


Abstract
Myelofibrosis (MF) can present with symptomatic splenomegaly and/or cytopenias including thrombocytopenia. Disease-related thrombocytopenia is a poor prognostic factor with a median overall survival of less than 2 years. Currently approved JAK1/2 inhibitors have not been evaluated in patients with platelets ≤ 50 × 109/L and in fact could potentiate thrombocytopenia because of their combined JAK1/2 inhibitory activity. Pacritinib (PAC), a selective JAK2, fms-like tyrosine kinase 3, interleukin-1 receptor-associated kinase 1 multikinase inhibitor was developed to meet this unmet need. PAC was evaluated in 2 randomized phase 3 trials in the frontline setting (PERSIST-1, PAC 400 mg daily vs best available therapy) and second-line setting in patients with MF with platelets ≤ 100 × 109/L (PERSIST-2, PAC 400 mg daily or 200 mg twice daily vs best available therapy). PERSIST-1 met its primary end point; however, the development of PAC hit a brief pause because of a US Food and Drug Administration–mandated clinical hold for excess of bleeding and cardiac events in the PAC 400 mg daily arm in the PERSIST-1 study. Although the PERSIST-2 study was terminated abruptly because of this clinical hold, it met its splenic response end point and demonstrated a trend toward symptom improvement. Subsequent, diligent review of the PERSIST-1 and PERSIST-2 studies did not confirm an excess of severe bleeding or cardiac events on the PAC arm. Additionally, the dose finding PAC203 study endorsed the safety and efficacy of 200 mg twice daily, leading to the approval of PAC for the treatment of patients with MF with platelets ≤ 50 × 109/L.
“Odyssey” derives from Odysseus, the hero of Homer’s Odyssey, who spent 20 years traveling home from the Trojan war experiencing astonishing adventures en route. Thus, an odyssey is any long, complicated journey, with a quest for a goal often marked by changing fortune.
—Merriam-Webster Dictionary
Introduction
Pacritinib (PAC), a selective JAK2, fms-like tyrosine kinase 3 (FLT3), interleukin-1 receptor-associated kinase 1 (IRAK1) inhibitor, was approved by the US Food and Drug Administration (FDA) on 28 February 2022 for the treatment of adult patients with intermediate-2 or high-risk myelofibrosis (MF) and platelets ≤ 50 × 109/L.1 As such, PAC holds the distinction of being the only drug approved in the cytopenic MF2 population marked by severe thrombocytopenia (platelets ≤ 50 × 109/L). MF (primary or secondary to polycythemia vera or essential thrombocythemia) is a clonal hematopoietic myeloproliferative neoplasm (MPN) fraught with molecular and clinical heterogeneity,3 which in particular has propelled researchers to seek individualized treatment options for patients with symptomatic splenomegaly and cytopenias including thrombocytopenia. Thrombocytopenia (platelets ≤ 100 × 109/L) in MF is an independent negative predictor of overall survival,4,5 and in particular, outcomes in patients with MF and severe thrombocytopenia (ie, platelets ≤ 50 × 109/L) were lugubrious6 because these patients were unable to receive the standard-of-care JAK1/2 inhibitors (ruxolitinib and fedratinib), as neither of these drugs were systematically evaluated in patients with MF and platelets ≤ 50 × 109/L.7,8 Furthermore, the approved labeling for both drugs recommend initiating these agents only in patients with baseline platelet counts of at least 50 × 109/L. Therefore, there is an urgent unmet therapeutic need for such patients, and PAC fulfills this significant treatment void in MF and platelets ≤ 50 × 109/L. This review will discuss the development of PAC, which was not dissimilar to the journey of Odysseus,9 spanning 16 years (2006-2022), a change in commercial partners, a brief FDA hold (2016-2017), and its final approval in a unique treatment space in MF.
Pharmacology of PAC (SB1518)
Discovery of JAK2 V617F, a key pathogenic driver of MPNs in 2005, galvanized the targeted therapeutic development of JAK2 inhibitors in MF.10-12 In 2006, SB1518 (named 21c) was discovered in Singapore, Asia (S*Bio pharma) during a high-throughput screen against Aurora kinase A, in which several novel small molecule aminopyrimidine inhibitors demonstrated activity against kinases such as JAK2 and FLT3.13 Among these small molecule inhibitors of JAK2/FLT3, SB1518,14 a highly selective JAK2/FLT3 inhibitor (JAK2 IC50 = 1 nM; JAK2 V617F IC50 = 3 nM; FLT3 IC50 = 3.9 nM; JAK1 IC50 = 1280 nM, and JAK3 IC50 = 18.3 nM) with its excellent pharmacokinetic profile emerged as the potential candidate for therapeutic development in MF.15 Subsequent kinome-wide profiling of 439 kinases confirmed the distinct inhibitory activity of PAC against JAK2, FLT3, and their respective common mutations, and most importantly, PAC spared JAK1 compared with other JAK inhibitors (ruxolitinib, fedratinib), possibly accounting for its less myelosuppressive effect. Serendipitously, PAC was also found to inhibit several other kinases identified as potential therapeutic targets in oncology, including IRAK1 (IC50 = 13.6 nM)16 (Figure 1).
