

Key Points
The aptamer rondoraptivon pegol improved VWF/FVIII levels, multimer patterns, and thrombocytopenia in type 2B von Willebrand disease.
Once weekly subcutaneous injections of rondoraptivon pegol may in the future be used for prophylaxis or treatment of VWD.

Abstract
Type 2B von Willebrand disease (VWD) is characterized by an increased binding affinity of von Willebrand factor (VWF) to platelet glycoprotein Ib. This can lead to clearance of high-molecular-weight (HMW) multimers and thrombocytopenia with a resulting moderate-severe bleeding phenotype. Rondoraptivon pegol (BT200) is a pegylated aptamer binding to the A1 domain of VWF with a novel mechanism of action: it enhances VWF/factor VIII (FVIII) levels by decreasing their clearance. To study the potential benefit of rondoraptivon pegol in patients with type 2B VWD, we conducted a prospective phase 2 trial. Patients with type 2B VWD received 3 mg rondoraptivon pegol subcutaneously on study days 1, 4, and 7, followed by 6 to 9 mg every week until day 28. Five patients (male:female ratio = 3:2) were included. Rondoraptivon pegol rapidly tripled platelet counts from a median of 60 to 179 × 10E9/L (P < .001). Circulating VWF antigen increased from a median of 64% to 143%, which doubled FVIII activity levels from 67% to 134%. In all thrombocytopenic patients, plasma levels of VWF:GPIbM normalized, VWF ristocetin cofactor and VWF collagen-binding activity increased, and HMW multimers appeared. These pronounced improvements reversed during washout of the drug, thus demonstrating causality. The A1 domain binding aptamer directly corrects the underlying defect of type 2B VWD, thus providing a novel potential option for prophylaxis and treatment of patients with this VWD type. These data provide the basis for a phase 2b/3 trial in such patients. This trial was registered at www.clinicaltrials.gov as #NCT04677803.
Introduction
von Willebrand disease (VWD) is classified based on clinical findings and laboratory results1 according to the recommendations of the von Willebrand factor (VWF) Scientific Standardization Committee of the International Society on Thrombosis and Haemostasis,2 as revised in 2021.
Type 1 VWD and type 3 VWD are partial and almost complete quantitative VWF deficiencies, respectively, and type 2 VWD is characterized by qualitative VWF abnormalities causing functional defects.2,3 In type 2B VWD, gain-of-function mutations are typically found in the VWF A1 domain4 ; these mutations increase the binding of VWF to platelet glycoprotein Ib (GpIb) receptors, which normally requires shear stress, polymerizing fibrin, or immobilization of VWF in the subendothelial matrix.5 They also facilitate transition of the VWF A1 domain from a globular to a stretched conformation, even under low shear forces.6 Conformational changes induced by type 2B VWD mutations lead to spontaneous binding of VWF to GPIb receptor on the platelets, which may induce platelet aggregation or impair megakaryopoiesis in specific cases.7 Mutant 2B VWF also binds in a shear stress-independent manner to a VWF clearance receptor, macrophage low-density lipoprotein receptor related protein (LRP-1).8 Additionally, this change in VWF conformation enhances the interaction with its cleavage enzyme A Disintegrin and Metalloprotease with Thrombospondin Type 1 motif 13 (ADAMTS-13),9 because VWF-platelet complexes are preferred substrates of ADAMTS-13 under shear stress.10,11 The resulting accelerated proteolysis of VWF by ADAMTS-13 induces a rapid clearance12 and loss of intermediate and high-molecular-weight (HMW) multimers.13 These VWF/platelet complexes are taken up by macrophages of the reticuloendothelial system,6 and increased clearance of labeled platelets and reduced platelet survival have been reported in a patient with type 2B VWD.14 Thrombocytopenia only occurs in a subgroup of patients,15 but when present, it enhances bleeding risk 4.5-fold.13 This results in a clear unmet medical need, because thrombocytopenia is corrected by neither VWF substitution nor desmopressin (DDAVP); DDAVP releases endogenous (mutant) VWF into circulation, which can worsen the thrombocytopenia.14,16,17 This also occurs in response to other stressors such as pregnancy18,19 and surgery.20
A VWF A1 domain binding aptamer (ARC1779) was previously found to increase plasma levels of VWF/factor VIII (FVIII) and platelet counts in patients with type 2B VWD.21 Its short half-life and poor bioavailability after subcutaneous injections did not lead to therapeutic plasma levels.22 This precluded further use of that first-generation aptamer in an outpatient setting and led to the development of rondoraptivon pegol (previously BT200), a novel, potent, and long-acting VWF A1 domain binding aptamer.23 In the first in-human trial, rondoraptivon pegol dose-dependently decreased the clearance of VWF, the chaperone and carrier of coagulation FVIII,24 and thereby elevated VWF/FVIII levels several-fold in healthy volunteers.25 The mechanistic explanation for this effect was provided in a recent study, where pegylation of the VWF A1 domain reduced the clearance of VWF by the macrophage low-density lipoprotein receptor related protein (LRP-1).26 As rondoraptivon pegol binds to the A1 domain with high affinity, it can be regarded as reversible pegylation of the A1 domain that consequently increases VWF and FVIII levels. This feature of rondoraptivon pegol could be particularly useful in type 2B VWD, where binding the A1 domain of VWF may not only increase VWF concentrations because of decreased clearance but also block the pathological interaction with ADAMTS13 and platelets.
