as positive control for animals that received eGFP-tagged iHPs. Tissues were collected 25 days (cy0959) or 1 year (remaining animals) after iHP infusion. Day-10 iHPs expressing tdTomato were used as positive control for animals that received iHPs tagged with tdTomato.
Evaluation of immunogenicity of iHPs derived from MHC homozygous iPSCs
To evaluate iHP immunogenicity, we infused nonmodified MHC homozygous iHPs across MHC-matched and fully mismatched barriers using unmanipulated immunocompetent animals (Figure 6A). In group 1, animals received autologous iHPs; in group 2, MHC homozygous animals received allogeneic MHC-identical iHPs (homozygous to matched homozygous cells); in group 3, MHC heterozygous animals received MHC-matched iHPs from MHC homozygous iPSCs (homozygous to matched heterozygous); in group 4, MHC-mismatched animals received iHPs from MHC-mismatched homozygous iPSCs (homozygous to MHC-mismatched heterozygous; supplemental Table 2). In these experiments, iHPs were infused IV 3 times at weekly intervals. Blood samples obtained before and 1 week after each infusion and 2 and 5 months after final infusion were collected to screen for anti-MHC antibodies using immortalized BLCLs with MHC genotypes corresponding to the infused iHPs. Serum from an MCM alloimmunized with PHA-activated PBMCs (positive control) contained antibodies binding BLCLs with nMFIs ≥2 (Figure 6B), even out to a 1:640 dilution. Conversely, we detected no alloantibodies in the serum of any of the MCMs infused with iHPs (Figure 6C). Therefore, we hypothesized that the low alloimmunogenicity of the iHPs was due to the weak MHC class I expression and lack of MHC class II expression (Figure 1E). Therefore, we reevaluated the alloimmune responses toward iHPs after culturing them with IFN-γ to upregulate MHC expression (Figure 1E). However, even after 3 rounds of transfusion of IFN-γ–stimulated iHPs, none of the serum samples demonstrated nMFI >2 in any group of animals, suggesting that even MHC-mismatched iHPs are hypoimmunogenic (Figure 6D).
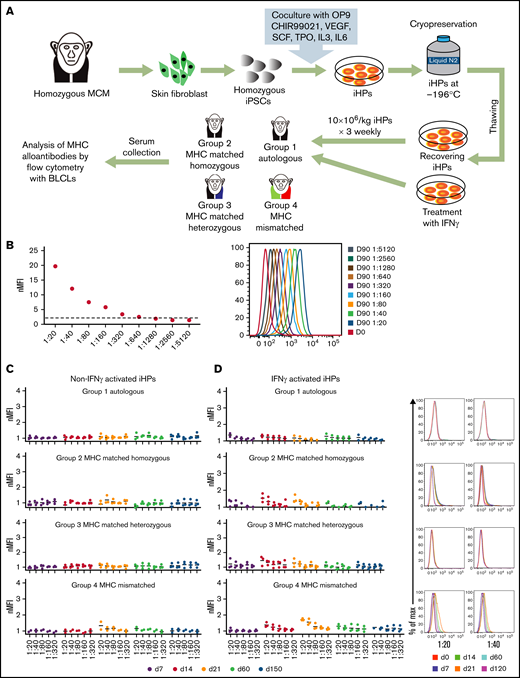
Evaluation of immunogenicity of iHPs derived from MHC homozygous iPSCs. (A) Schematic diagram for the assessment of alloimmune response to iPSC-derived iHPs. (B) Titration of serum from MCMs infused with MHC-mismatched peripheral blood lymphocytes (positive control). Day-0 serum before lymphocyte infusion. Day-90 serial dilutions of serum collected 90 days after infusion. nMFI values and fluorescence-activated cell sorting plots are shown. (C) Titration graphs of serum samples from each group collected at different time points from animals that were infused with MHC homozygous iHPs that were not activated with IFN-γ. (D) Titration graphs of serum samples from each group collected at different time points from animals that were infused with MHC homozygous iHPs that were activated with IFN-γ and analyzed. Dot plots of 1/20 and 1/40 dilutions of serum collected at different times after iHP infusion from representative animals are shown. In panels C and D titration graphs, each dot represents a different animal from each group. The different days of the serum collection after the first iHP infusion are indicated by different colors (day 0-120).
Evaluation of immunogenicity of iHPs derived from MHC homozygous iPSCs. (A) Schematic diagram for the assessment of alloimmune response to iPSC-derived iHPs. (B) Titration of serum from MCMs infused with MHC-mismatched peripheral blood lymphocytes (positive control). Day-0 serum before lymphocyte infusion. Day-90 serial dilutions of serum collected 90 days after infusion. nMFI values and fluorescence-activated cell sorting plots are shown. (C) Titration graphs of serum samples from each group collected at different time points from animals that were infused with MHC homozygous iHPs that were not activated with IFN-γ. (D) Titration graphs of serum samples from each group collected at different time points from animals that were infused with MHC homozygous iHPs that were activated with IFN-γ and analyzed. Dot plots of 1/20 and 1/40 dilutions of serum collected at different times after iHP infusion from representative animals are shown. In panels C and D titration graphs, each dot represents a different animal from each group. The different days of the serum collection after the first iHP infusion are indicated by different colors (day 0-120).