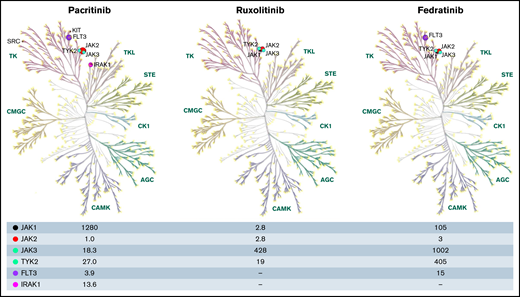
The kinase profile was constructed through KinMap, a web-based interactive tool with built-in human kinome data.41 Kinome profile of PAC adapted from Singer et al16 and the kinase profiles of ruxolitinib and fedratinib from Mullally et al.42 The numbers represent the half maximal inhibitory concentration (IC50) of the respective kinases on the far-left corner. AGC, protein kinase A; G, C group; CAMK, calcium/calmodulin-dependent protein kinases; CK1, casein kinase; CMGC, includes cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and CDK-like kinases; FLT3, fms-like receptor tyrosine kinase 3; IRAK1, interleukin-1 receptor-associated kinase 1; STE, homolog of sterile; TK, tyrosine kinase; TKL, tyrosine kinase-like group of kinases, TYK2-tyrosine kinase 2.
The kinase profile was constructed through KinMap, a web-based interactive tool with built-in human kinome data.41 Kinome profile of PAC adapted from Singer et al16 and the kinase profiles of ruxolitinib and fedratinib from Mullally et al.42 The numbers represent the half maximal inhibitory concentration (IC50) of the respective kinases on the far-left corner. AGC, protein kinase A; G, C group; CAMK, calcium/calmodulin-dependent protein kinases; CK1, casein kinase; CMGC, includes cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and CDK-like kinases; FLT3, fms-like receptor tyrosine kinase 3; IRAK1, interleukin-1 receptor-associated kinase 1; STE, homolog of sterile; TK, tyrosine kinase; TKL, tyrosine kinase-like group of kinases, TYK2-tyrosine kinase 2.
Role of IRAK1 in MF
IRAK1, a serine/threonine kinase, is a key downstream mediator of the pivotal Toll-like (TLR) and interleukin-1 (IL1R) receptor signaling pathway in inflammatory response.17 TLR/IL1R activation recruits specific adaptor proteins such as TIRAP and MyD88, leading to phosphorylation of IRAK1, which sequentially activates TRAF6 resulting in nuclear factor κB (NF-κB) activation.18 NF-κB, a key transcriptional regulator in MPN-associated inflammation, is negatively regulated by microRNA(miR)-146a through IRAK1 inhibition.19,20 Previously, miR array studies have shown decreased levels of miR-146a in neutrophils from patients with MF,21 and recently the Grupo Español de Enfermedades Mieloproliferativas Filadelfia Negativas (GEMFIN) study group showed that functional polymorphisms of miR-146a result in decreased miR-146a expression and can engender MF through increased STAT3 signaling.22 Most importantly, miR-146a knockout mice without any driver MPN mutations demonstrated age-related increase in Stat3 activation resulting in MF phenotype with anemia, myeloproliferation, bone marrow fibrosis, progressive splenomegaly, and extramedullary hematopoiesis, and as expected, IRAK1 protein levels were dramatically increased in the hematopoietic compartment.19,22 Furthermore, endogenous TLR ligands, such as S100A8/A9, have shown to be overexpressed in MPNs, attesting to its inherently inflammatory milieu.23 Recently, Leimkühler et al24 observed that upregulation of S100A8/A9 harbinger disease progression with worsening fibrosis, and demonstrated that inhibition of S100A8/A9 signaling ameliorated myeloproliferation and reduced bone marrow fibrosis in a Jak2 V617F-driven murine model. Of specific interest, PAC treatment inhibited IRAK1 and in turn decreased the expression of S100A8/A9 in a solid tumor cell line.25 Taken together, these data implicate miR-146a and IRAK1-associated inflammation in the pathogenesis and progression of MPN, and PAC, by virtue of its JAK2/IRAK1/FLT3 inhibition, may have multipronged therapeutic activity in MF independent of driver mutation status.
Preclinical evaluation of PAC
Sound preclinical rationale dictated the clinical development of PAC in MF. In cell lines expressing endogenous JAK2 V617F (HEL92.1.7, and SET-2), PAC potently blocked JAK/STAT signaling, induced apoptosis, and effectively curtailed neoplastic cell proliferation in a dose-dependent manner. PAC recapitulated these results in a JAK2 V617F-dependent SET-2 and BaF3 xenograft model with resolution of hepatosplenomegaly in the absence of hematologic toxicities such as anemia or thrombocytopenia or leukopenia and prolonged survival. Furthermore, PAC promoted apoptosis and cell cycle arrest through JAK/STAT inhibition in JAK2 wild-type cell lines as well.26 Collectively, these data corroborated the activity of PAC in MF with or without any driver mutations, the latter possibly attributed to its then unknown IRAK1 inhibitory activity.