In this study, we tested the hypothesis that rondoraptivon pegol might directly correct the underlying pathophysiology of type 2B VWD, increase VWF antigen and HMW multimers levels, and improve FVIII activity and platelet counts in type 2B VWD.
Methods
Population and study design
This phase 2a trial (EUDRA-CT 2020-003807-32 and #NCT04677803) was approved by the Austrian Federal Office for Safety in Health Care and the Ethics Committee of the Medical University of Vienna and was conducted at the Department of Clinical Pharmacology at the Medical University of Vienna. Patients with inherited bleeding disorders (hemophilia A without inhibitors, or VWD (type 1, type 2B, type 3 under substitution therapy) or acquired von Willebrand syndrome without inhibitors) irrespective of bleeding severity were eligible for this basket trial, and this report focuses on the VWD cohort. An intraindividual dose escalation design was used. We contacted all patients with known type 2B VWD diagnosed at the Medical University of Vienna. Only about half of the patients whose addresses had been available responded to the written invitation. There were no restrictions in the inclusion criteria regarding genotypes, bleeding scores, or HMW multimers. All study participants provided oral and written informed consent and were at least 18 years old. Key exclusion criteria included clinically significant medical history or ongoing chronic illness that might jeopardize the safety of the patient or compromise the quality of the data; previous spontaneous thromboembolic events; significant drug allergies or anaphylactic reactions; substance abuse; and severe mental illness. Additionally, female patients needed to be of non-childbearing potential (postmenopausal or surgically sterilized). Patients were enrolled between January and May 2021.
The primary objective was to assess the effect of subcutaneous rondoraptivon pegol injections on platelet counts and FVIII levels and the safety thereof. As a secondary objective we investigated the pharmacokinetics and pharmacodynamics of rondoraptivon pegol.
Study treatment
The structure and function of the VWF A1 domain binding aptamer rondoraptivon pegol were described recently. The core aptamer has a molecular weight of 10 kDa, which is pegylated (40 kDa) and binds at multiple binding sites (amino acids: 1367-1402; old nomenclature: 604-639) of the botrocetin binding region,23 which is also in close proximity to lysine 1408, a critical binding site for LRP-1.27
Rondoraptivon pegol was supplied at a concentration of 15 mg/mL in sterile saline for injection (all doses refer to the core, unpegylated aptamer). Patients received subcutaneous injections of 3 mg of rondoraptivon pegol on days 0, 4, and 7, followed by weekly injections of 3 to 9 mg until day 28 (inclusive) during a dose titration phase (Figure 1). More precisely, patients received injections of 6 mg on days 14 and 21, and on day 28, all patients received 9 mg of rondoraptivon pegol, except for patient 5 who received 6 mg. Dose titration and safety were monitored by measuring VWF-dependent platelet function using multiple electrode aggregometry, and the dose was not adjusted based on the body weight. Because of the high bleeding tendency in our patients on VWF prophylaxis, they were not asked for a complete washout and instead were asked to come in for blood sampling immediately before the planned administration of each dose. In addition to rondoraptivon pegol, these patients maintained their regular schedules of VWF infusion during the trial (every other day for patient 1, and Monday, Wednesday, Friday for patient 4). Therefore, the measured VWF parameters represent an interaction between rondoraptivon pegol and the recombinant Von Willebrand Factor (rVWF) infusions. Rondoraptivon pegol binds to wild-type VWF as demonstrated in the phase 1 trial in healthy volunteers25 and to mutated VWF as shown for the mutations investigated in the current trial. However, we cannot exclude that some mutations in VWF can distort the structure of VWF, which could lead to decreased binding of rondoraptivon pegol.

Dose escalation scheme of rondoraptivon pegol. Study scheme: the last dose was escalated to 9 mg only in patients with thrombocytopenia at baseline. biw, biweekly; ew, every week.
Dose escalation scheme of rondoraptivon pegol. Study scheme: the last dose was escalated to 9 mg only in patients with thrombocytopenia at baseline. biw, biweekly; ew, every week.
Analytical methods of biomarkers
FVIII activity was determined via a 1-stage clotting assay using soy phosphatides and rabbit brain phosphatides with ellagic acid as an activator (Siemens Dade Actin FS reagent). This was done to avoid putative interference of PEG with silica-based clotting assays. The following additional assays were used: VWF Antigen (STA LIATEST VWF:Ag; Diagnostica Stago, France); ristocetin induced platelet aggregation with multiple electrode aggregometry (Multiplate, Roche, Vienna, Austria); shear-induced platelet function with the platelet function analyzer (PFA-200, Siemens Healthcare Diagnostics) VWF Ristocetin Co-Factor and VWF GpIbM activity (Siemens HealthCare Diagnostics), VWF propeptide (Haemochrom Diagnostics, FRG), and VWF collagen binding activity (VWF:CBA; Technoclone, Vienna, Austria). Units for VWF and FVIII are given as percentage (equal to IU/dL) because the units relate to a normal range established in-house. Plasma rondoraptivon pegol concentrations were measured under Good Laboratory Practice at QPS (Newark, DE) using high-performance liquid chromatography after hybridization of rondoraptivon pegol to a fluorescent labeled complementary oligonucleotide. The method was qualified and fully validated according to regulatory standards with a lower limit of quantification of 1 ng/mL.25 Multimer analyses were carried out as previously described.11
Study oversight
The clinical trial was conducted in accordance with the principles set forth in the Declaration of Helsinki and local drug law. This investigator-initiated trial was sponsored by the Medical University of Vienna and received financial support from Band Therapeutics LLC, a subsidiary of Guardian Therapeutics LLC. Compliance with Good Clinical Practice was confirmed by the Clinical Coordination Center for Studies. All authors had access to the primary clinical trial data and approved the decision to submit the manuscript for publication.