Discussion
In the present study, we used an NHP model to demonstrate that infusing iPSC-derived iHPs is well tolerated and safe. None of the animals involved in this study developed teratomas or evidence of malignancy. Although iPSC-derived cells were detected in peripheral blood 3 to 11 days postinfusion, we did not observe significant improvements in peripheral blood counts in iHP-infused animals compared with the control group. These findings are consistent with clinical trials and mouse studies using ex vivo expanded somatic myeloid progenitors, which also failed to detect improvements in peripheral blood neutrophil counts or significant contribution from progenitor-derived cells to peripheral blood neutrophils.12,20,39 Nevertheless, infusion of these cells reconstituted the myeloid compartment within the spleen and protected mice from Aspergillosis and Pseudomonas aeruginosa infection after hematopoietic stem cell transplantation or chemotherapy.9,12 Given these findings, the conclusion was made that the protective effects of myeloid progenitors were due to their migration to peripheral tissues and sites of infection.9 In our study, we detected iPSC derivatives in 2 of 6 animals in peripheral tissues several months postinfusion. In 1 animal, we found eGFP DNA in multiple organs 25 days postinfusion of eGFP-tagged iHPs. Interestingly, we noticed frequent development of intestinal candidiasis in control animals undergoing autologous HSPC transplantation, whereas no Candida species were detected in routine bacterial fecal cultures of animals that received iHP infusion. These findings support the hypothesis that iHPs can migrate to tissues and contribute to short-term reconstitution of myeloid effectors, similar to somatic expanded myeloid progenitors. However, the small sample size and lack of historical data on frequency of Candida infection after HSPC transplantation in MCMs limit our ability to draw a definitive conclusion about the effect of iHP infusion on intestinal candidiasis.
Alloimmunization after infusion of myeloid progenitors is another major concern in potential clinical application. Because generation and banking of iPSCs from rare individuals homozygous at HLA-A, -B, and -DR loci can be used to produce iHPs with beneficial HLA matching to avoid alloimmunization,21-25 we performed multiple rounds of MHC homozygous iHP infusions across different MHC backgrounds to assess alloimmunogenicity. Our study demonstrated that iHPs were hypoimmunogenic and did not cause a significant alloimmune response, even when infused into completely MHC-mismatched animals after pretreatment with IFN-γ. These findings suggest that iHP products can be safely administered with a low risk of alloimmunization. The low immunogenicity of iHPs can be at least partially explained by weak MHC class I and lack of MHC class II expression. However, we also observed low immunogenicity of iHPs even after upregulation of MHC expression with IFN-γ. This could be attributed to the known intrinsic immunosuppressive properties of immature myeloid cells.16,40,41
Infusion of ex vivo expanded myeloid progenitors has been proposed and tested in clinical trials as an alternative strategy to mitigate neutropenia and reduce incidence of infection after myeloablative chemotherapy and radiotherapy.9 ,,,-14,17 -20 Myeloid progenitors used in these trials were manufactured by ex vivo expansion of cord blood or mobilized peripheral blood CD34+ cells.17-20 However, sourcing of CD34+ progenitors from donors has several limitations, including the added burden to donors as a result of complex leukapheresis procedures required for obtaining cellular product, cellular product variability that results from sourcing cells from different donors, and constraints in obtaining enough therapeutically active cells for infusion. We propose that iPSCs could become a logical alternative to somatic sources of myeloid progenitors. Moreover, iHPs correspond to the transient wave of hematopoietic progenitors formed before the emergence of hematopoietic stem cells in the embryo. Although these cells are not capable of long-term hematopoietic reconstitution, they efficiently produce terminally differentiated blood cells and are suitable for transient hematopoietic reconstitution. Given that iPSC lines can be expanded indefinitely, this would allow unlimited numbers of iHPs to be manufactured. A desired cellular product could be produced on demand regardless of donor availability, thus alleviating supply shortages. Moreover, manufacturing iHPs from iPSCs would greatly reduce the risk of viral, prion, or bacterial transmission. iPSCs could be genetically engineered to produce iHPs with more potent antimicrobial properties or features that may improve outcomes of myeloablative therapies or radiation exposure from radiologic attacks. iHPs are naturally free of T cells and are not expected to cause graft-versus-host disease. Thus, iHPs could be an ideal cell source to reduce sequelae of cytopenia after myeloablative BM transplantation or chemotherapy. Although we demonstrated the safety and hypoimmunogenicity of iHPs, focused studies are required to assess the efficacy of iHPs in experimental fungal and bacterial infection models.
Acknowledgments
The authors thank Theodora Hatziioannou (Aaron Diamond AIDS Research Center) for providing a packaging plasmid containing SIV capsid protein.
Antirhesus immunoglobulin G1/3 [1B3]-FITC monoclonal antibody was obtained from the National Institutes of Health (NIH) Nonhuman Primate Reagent Resource supported by HHSN272200900037C and OD010976. This research was funded by NIH, National Heart, Lung, and Blood Institute grants R01HL132891 and U01HL134655 and NIH, Office of the Director grant P51 OD011106 to the Wisconsin National Primate Research Center.
Authorship
Contribution: S.S.D. processed bone marrow aspirates for hematopoietic stem cell (HSC) enrichment, generated iPSC-derived iHPs, performed all the experiments, interpreted experimental data, created figures, and wrote the manuscript; A.K. processed bone marrow aspirates for HSC enrichment, generated iHPs, and created figures; J.M. and E.P. generated iPSCs from skin fibroblasts and characterized them; J.T.W. generated the BLCLs and provided the positive serum for the alloimmunization studies; M. Raymond performed flow cytometry for alloimmunization studies and edited the manuscript; N.S.S. performed bone marrow aspirates and bone marrow transplantation in the control animals; J.C. performed the animal experiments and took care of all the animals used for the study; A.M. and H.A.S. performed the necropsy and analysis of the animal tissues posteuthanasia; B.E.T. provided the eGFP lentiviral vector and advice on the use of rapamycin for transfection of CD34+ bone marrow cells; M. Reynolds assisted in the alloimmunization studies; and J.A.T. and I.I.S. developed the concept, led and supervised the study, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Igor I. Slukvin, 1220 Capitol Court, Madison, WI 53715; e-mail: islukvin@wisc.edu.