Clinical development of PAC
Early-phase studies
The initial phase of PAC development spanned 2 continents. Between 2008 and 2010, 2 phase 1 dose escalation studies enrolled patients with advanced MF (United States: n = 36; Australia: n = 20) and tested 6 dose levels of PAC ranging between 100 and 600 mg daily, administered in 28-day cycles.27,28 At baseline, grade ≥ 2 anemia and/or thrombocytopenia was observed in 65.1% and 48.8% of patients, respectively. Any grade diarrhea (33%) emerged as the most common nonhematologic treatment emergent adverse event (TEAE), and grade ≥ 3 diarrhea (4%) was more frequently observed in PAC > 400 mg daily, likely because of on-target FLT3 inhibition. As patients with baseline cytopenias were included in the safety analysis, occurrences of the hematologic AEs were captured before study drug initiation and were attributed to underlying MF. Although TEAE grade ≥ 3 anemia and thrombocytopenia were observed in 16.3% and 14.0% of patients, respectively, they neither warranted treatment interruption nor discontinuation. PAC exhibited on-target activity (inhibition of pSTAT3/5) from the starting dose level of 100 mg daily. Among the several dose levels tested, there was a less than proportionate increase in systemic exposure at doses from 100 to 400 mg, with negligible increase in exposure > 400 mg daily; therefore, 400 mg daily was identified as the recommended phase 2 dose for further evaluation.27,28
In the phase 1 dose escalation study by Verstovsek et al,27 1 patient with significant preexisting hypokalemia and cardiac comorbidities (atrial fibrillation and coronary artery disease) receiving concomitant QTc potentiating drugs (fluconazole, solifenacin, and amiodarone) experienced grade 3 QTc prolongation that resolved with stopping PAC. This led to a protocol amendment in the phase 2 study eligibility criteria mandating a baseline QTc ≤ 0.47 seconds (Bazett formula) and avoidance of CYP3A4 inducers or inhibitors up to 1 week before PAC initiation.29
Given the favorable myelosuppressive profile of PAC observed in the phase 1 portion, the phase 2 portion enrolled patients with intermediate- or high-risk (per Lille prognostic scoring system)30 newly diagnosed or treatment refractory MF with any baseline platelet count or hemoglobin value regardless of red blood cell or platelet transfusion dependence, with the primary and secondary end points of ≥35% reduction in spleen volume (SVR35%) at week 24 as determined by magnetic resonance imaging and the proportion of treated patients with a ≥50% reduction in symptom score (TSS50) as assessed by the MF-SAF up to week 24, respectively.31 Between January and June 2010, 35 patients were enrolled, of which 43% and 20% of the patients had platelet counts <100 × 109 /L, and <50 × 109 /L, respectively, at baseline. PAC achieved its primary end point with SVR35% in 31% and TSS50 in 48.4% of patients at week 24. Common reasons for treatment discontinuation included TEAEs (26%; of which 17% were deemed related to PAC) and study termination by the sponsor (23%; S*BIO Pte Ltd). There were no new safety signals observed, and in line with the phase 1 studies, the most common nonhematologic TEAE grade ≤ 2 were diarrhea (69%), nausea (46%), and vomiting (31%). Among grade ≥ 3 hematologic toxicities, anemia and thrombocytopenia occurred in 26% and 20% of patients, respectively, with minimal change in baseline counts. Specifically, thrombocytopenia-related treatment discontinuation or interruption occurred in only 9% of patients treated with PAC.29 Subsequently, a pooled safety and efficacy analysis of PAC in patients with MF with platelet counts ≤ 100 × 109 /L reported 37% SVR35% with minimal change in baseline platelet counts or hemoglobin.32 This clinically minimal impact of PAC on hematologic parameters provided the impetus to develop PAC in cytopenic MF.
Initial pharmacokinetic/pharmacodynamic studies of PAC
Given the less than proportional increase in systemic exposure with the administration of once daily dosing regimen of PAC, pharmacokinetic (PK)/pharmacodynamic (PD) population modeling studies on the exposure-efficacy relationship were conducted to explore the possibility of twice daily dosing, to improve on the efficacy of PAC. A pooled exposure-efficacy and exposure AE distribution model was constructed from the PK data (once daily dosing of PAC) obtained from the combined phase 1/2 clinical trials (n = 129),29,32 and these key PK parameters were simulated for the 200 mg twice daily regimen that showed that the 200 mg twice daily regimen is likely to result in a higher (41%) systemic exposure than the 400 mg daily regimen. Furthermore, patients with PK data were divided into quartiles (Q1-Q4) based on their model-predicted area under the curve at steady state, and the corresponding PD data showed that patients in the highest quartile (Q4) exhibited maximal and durable clinical response over time compared with those in lower exposure quartiles. Most importantly, the simulation modeling data projected that approximately 48% of patients receiving the 200 mg twice daily regimen will fall in the Q4 exposure zone as opposed to 25% in the 400 mg daily regimen suggesting that 200 mg twice daily regimen may yield higher area under the curves and lower maximum concentration, which in turn may translate to higher response rates and improve tolerability.33 Together, this data supported further evaluation of 400 mg daily and 200 mg twice daily doses of PAC in MF.