Statistical analysis
The sample size estimation was based on a 120% increase in VWF antigen levels in healthy volunteers after 6 mg rondoraptivon pegol, the day-to-day variability of 17% in VWF antigen levels,25 and an expected strong effect size on platelet counts from our previous experience with a short-acting anti-VWF aptamer,28 indicating that 5 patients would suffice to detect such a rise (α-error = 0.025; β = 0.8). Pharmacokinetic parameters of rondoraptivon pegol were calculated using Phoenix WinNonlin v.8.3 by Certara (Montreal, Canada). Data were summarized by visit. Descriptive statistics (number of observations, median, minimum, and maximum) were provided for continuous variables. Frequency counts and percentages were presented for categorical variables. Data listings included all enrolled subjects. A Friedman analysis of variance was applied to test the time course of the changes in co-primary end points (FVIII levels and platelet counts) from baseline to day 35 (peak effect 1 week after the last administration) for significance, and 2-sided significance levels of 5% were applied. The baseline value of patient 3 was imputed for a single missing platelet count on day 4. No correction for multiplicity was carried out for the other variables because the different parameters are dependent variables, based on the pathophysiology of disease.
Results
We contacted 6 potential patients with type 2B VWD; 1 patient (>80 years) chose not to participate. Five patients were included, all of whom completed the trial. Data are complete except for 1 missing platelet count data point on day 4 in patient 3. Demographic characteristics and baseline VWF levels are shown in Table 1. Genotyping previously confirmed the diagnosis in each patient with type 2B VWD (Table 1); 1 patient (5) additionally carried the V1565L polymorphism.
Demographics and laboratory values of patients with type 2B VWD on day 1
| Units | Patient ID | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| Mutation | R1308C | R1306W | R1306W | V1316M | W1313C | |
| Prophylaxis | rVWF | No | No | rVWF | No | |
| Sex | Female | Male | Male | Male | Female | |
| Age | y | 64 | 61 | 24 | 72 | 61 |
| Weight | kg | 94 | 100 | 100 | 91 | 77 |
| Height | cm | 163 | 177 | 180 | 175 | 158 |
| Platelets | ×10E9/L | 41 | 50 | 120 | 60 | 167 |
| VWF:Ag | % | 106 | 64 | 32 | 69 | 40 |
| FVIII | % | 91 | 67 | 44 | 84 | 64 |
| VWF:GpIbM | % | 42 | 25 | 19 | 18 | 3 |
| VWF:RCo | % | 30 | 31 | 17 | 17 | 9 |
| VWF:CB | % | 72 | 50 | 19 | 52 | 15 |
| VWF:RCo/VWF:Ag ratio | 0.28 | 0.48 | 0.53 | 0.25 | 0.23 | |
| VWFpp/VWF:Ag ratio | 21 | 25 | 47 | 18 | 27 | |
| VWFpp | mIU/mL | 2229 | 1614 | 1506 | 1252 | 1095 |
| CADP-CT | s | 301 | 301 | 301 | 301 | 301 |
| Ristocetin induced aggregation | U | 55 | 34 | 31 | 120 | 29 |
| VWF intermediate molecular weight multimers | Reduced | Reduced | Reduced | Reduced | Reduced | |
| VWF high molecular weight multimers | Reduced | Reduced | Reduced | Reduced | Reduced | |
| Bleeding score | 30 | 20 | 16 | 19 | 21 | |
| Units | Patient ID | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| Mutation | R1308C | R1306W | R1306W | V1316M | W1313C | |
| Prophylaxis | rVWF | No | No | rVWF | No | |
| Sex | Female | Male | Male | Male | Female | |
| Age | y | 64 | 61 | 24 | 72 | 61 |
| Weight | kg | 94 | 100 | 100 | 91 | 77 |
| Height | cm | 163 | 177 | 180 | 175 | 158 |
| Platelets | ×10E9/L | 41 | 50 | 120 | 60 | 167 |
| VWF:Ag | % | 106 | 64 | 32 | 69 | 40 |
| FVIII | % | 91 | 67 | 44 | 84 | 64 |
| VWF:GpIbM | % | 42 | 25 | 19 | 18 | 3 |
| VWF:RCo | % | 30 | 31 | 17 | 17 | 9 |
| VWF:CB | % | 72 | 50 | 19 | 52 | 15 |
| VWF:RCo/VWF:Ag ratio | 0.28 | 0.48 | 0.53 | 0.25 | 0.23 | |
| VWFpp/VWF:Ag ratio | 21 | 25 | 47 | 18 | 27 | |
| VWFpp | mIU/mL | 2229 | 1614 | 1506 | 1252 | 1095 |
| CADP-CT | s | 301 | 301 | 301 | 301 | 301 |
| Ristocetin induced aggregation | U | 55 | 34 | 31 | 120 | 29 |
| VWF intermediate molecular weight multimers | Reduced | Reduced | Reduced | Reduced | Reduced | |
| VWF high molecular weight multimers | Reduced | Reduced | Reduced | Reduced | Reduced | |
| Bleeding score | 30 | 20 | 16 | 19 | 21 | |
Abnormal values are highlighted in bold. Mutations are indicated by amino acid substitutions (https://www.omim.org/entry/613160#73, www.VWF.group.shef.ac.uk; last accessed 18 December 2021). Patients 2 and 3 are father and son; patient 5 has an additional amino acid polymorphism p.(V1565L) in the A2 domain.