Late-phase development
In the frontline setting, PERSIST-1 (NCT01773187), a multicenter (67 centers worldwide) phase 3 randomized trial evaluated the efficacy and safety of PAC 400 mg daily (vs best available therapy [BAT]: watchful waiting or any symptom directed therapy excluding JAK2 inhibitors in patients with MF with the primary end point of SVR35% at week 24 and TSS50 assessed by MPN-SAF v2.0 as a key secondary end point).34 Patients were randomized in a 2:1 fashion and stratified according to platelet counts (<50 × 109/L vs ≥50 to <100 × 109/L vs ≥100 × 109/L). Between 2013 and 2014, 327 patients were enrolled (PAC: n = 220, BAT: n = 107), and at baseline, 32% and 15% of patients had baseline platelet counts of <100 × 109/L and <50 × 109/L, respectively. In the intent-to-treat population, SVR35% was significantly higher in the PAC-treated patients (19.1% vs 4.7% with BAT, P = .003), and this benefit was consistent regardless of baseline platelet counts. In patients evaluable for response, 36% of patients treated with PAC achieved TSS50 vs 14% in the BAT group (P = .029). Most common nonhematologic toxicities were primarily gastrointestinal, namely grade ≤ 2 diarrhea (58%) and nausea (31%) managed with supportive care. All grade anemia and thrombocytopenia were the most common hematologic toxicities, but the incidence rates were similar between both treatment groups, including grade 3 to 4 anemia (17% vs 15% in the BAT arm) and thrombocytopenia (12% vs 11% in the BAT arm); in fact, treatment with PAC resulted in an increase in platelet counts (week 12: mean +15% [SEM +10% to +25%] week 24: mean +35% [SEM, +18% to +55%]; P = .055) in patients with baseline platelets < 50 × 109/L. Of note, the US FDA mandated a clinical hold on 8 February 2016 because of concerns over bleeding and cardiovascular events and deaths observed in the PAC arm in the PERSIST-1 study, leading to an abrupt closure of the PERSIST-235 study, resulting in an underpowered trial and incomplete data.
PERSIST-2 (NCT02055781), a global, multicenter, randomized, phase 3 trial, evaluated 2 different doses of PAC (200 mg twice daily or 400 mg daily) vs BAT (including ruxolitinib in 45%) in 311 patients with intermediate-1, intermediate-2, and high-risk MF and platelets ≤ 100 × 109/L (Table 1).35 The primary objective was to compare the efficacy of the pooled PAC arms with BAT with co-primary end points of SVR35% and TSS50 at week 24, and the secondary objective was to compare the efficacy of PAC once daily or twice daily vs BAT. At the time of database lock (19 August 2016), PAC (arms combined) was more effective than BAT in terms of SVR35% (18% vs 3%, P = .001) and demonstrated a trend toward improved TSS50 (25% vs 14%, P = .08). Specifically, PAC 200 mg twice daily led to significant improvements in both end points over BAT (SVR35%: 22% vs 3%, P = .001; TSS50: 32% vs 14%, P = .01), and this benefit was observed regardless of prior ruxolitinib therapy (SVR35%: 13% vs 3%,TSS50: 32% vs 15%, respectively) or platelets ≤50 × 109/L (SVR35%: 29% vs 3%, TSS50: 23% vs 13%, respectively) or platelets ≥ 50 × 109/L (SVR35%: 14.3% vs 3%, TSS50: 38.1% vs 15.4%, respectively). When compared with 400 mg once daily, PAC 200 mg twice daily demonstrated numerically improved rates of SVR35% and TSS50 overall and in those treated with prior ruxolitinib or those with platelets ≤ 50 × 109/L. PK studies corroborated the findings from the population modeling studies and affirmed that PAC twice daily was associated with a trend toward increased SVR and TSS reduction and higher systemic exposure.33 TEAE grade ≤ 2 diarrhea (53%) was the most common nonhematologic AE that was less pronounced in the twice daily arm (48%) and resolved in the majority of patients with continued treatment by week 2. The most common AE leading to discontinuation was thrombocytopenia (4%) in the PAC once daily arm and anemia (3%) in the twice daily arm. Despite the truncation of the study because of the US FDA mandated clinical hold, PAC 200 mg twice daily was more effective in achieving spleen and symptom benefit than BAT with favorable benefit-risk profile compared with PAC once daily and BAT35 (Table 1).