CADP-CT, collagen adenosine diphosphate-closure time (s); NA, not assessed; VWF:Ag, von Willebrand factor antigen (%); FVIII, factor VIII clotting activity (%); VWF:CB, VWF collagen binding activity (%); VWF:GpIbM, VWF glycoprotein Ib mutation activity (%); VWFpp, VWF propeptide (mIU/mL); VWF:RCo, VWF ristocetin cofactor activity (%).
All patients had low levels of VWF:RCo and VWF:GpIb activity, resulting in VWF activity/antigen ratios of <0.6 for both assays, well below recently recommended cutoffs (<0.7).1,29 All patients lacked intermediate and HMW VWF multimers (Figure 2). Four of the 5 patients were thrombocytopenic, 2 of the 4 (patients 1 and 4) despite being on regular prophylactic therapy with rVWF. Chronic substitution was necessary in those 2 patients because of recurrent severe bleeding requiring frequent hospitalizations: patient 4 mainly suffered recurrent severe epistaxis, whereas patient 1 had recurrent gastrointestinal bleeds caused by angiodysplasia. Both patients bled immediately before their scheduled first days in this trial. Patient 4 had another episode of severe epistaxis requiring treatment at an otolaryngology department in a hospital, and patient 1 had mild hemoptysis. Thus, no attempt was made to wash rVWF out.

Rondoraptivon pegol effects on VWF multimers. Rondoraptivon pegol increases intermediate and HMW multimers of VWF. Three milligrams of rondoraptivon pegol was injected subcutaneously on days 1, 4, and 7, followed by up-titration to 6 to 9 mg at weekly intervals. (A) Time course in patient 3. (B) Comparison between baseline and day 35 (1 week after the last rondoraptivon pegol dose) for the other patients. Patients 1 and 4 were on substitution therapy with recombinant VWF.
Rondoraptivon pegol effects on VWF multimers. Rondoraptivon pegol increases intermediate and HMW multimers of VWF. Three milligrams of rondoraptivon pegol was injected subcutaneously on days 1, 4, and 7, followed by up-titration to 6 to 9 mg at weekly intervals. (A) Time course in patient 3. (B) Comparison between baseline and day 35 (1 week after the last rondoraptivon pegol dose) for the other patients. Patients 1 and 4 were on substitution therapy with recombinant VWF.
Dosing and pharmacokinetics
Rondoraptivon pegol was injected subcutaneously at a dose of 3 mg on days 0, 4, and 7 and further up-titrated to 6 mg on days 14 and 21; on day 28, all patients received 9 mg of rondoraptivon pegol, except for patient 5 who received 6 mg. The 9-mg dose was given to patients who demonstrated thrombocytopenia at baseline. This resulted in maximum plasma levels of 777 ng/mL (range, 423-999 ng/mL; Figure 3) on day 35 (ie, 1 week after the last dose). The mean apparent half-life of rondoraptivon pegol was 5.84 days (coefficient of variation%: 24%).
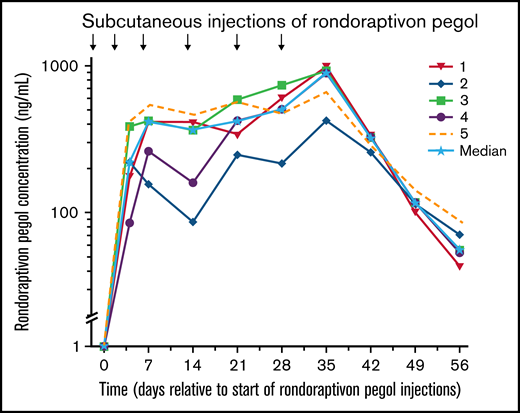
Pharmacokinetics of rondoraptivon pegol. Pharmacokinetics of rondoraptivon pegol after subcutaneous injections in patients with type 2B VWD. Rondoraptivon pegol was given as 3 mg on days 0, 4, and 7, followed by weekly doses of 6 or 9 mg (only in patients with thrombocytopenia at baseline). The solid line with the stars indicates the median.
Pharmacokinetics of rondoraptivon pegol. Pharmacokinetics of rondoraptivon pegol after subcutaneous injections in patients with type 2B VWD. Rondoraptivon pegol was given as 3 mg on days 0, 4, and 7, followed by weekly doses of 6 or 9 mg (only in patients with thrombocytopenia at baseline). The solid line with the stars indicates the median.
Pharmacodynamic effects
Platelet counts.
Thrombocytopenia is observed in about one third of patients with type 2B VWD with the R1306W, R1308C, I1309V, or V1316M mutations (supplemental Figure 1),13,15 and our thrombocytopenic patients had missense mutations at these positions. Patient 5 carried an additional polymorphism (p.V1565L) that is associated with increased susceptibility to proteolysis by ADAMTS13,30 resulting in significantly decreased VWF:CB/VWF:Ag ratios. The particular combination of these genotypes may lead to overwhelming proteolysis of VWF without apparent reduction in platelet turnover. Rondoraptivon pegol elevated platelet counts from a median of 60 (range: 44-166) to a peak of 179 × 109/L (range: 89-264) on day 35 (P < .001; Figure 4). All 4 thrombocytopenic patients experienced a rapid and sustained increase in platelet counts with normalization in 3 of the 4 patients. The only thrombocytopenic patient whose platelet count did not normalize had the lowest trough levels of rondoraptivon pegol. The patient (5) with normal platelet counts at baseline showed no clinically relevant increase in platelet counts.
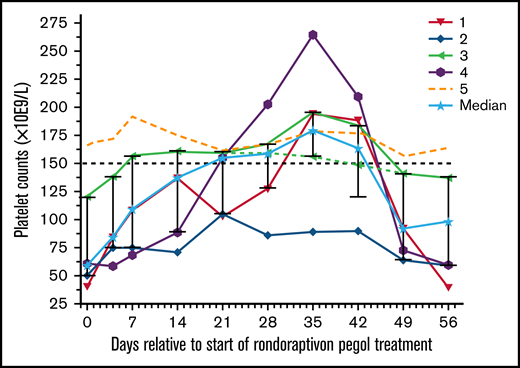
Effects of rondoraptivon pegol on platelet counts. Rondoraptivon pegol increases platelet counts (×10E9/L) in patients with VWD type 2B. Rondoraptivon pegol was given as 3 mg on days 0, 4, and 7, followed by up-titration to 6 to 9 mg at weekly intervals. The highest dose was only given to patients with thrombocytopenia at baseline (open symbols). The horizontal dashed line indicates the lower normal range. The solid line with the stars indicates the median with standard deviation error bars.
Effects of rondoraptivon pegol on platelet counts. Rondoraptivon pegol increases platelet counts (×10E9/L) in patients with VWD type 2B. Rondoraptivon pegol was given as 3 mg on days 0, 4, and 7, followed by up-titration to 6 to 9 mg at weekly intervals. The highest dose was only given to patients with thrombocytopenia at baseline (open symbols). The horizontal dashed line indicates the lower normal range. The solid line with the stars indicates the median with standard deviation error bars.
VWF:Ag, FVIII, and activated partial thromboplastin time.
Circulating VWF:Ag increased by a median threefold (range: 2.0-4.5) from a median of 64% (range: 32%-106%) to 143% (range: 103%-351%, P < .001; Figure 5). As VWF is the carrier of FVIII,24 this led to a doubling of FVIII from a median of 67% (range: 44%-91%) to 134% (range: 114%-200%; P = .002). This was mirrored by a reciprocal decrease in activated partial thromboplastin time from 43 seconds (range: 39-44 seconds) to 31 seconds (range: 30-33 seconds; P < .001) on day 35. As expected,25 plasma levels of VWF propeptide did not change (data not shown).
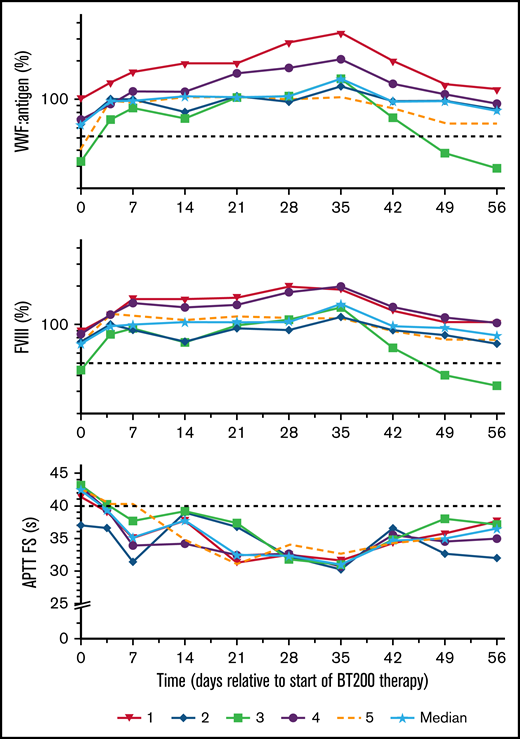
Effects of rondoraptivon pegol on VWF antigen, FVIII activity, and aPTT. Rondoraptivon pegol increases circulating von Willebrand factor mass (%) and coagulation factor FVIII activity (%), with a reciprocal normalization of activated partial thromboplastin time (s) (aPTT actin factor specific). Medians are indicated by solid lines and stars. The horizontal dashed lines indicate the lower normal range for VWF and FVIII and the upper normal range for the aPTT. Note logarithmic scale of y axis for VWF and FVIII layers.