Snapshot of PERSIST-2 and PAC-203 clinical trials
| PERSIST-235 | PAC20338 | |
|---|---|---|
| Design | Phase 3/randomized 1:1:1 | Phase 2/randomized 1:1:1 |
| Inclusion criteria | ||
| Disease | Primary, post-ET/PVMF | Primary, post-ET/PVMF |
| Risk | DIPSS INT-1, INT-2, high risk | DIPSS INT-1, INT-2, high risk |
| Prior RX | 1 or 2 JAK-inhibitors | Ruxolitinib |
| Platelet count | ≤100 × 109/L | Any |
| Ruxolitinib (RUX) Intolerance | N/A | RUX<20 mg twice day for ≥ 28 complicated by a requirement for red blood cell transfusion, grade ≥ 3 anemia, thrombocytopenia, hematoma, and/or hemorrhage |
| Ruxolitinib failure | N/A | RUX treatment for ≥ 3 mo with < 10% SVR or < 30% decrease in spleen length or regrowth |
| Key exclusion criteria | ||
| To mitigate bleeding risk | Active bleeding | a) Grade ≥ 2 bleeding events within the previous 3 mo b) Treated with anticoagulation or antiplatelet agents within 14 d prior to starting treatment |
| To mitigate cardiovascular risk | a) Symptomatic/uncontrolled cardiac disease b) NYHA class 3-4 c) MI or unstable angina or CHF 6 mo prior to randomization d) Grade ≥3 cardiac arrythmias e) QTc ≥ 450 ms | a) NYHA class≥2 heart failure b) Grade ≥ 2 cardiac conditions within 6 mo prior to starting treatment c) QTc > 450 ms d) LVEF < 45% |
| Intervention | ||
| Dosing/arms | PAC 400 mg once daily | PAC 100 mg once daily |
| PAC 200 mg twice daily | PAC 100 mg twice daily | |
| BAT (physician choice, symptomatic treatment, observation) | PAC 200 mg twice daily | |
| End points | Co-primary end points | Primary objective: to determine the recommended dose of PAC |
| – SVR 35% and TSS50 at week 24 | Secondary objectives: | |
| a) examine the dose-response relationship for efficacy and safety b) characterize pharmacokinetics and pharmacodynamics of PAC | ||
| SVR35% at week 24 | ||
| Overall | PAC 400 mg daily – 15% | PAC 100 mg once daily – 0% |
| PAC 200 mg twice daily – 22% | PAC 100 mg twice daily – 1.8% | |
| BAT – 3% | PAC 200 mg twice daily – 9.3% | |
| Platelet count <50 × 109/L | PAC 400 mg daily – 18% | PAC 100 mg once daily – 0% |
| PAC 200 mg twice daily–29% | PAC 100 mg twice daily – 0% | |
| BAT – 3% | PAC 200 mg twice daily – 16.7% | |
| TSS50 | ||
| Overall | PAC 400 mg daily – 17% | PAC 100 mg once daily – 7.7% |
| PAC 200 mg twice daily – 32% | PAC 100 mg twice daily – 7.3% | |
| BAT – 14% | PAC 200 mg twice daily – 7.4% | |
| Platelet count <50 × 109/L | PAC 400 mg daily – 16% | PAC 100 mg once daily – 8.7% |
| PAC 200 mg twice daily – 23% | PAC 100 mg twice daily – 0% | |
| BAT – 13% | PAC 200 mg twice daily – 8.3% | |
| Most common TEAEs | ||
| Nonhematologic | ||
| Diarrhea Gr 1 or 2 | PAC 400 mg daily – 67% | PAC 200 mg twice daily – 29.6% |
| PAC 200 mg twice daily – 48% | ||
| BAT – 15% | ||
| Hematologic | ||
| Thrombocytopenia | PAC 400 mg daily – 33% | PAC 200 mg twice daily – 40.7% |
| PAC 200 mg twice daily – 34% | ||
| BAT–23% | ||
| Anemia | PAC 400 mg daily – 33% | PAC 200 mg twice daily – 24.9% |
| PAC 200 mg twice daily – 34% | ||
| BAT – 23% | ||
| TEAE of special interest in patients with platelets ≤ 50 × 109/L 39 | ||
| Cardiac event grade ≥ 3 | PAC 400 mg daily – 13% | PAC 200 mg twice daily – 8% |
| PAC 200 mg twice daily – 9% | ||
| BAT – 19% | ||
| Any bleeding grade ≥ 3 | PAC 400 mg daily – 13% | PAC 200 mg twice daily – 13% |
| PAC 200 mg twice daily – 17% | ||
| BAT – 12% | ||
| PERSIST-235 | PAC20338 | |
|---|---|---|
| Design | Phase 3/randomized 1:1:1 | Phase 2/randomized 1:1:1 |
| Inclusion criteria | ||
| Disease | Primary, post-ET/PVMF | Primary, post-ET/PVMF |
| Risk | DIPSS INT-1, INT-2, high risk | DIPSS INT-1, INT-2, high risk |
| Prior RX | 1 or 2 JAK-inhibitors | Ruxolitinib |
| Platelet count | ≤100 × 109/L | Any |
| Ruxolitinib (RUX) Intolerance | N/A | RUX<20 mg twice day for ≥ 28 complicated by a requirement for red blood cell transfusion, grade ≥ 3 anemia, thrombocytopenia, hematoma, and/or hemorrhage |
| Ruxolitinib failure | N/A | RUX treatment for ≥ 3 mo with < 10% SVR or < 30% decrease in spleen length or regrowth |
| Key exclusion criteria | ||
| To mitigate bleeding risk | Active bleeding | a) Grade ≥ 2 bleeding events within the previous 3 mo b) Treated with anticoagulation or antiplatelet agents within 14 d prior to starting treatment |
| To mitigate cardiovascular risk | a) Symptomatic/uncontrolled cardiac disease b) NYHA class 3-4 c) MI or unstable angina or CHF 6 mo prior to randomization d) Grade ≥3 cardiac arrythmias e) QTc ≥ 450 ms | a) NYHA class≥2 heart failure b) Grade ≥ 2 cardiac conditions within 6 mo prior to starting treatment c) QTc > 450 ms d) LVEF < 45% |
| Intervention | ||
| Dosing/arms | PAC 400 mg once daily | PAC 100 mg once daily |
| PAC 200 mg twice daily | PAC 100 mg twice daily | |
| BAT (physician choice, symptomatic treatment, observation) | PAC 200 mg twice daily | |
| End points | Co-primary end points | Primary objective: to determine the recommended dose of PAC |
| – SVR 35% and TSS50 at week 24 | Secondary objectives: | |
| a) examine the dose-response relationship for efficacy and safety b) characterize pharmacokinetics and pharmacodynamics of PAC | ||
| SVR35% at week 24 | ||
| Overall | PAC 400 mg daily – 15% | PAC 100 mg once daily – 0% |
| PAC 200 mg twice daily – 22% | PAC 100 mg twice daily – 1.8% | |
| BAT – 3% | PAC 200 mg twice daily – 9.3% | |
| Platelet count <50 × 109/L | PAC 400 mg daily – 18% | PAC 100 mg once daily – 0% |
| PAC 200 mg twice daily–29% | PAC 100 mg twice daily – 0% | |
| BAT – 3% | PAC 200 mg twice daily – 16.7% | |
| TSS50 | ||
| Overall | PAC 400 mg daily – 17% | PAC 100 mg once daily – 7.7% |
| PAC 200 mg twice daily – 32% | PAC 100 mg twice daily – 7.3% | |
| BAT – 14% | PAC 200 mg twice daily – 7.4% | |
| Platelet count <50 × 109/L | PAC 400 mg daily – 16% | PAC 100 mg once daily – 8.7% |
| PAC 200 mg twice daily – 23% | PAC 100 mg twice daily – 0% | |
| BAT – 13% | PAC 200 mg twice daily – 8.3% | |
| Most common TEAEs | ||
| Nonhematologic | ||
| Diarrhea Gr 1 or 2 | PAC 400 mg daily – 67% | PAC 200 mg twice daily – 29.6% |
| PAC 200 mg twice daily – 48% | ||
| BAT – 15% | ||
| Hematologic | ||
| Thrombocytopenia | PAC 400 mg daily – 33% | PAC 200 mg twice daily – 40.7% |
| PAC 200 mg twice daily – 34% | ||
| BAT–23% | ||
| Anemia | PAC 400 mg daily – 33% | PAC 200 mg twice daily – 24.9% |
| PAC 200 mg twice daily – 34% | ||
| BAT – 23% | ||
| TEAE of special interest in patients with platelets ≤ 50 × 109/L 39 | ||
| Cardiac event grade ≥ 3 | PAC 400 mg daily – 13% | PAC 200 mg twice daily – 8% |
| PAC 200 mg twice daily – 9% | ||
| BAT – 19% | ||
| Any bleeding grade ≥ 3 | PAC 400 mg daily – 13% | PAC 200 mg twice daily – 13% |
| PAC 200 mg twice daily – 17% | ||
| BAT – 12% | ||
DIPSS, Dynamic International Prognostic Scoring System; ET, essential thrombocythemia; PV, polycythemia vera.