Effects of rondoraptivon pegol on VWF antigen, FVIII activity, and aPTT. Rondoraptivon pegol increases circulating von Willebrand factor mass (%) and coagulation factor FVIII activity (%), with a reciprocal normalization of activated partial thromboplastin time (s) (aPTT actin factor specific). Medians are indicated by solid lines and stars. The horizontal dashed lines indicate the lower normal range for VWF and FVIII and the upper normal range for the aPTT. Note logarithmic scale of y axis for VWF and FVIII layers.
VWF platelet-dependent activity levels and multimer patterns.
Plasma levels of VWF:GPIbM increased from a baseline median 18% (range: 3%-40%) to a peak level of 72% (range: 3%-95%; P < .001; Figure 6), VWF:RCo increased from 17% (range: 9%-21%) to 48% (range: 15%-69%; P < .001) and VWF:CB from 50% (range: 15%-72%) to 100% (range: 32%-133%; P = .002) on day 35. In contrast to the thrombocytopenic patients, the nonthrombocytopenic patient showed only minor changes in VWF activity levels. Although her VWF:Ag levels increased 2.5-fold, her VWFpp remained unchanged. Ristocetin-induced platelet aggregation increased in the thrombocytopenic patients from a baseline median of 45 U (range: 31-120 U) to a peak level of 83 U (range: 55-182 U; P = .017), reflecting the increase in platelet counts.
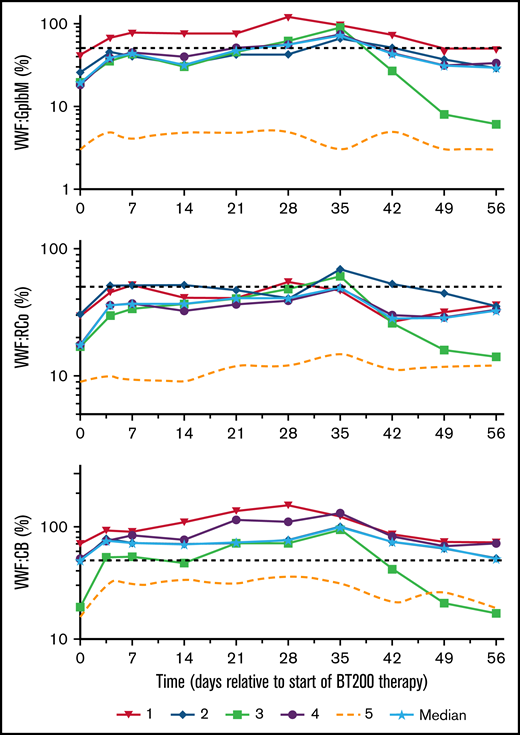
Effects of rondoraptivon pegol on VWF parameters. Rondoraptivon pegol increases plasma levels of VWF–GpIbM (%), ristocetin cofactor (VWF:RCo) (%), and collagen binding activity (VWF:CB) (%) in patients with type 2B VWD. The dotted line represents the nonthrombocytopenic patient, and the horizontal dashed line is the lower normal limit. Note logarithmic scale of y axis.
Effects of rondoraptivon pegol on VWF parameters. Rondoraptivon pegol increases plasma levels of VWF–GpIbM (%), ristocetin cofactor (VWF:RCo) (%), and collagen binding activity (VWF:CB) (%) in patients with type 2B VWD. The dotted line represents the nonthrombocytopenic patient, and the horizontal dashed line is the lower normal limit. Note logarithmic scale of y axis.
Multimer patterns were studied in more detail in 1 patient (3): he showed a gradual normalization of intermediate and HMW multimers until day 35, with a subsequent loss of those intermediate and HMW multimers starting 2 weeks after the final rondoraptivon pegol dose (day 42; supplemental Figure 1a). Subsequently, we compared multimer patterns obtained at baseline and day 35 in the remaining patients: rondoraptivon pegol improved multimer patterns in all thrombocytopenic patients (Figure 2A-B). In contrast, rondoraptivon pegol did not increase intermediate and HMW multimers in the 1 nonthrombocytopenic patient, which is consistent with a limited increase in the VWF platelet-dependent activity levels, although her FVIII, VWF:Ag, and VWF:CB activity levels increased approximately twofold.
Tolerability and safety.
Local and systemic tolerability were excellent. There were no serious or severe adverse events, and none of the adverse effects led to treatment interruption or discontinuation. A full list of adverse events is shown in Table 2.