Resolved US FDA hold of PAC
In the pooled retrospective efficacy analysis of 189 patients with severe thrombocytopenia (platelets ≤ 50 × 109/L) treated in the PERSIST-1 and PERSIST-2 studies, the median platelet count at baseline was 28 × 109/L, 63.5% had hemoglobin < 10 g/dL, and 35% had prior exposure to another JAK inhibitor. In this high-risk population, PAC-treated patients achieved significantly higher SVR35% (23.1% vs 2.1%, P = .0007) and modified TSS50 (25% vs 8.1%, P = .0441) at week 24 compared with those treated with BAT), with notably higher spleen response rates in the PAC 200 mg twice daily arm (29.0% vs 20.5% in 400 mg daily). The most common hematologic TEAEs in this thrombocytopenic population treated with PAC were thrombocytopenia (34.8%) and anemia (31.8%). Given the baseline cytopenias, hematologic TEAEs were generally grade 3 or 4, which generally did not warrant dose reduction (4.5% and 2.3% for thrombocytopenia and anemia, respectively) or discontinuation (3.8% and 3.8%, respectively). Despite the treatment associated thrombocytopenia with PAC, there was no excess of hemorrhagic events in the PAC arm (PAC vs BAT: grade ≥ 1, 51.5% vs 59.6%; grade 3-4, 13.6% vs 10.5%; fatal, 2% vs 0%). Similarly, there was no excess of cardiac events with PAC compared with BAT (grade 3-4, 9.1% vs 14%; fatal, 3% vs 1.8%), with similar survival among both arms (hazard ratio, 1.01; 95% confidence interval, 0.57-1.80).36 These data confirmed the safety of PAC in patients with MF and severe thrombocytopenia (platelets ≤ 50 × 109/L). After thorough review of mature PERSIST-1 data and complete PERSIST-2 data and submission of a study protocol comparing 3 doses of PAC (#NCT03165734) to determine whether a lower dose of PAC would be safer with maintained efficacy, the clinical hold was removed on 5 January 2017.
Resolute commitment to safety and efficacy
PAC203,37 a multicenter, phase 2 randomized dose-finding study, evaluated PAC in patients with advanced MF who are intolerant of or resistant to ruxolitinib with the primary objective of determining the recommended dose of PAC, and the secondary objective of characterizing the exposure-efficacy and exposure-safety relationship using all available PK/PD data for PAC-treated patients from PAC203 and previous studies (Table 1). Enhanced eligibility criteria, rigorous monitoring, and strict dose modifications were implemented to mitigate the risk of cardiac and hemorrhagic events. Between July 2017 and January 2019, 161 patients were randomized 1:1:1 to PAC 100 mg once daily, 100 mg twice daily, or 200 mg twice daily and stratified by baseline platelet counts. Baseline demographics were well matched between the treatment arms. In this high-risk cohort, 44.1% had a platelet counts < 50 × 109/L, 70.8% had hemoglobin < 10 g/dL, and 40% harbored high-molecular-risk mutations (ASXL1, SRSF2, EZH2, IDH1/2, U2AF1Q157). The SVR35% rate was highest with 200 mg twice daily (100 mg once per day, 0%; 100 mg twice per day, 1.8%; 200 mg twice per day, 9.3%) and was more pronounced in patients with severe thrombocytopenia (platelet counts < 50 × 109/L; −17%). Similarly, median percent reduction in TSS was higher in the 200 mg twice daily arm (–3%, −16%, and −27%, respectively). There was no excess of grade ≥ 3 hemorrhagic or cardiac events with PAC 200 mg twice daily, in line with the data from previous studies.36,38 Pooled PK data from 16 studies (n = 630) encompassing PAC doses of 100 to 600 mg daily established the population PKs for PAC. In particular, pooled dose-response data from the PAC203, PERSIST-1, and PERSIST-2 studies demonstrated a directly proportional relationship between dose and response (ie, greatest SVR and TSS50 with 200 mg twice daily dose compared with 100 mg twice daily or 100 mg once daily) that was substantiated by the PK/PD population modeling based exposure-response analysis. Collective PK/PD data33-35 and clinical data established PAC 200 mg twice daily as the recommended dose for the phase 3 study in patients with MF and platelets < 50 × 109/L.
A pooled safety analysis of patients with baseline platelet counts < 50 × 109/L treated with PAC 200 mg twice daily in PERSIST-2 (n = 47) and PAC203 (n = 24) or BAT (n = 42) in PERSIST-2 was recently presented (Table 1).39 In this analysis, compared with BAT (19%), fatal TEAEs were lower in the pooled PAC group (13%). Similarly, the incidence of serious cardiac events (10% vs 21%) and those grade ≥ 3 (9% vs 19%) were lower in the pooled PAC group compared with the BAT group; no major adverse cardiac event was observed in the pooled PAC group. Of note, the incidence of serious bleeding AEs was lower with PAC 200 mg twice daily (13%) in PAC203 than those reported with PAC 200 mg twice daily (17%) in PERSIST-2, likely attributed to the enhanced safety measures in place for the PAC203 study. On meticulous review of this collective data, the US FDA conferred accelerated approval of PAC 200 mg twice daily for the treatment of patients with MF and platelet counts ≤ 50 × 109/L.