Adverse events
| System organ class | Severity | Type 2 VWD (N = 5), n (%) m |
|---|---|---|
| Patients with at least one event | Mild | 2 (40.0) 3 |
| Moderate | 2 (40.0) 5 | |
| Blood and lymphatic system disorders | Moderate | 1 (20.0) 1 |
| Lymphadenopathy | Moderate | 1 (20.0) 1 |
| Cardiac disorders | Moderate | 1 (20.0) 1 |
| Tachycardia | Moderate | 1 (20.0) 1 |
| Gastrointestinal disorders | Mild | 2 (40.0) 3 |
| Moderate | 1 (20.0) 2 | |
| Diarrhea | Mild | 1 (20.0) 1 |
| Dyspepsia | Mild | 1 (20.0) 1 |
| Gastrointestinal hemorrhage | Moderate | 1 (20.0) 2 |
| Nausea | Mild | 1 (20.0) 1 |
| Moderate | 1 (20.0) 1 | |
| Urinary tract infection | Moderate | 1 (20.0) 1 |
| System organ class | Severity | Type 2 VWD (N = 5), n (%) m |
|---|---|---|
| Patients with at least one event | Mild | 2 (40.0) 3 |
| Moderate | 2 (40.0) 5 | |
| Blood and lymphatic system disorders | Moderate | 1 (20.0) 1 |
| Lymphadenopathy | Moderate | 1 (20.0) 1 |
| Cardiac disorders | Moderate | 1 (20.0) 1 |
| Tachycardia | Moderate | 1 (20.0) 1 |
| Gastrointestinal disorders | Mild | 2 (40.0) 3 |
| Moderate | 1 (20.0) 2 | |
| Diarrhea | Mild | 1 (20.0) 1 |
| Dyspepsia | Mild | 1 (20.0) 1 |
| Gastrointestinal hemorrhage | Moderate | 1 (20.0) 2 |
| Nausea | Mild | 1 (20.0) 1 |
| Moderate | 1 (20.0) 1 | |
| Urinary tract infection | Moderate | 1 (20.0) 1 |
Percentage is calculated as the number of patients with an event divided by N (N-number of patients in the study; n - number of patients with events, m - number of events).
Two moderate intensity bleeding events occurred in the female thrombocytopenic patient (1). The first one (25 days after the first rondoraptivon pegol injection) was a short episode of gastrointestinal bleeding. This patient had a medical history of severe gastrointestinal bleeding requiring repeated hospitalization often for many weeks at a time. The second event occurred 51 days after the first rondoraptivon pegol injection. Both events of gastrointestinal bleeding stopped soon after shortening her dosing interval of rVWF (from every other day to daily for 3 days). This intensified rVWF infusion in this patient did not markedly increase VWF:Ag levels, although such an effect can be expected after rVWF infusion. No transfusion of packed red blood cells was necessary. This patient also later experienced a retinal bleed after complete drug washout, shortly after the end of this trial.
Discussion
Once-weekly subcutaneous injection of rondoraptivon pegol substantially increased platelet counts and VWF/FVIII levels in thrombocytopenic patients with type 2B VWD. Platelet counts rose threefold in thrombocytopenic patients and normalized in 3 of 4 participants. This is in stark contrast to no effect of rondoraptivon pegol on platelet counts in healthy volunteers (supplemental Figures 2 and 3), showing that the increase in platelet counts is specific for type 2B VWD. This was accompanied by an increase in VWF antigen and platelet-dependent activity levels, restoration of intermediate and HMW multimers of VWF, and enhanced FVIII activity. The sustained correction of thrombocytopenia in type 2B VWD is particularly important because low platelet counts are associated with a 4.5-fold increase in bleeding risk in this population.13
There are 2 subgroups of type 2B VWD: those with and those without HMW multimers. One study showed that only those lacking HMW multimers were thrombocytopenic.15 Our patient with mutation W1313C lacked intermediate and HMW multimers but was not thrombocytopenic, which is in contrast to a single patient (7) reported previously.14 However, phenotypic heterogeneity may even exist between family members carrying the same molecular defect,31 although this may have been because of undetected polymorphisms that were not detected in these patients in the past. Heterogeneity in VWF levels and platelet counts was also observed between our patients 2 and 3, although the lower platelet counts of the father compared with the son may be age related.32 Patient 2 had the slowest rate of rise to steady-state in his plasma concentration of rondoraptivon pegol among all 5 in this patient population, and therefore he might have benefitted from a more aggressive dose titration scheme in such a short trial. Future clinical trials of rondoraptivon pegol in patients with type 2B VWD will separately focus on those with or without persistent thrombocytopenia, recognizing that these 2 subpopulations may differ in their overall pattern of response to treatment. Rondoraptivon pegol induced a sustained increase in the circulating intermediate and HMW multimers in all thrombocytopenic patients and normalized them in 2 of these patients. Again, the only patient (5) who showed no relevant improvement in multimer patterns was the one who had normal platelet counts. One may hypothesize that the patient’s phenotype was influenced by the p.V1565L mutation resulting in overwhelming proteolysis together with the p.W1313C mutation. Future nonresponders to rondoraptivon pegol should be examined for this particular polymorphism to confirm or refute this hypothesis in an extended target population. In contrast to rondoraptivon pegol treatment, plasma-derived VWF replacement products do not correct multimer patterns because they lack HMW multimers because of their cleavage by the presence of ADAMTS-13 during the manufacturing process.33 Furthermore, when rVWF is infused, the modest improvement in multimer patterns is only transient: the proportion of intermediate and HMW multimers was reported to decrease from 30% to 14% at 24 hours and further to 3% at 72 hours.34 This is in agreement with the absence of such HMW multimers in the baseline blood samples from our 2 patients with type 2B VWD despite substitution therapy with rVWF.