Prescribing information for PAC
PAC is the drug of choice in patients with MF and platelet counts ≤ 50 × 109/L. Before starting treatment with PAC, a complete blood count with differential, coagulation profile (prothrombin time/international normalized ratio, partial thromboplastin time, and thrombin time), and baseline EKG must be obtained to monitor for safety. As PAC is primarily metabolized by cytochrome P450 (CYP)3A4, concomitant use of strong CYP3A4 inhibitors or inducers are contraindicated. Avoid using PAC in patients with active bleeding or baseline QTc > 480 ms. Before any planned surgical procedure, hold PAC to decrease the risk of bleeding. Treatment emergent thrombocytopenia and diarrhea can be managed by dose reductions and/or interruptions (Table 2). For clinically significant treatment emergent thrombocytopenia lasting ≥ 7 days, PAC should be held and restarted at 50% dose reduction on resolution. Diarrhea is managed with antidiarrheals and mostly abates within 2 weeks without warranting treatment modification. Correct electrolyte disturbances at the earliest to avoid the risk of QTc prolongation. Continue to monitor the patients for ongoing symptom benefit and proactively manage hematologic and nonhematologic AEs.38
Dose modifications of PAC in specific situations
| Symptoms/signs | Dose modification |
|---|---|
| Diarrhea | |
| New onset | Initiate antidiarrheal medications |
| Increase of at least 7 stools per day over baseline | Hold PAC until diarrhea resolves |
| Recurrence | Hold PAC until diarrhea resolves and restart at 50% of the original dose |
| Thrombocytopenia | |
| Clinically significant worsening of platelet counts >7 d | Hold PAC until platelets recover to baseline, and restart at 50% of the original dose |
| Bleeding | |
| Moderate bleeding necessitating intervention | Hold PAC until bleeding resolves and restart at the last given dose |
| Severe bleeding requiring transfusion, or hospitalization | Hold PAC until bleeding resolves and restart at 50% the last given dose |
| Life-threatening bleeding or recurrence of severe bleeding | Discontinue PAC |
| QTc prolongation | |
| >500 ms or >60 ms from baseline | Hold PAC |
| If QTc prolongation resolves to ≤480 ms or baseline ≤7 d, restart PAC at the same dose | |
| If time to resolution >7 d, restart PAC at 50% of the original dose |
| Symptoms/signs | Dose modification |
|---|---|
| Diarrhea | |
| New onset | Initiate antidiarrheal medications |
| Increase of at least 7 stools per day over baseline | Hold PAC until diarrhea resolves |
| Recurrence | Hold PAC until diarrhea resolves and restart at 50% of the original dose |
| Thrombocytopenia | |
| Clinically significant worsening of platelet counts >7 d | Hold PAC until platelets recover to baseline, and restart at 50% of the original dose |
| Bleeding | |
| Moderate bleeding necessitating intervention | Hold PAC until bleeding resolves and restart at the last given dose |
| Severe bleeding requiring transfusion, or hospitalization | Hold PAC until bleeding resolves and restart at 50% the last given dose |
| Life-threatening bleeding or recurrence of severe bleeding | Discontinue PAC |
| QTc prolongation | |
| >500 ms or >60 ms from baseline | Hold PAC |
| If QTc prolongation resolves to ≤480 ms or baseline ≤7 d, restart PAC at the same dose | |
| If time to resolution >7 d, restart PAC at 50% of the original dose |
Conclusion
The accelerated approval of PAC will facilitate robust collection of real-world data that would further enable us to understand the fullest extent of efficacy and safety of PAC in patients with MF and severe thrombocytopenia. To confirm the clinical benefit and safety of PAC in the patient population with platelets < 50 × 109/L, a postmarketing confirmatory phase 3 trial is underway. PACIFICA (#NCT03165734), a randomized phase 3 study, is currently evaluating PAC 200 mg twice daily against physician's choice (corticosteroids, hydroxyurea, danazol, or low-dose ruxolitinib) in patients with MF and severe thrombocytopenia (platelet ≤ 50 × 109/L) who have had up to 180 days of prior treatment with a JAK2 inhibitor or are JAK2 inhibitor naïve. The primary objective is to compare the efficacy of PAC vs physician's choice based on the proportion of patients achieving SVR35% at week 24, and the secondary objectives include comparisons of the proportion of patients achieving TSS50 at week 24, assessment as measured by the patient global impression of change, and overall survival.40 In addition to MF, PAC may be explored in related myeloid neoplasms like chronic myelomonocytic leukemia, or other malignant and inflammatory conditions including prostate cancer, breast cancer, and chronic graft-versus-host disease. In summary, PAC approval ushers in the new era of individualized treatment options in patients with MF and cytopenias, and most importantly, PAC is a welcome therapeutic option for those patients with MF with platelets ≤ 50 × 109/L who until recently have been unable to receive appropriate therapies.
Authorship
Contribution: S.V. and J.M. conceptualized the article; S.V. did the literature search, wrote the original draft, and created the figures; J.M. provided significant edits and contributed crucial editorial oversight; and both authors critically reviewed the manuscript for intellectual content and approved the final manuscript.
Conflict-of-interest disclosure: J.M. receives research funding paid to the institution from Incyte, Novartis, Roche, Merck, Geron, CTI Bio, BMS, Abbvie, Celgene, Kartos, and PharmaEssentia and consulting fees from Incyte, CTI Bio, Constellation, Geron, Kartos, BMS, Celgene, Novartis, Karyopharm, Sierra Oncology, Abbvie, PharmaEssentia, and Galecto. S.V. declares no competing financial interests.
Correspondence: John Mascarenhas, Division of Hematology/Oncology, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1079, New York, NY 10029; e-mail: john.mascarenhas@mssm.edu.