The absence of such multimers is pathogenetically involved in the development of angiodysplasia, which can cause recurrent, intractable, severe gastrointestinal bleedings. As reviewed in detail by Mannucci,33 one of the major issues with VWF products in treating patients with VWD is the low efficacy of both on-demand and prophylactic therapy in gastrointestinal bleedings because of angiodysplasia. One possible explanation for this is that a fully intact VWF multimers structure is needed to prevent this kind of bleeding. We have observed an improvement in both intermediate and HMW VWF multimers levels in our study, which could potentially be beneficial in that specific situation. Although there were 2 gastrointestinal bleeding events in patient 1 because of angiodysplasia, this was a patient who had a history of severe gastrointestinal bleedings, requiring hospitalization. During our study, this patient had no severe gastrointestinal bleeding, nor did she require hospitalization, and bleeding stopped after shortening her dosing interval of rVWF. Consistent with observations in the large number of healthy subjects previously studied in the phase 1 trial,25 subcutaneous injections of rondoraptivon pegol were well tolerated in these patients with type 2B VWD without signs of the hypersensitivity reactions that have been described in association with some pegylated drugs.35 In this regard, it is possible that the subcutaneous route of administration and subsequent slow absorption contributes to the safety of rondoraptivon pegol, and aptamers, in contrast to biologics, are nonimmunogenic.35 There was no specific safety signal attributable to rondoraptivon pegol. Although rondoraptivon pegol increases FVIII levels, it is of note that they never exceeded 200%, even in patients under substitution therapy with rVWF. It is assumed that FVIII levels >200% or >250% should be avoided because of the potential risk of thrombosis.36
The unmet medical need for improved treatment of type 2B VWD is clearly shown by our 2 patients on substitution therapy. Although therapy of type 2B VWD is considered similar to other types of VWD,37 situations that can worsen thrombocytopenia need special consideration. Whereas the desmopressin-induced release of endothelial VWF can be effective in other types of VWD (except VWD type 3), use of desmopressin is controversial in type 2B VWD.37 As desmopressin fails to demonstrate beneficial effects on hemostatic parameters in the majority of patients with type 2B VWD or may, in some cases, even worsen them,14 it is usually contraindicated for these patients.38 It is of potential interest that in healthy volunteers rondoraptivon pegol acts synergistically with desmopressin to increase VWF/FVIII.25 Blocking the VWF A1 domain prevented the characteristic stress-induced drop in platelet counts by desmopressin in type 2B VWD.28 In clinical practice, rondoraptivon pegol monotherapy could potentially be used as continuous prophylaxis in thrombocytopenic patients with type 2B VWD and other VWD types. Rondoraptivon pegol could also be used to extend the half-life of substituted VWF/FVIII products in patients with VWD or hemophilia A.39
Limitations of this study include the small sample size, but the trial was adequately powered, and effect sizes were large and are supported by a previous trial with a different A1 domain binding aptamer,21 indicating external validity. Moreover, type 2B VWD is a very rare disease. The 1-month trial duration did not allow us to investigate rondoraptivon pegol under steady-state conditions. For example, the patient whose platelet counts increased, but did not normalize, could possibly have benefited from further dose escalation because his rondoraptivon pegol plasma levels were the lowest. Although it can be estimated from the observed rise in VWF:Ag25 that rondoraptivon pegol can extend the half-life of VWF two- to threefold, a trial in patients with severe VWD would be necessary to formally confirm this. These patients often require home therapy because of severe bleeding,40 and rondoraptivon pegol could allow an increase in dosing intervals and reduction in the frequency of infusions of VWF products.
In conclusion, rondoraptivon pegol addresses the underlying pathophysiology of thrombocytopenic type 2B VWD and significantly increases platelet counts and VWF/FVIII in such patients. These data build the basis for a subsequent phase 2b/3 trial in this patient population.
Acknowledgments
The authors thank the patients for participation in the trial and Dr. Christa Firbas, Sabine Schranz, Sarah Ely PgDip, and Christa Drucker for helping to conduct the clinical trial. Additionally, they are grateful to Sarah for final linguistic improvements.
This trial was sponsored by the Medical University of Vienna, which received financial support from Band Therapeutics Inc., a Guardian Therapeutics company.
Authorship
Contribution: C.A., I.P., and U.D. informed and included patients; U.D. was the principal investigator responsible for conducting the trial; K.D.K., P.Q., and P.J.-S. were responsible for laboratory analyses and quality checking methods; R.S.-P. performed the genetic analysis; I.P., J.C.G., B.J., and U.D. designed the trial and wrote the protocol; S.Z. developed the analytical methods; and all authors contributed to both writing and critically reviewing the manuscript.
Conflict-of-interest disclosure: C.A. received honoraria from Bayer, CSL Behring, NovoNordisk, Pfizer, Roche, Sobi, and Takeda for lectures and participation in advisory board meetings. I.P. received honoraria from Bayer, Biomarin, CSL Behring, NovoNordisk, Pfizer, Roche, Sobi, and Takeda for lectures and advisory board meetings. J.C.G. and S.Z. are employees of Guardian Therapeutics. B.J. is a consultant for Band Therapeutics Inc. The remaining authors declare no competing financial interests.
Correspondence: Bernd Jilma, Department of Clinical Pharmacology, Medical University of Vienna, Spitalgasse 23, 1090 Vienna, Austria; e-mail: bernd.jilma@meduniwien.ac.at; and Ingrid Pabinger, Department of Medicine I, Medical University of Vienna, Spitalgasse 23, 1090 Vienna, Austria; e-mail: ingrid.pabinger@meduniwien.ac.at.

