

Abstract
Following the discovery of the JAK2V617F mutation in myeloproliferative neoplasms in 2005, fedratinib was developed as a small molecular inhibitor of JAK2. It was optimized to yield low-nanomolar activity against JAK2 (50% inhibitory concentration = 3 nM) and was identified to be selective for JAK2 relative to other JAK family members (eg, JAK1, JAK3, and TYK2). It quickly moved into clinical development with a phase 1 clinical trial opening in 2008, where a favorable impact on spleen and myelofibrosis (MF) symptom responses was reported. A phase 3 trial in JAK2 inhibitor treatment-naive MF patients followed in 2011 (JAKARTA); a phase 2 trial in MF patients resistant or intolerant to ruxolitinib followed in 2012 (JAKARTA-2). Clinical development suffered a major setback between 2013 and 2017 when the US Food and Drug Administration (FDA) placed fedratinib on clinical hold due to the development of symptoms concerning for Wernicke encephalopathy (WE) in 8 of 608 subjects (1.3%) who had received the drug. It was ultimately concluded that there was no evidence that fedratinib directly induces WE, but clear risk factors (eg, poor nutrition, uncontrolled gastrointestinal toxicity) were identified. In August 2019, the FDA approved fedratinib for the treatment of adults with intermediate-2 or high-risk MF. Notably, approval includes a “black box warning” on the risk of serious and fatal encephalopathy, including WE. FDA approval was granted on the basis of the JAKARTA studies in which the primary end points (ie, spleen and MF symptom responses) were met in ∼35% to 40% of patients (JAKARTA) and 25% to 30% of patients (JAKARTA-2), respectively.
Introduction
Fedratinib is a JAK2-selective kinase inhibitor recently approved by the US Food and Drug Administration (FDA) for the treatment of adult patients with intermediate-2 or high-risk myelofibrosis (MF). As such, it becomes the second approved JAK2 inhibitor in MF (the first was ruxolitinib in 2011) and can now be prescribed in the first line in JAK inhibitor–naive patients with MF or in the second line in patients with MF who are resistant or intolerant to ruxolitinib. The path to FDA approval has been complex, including a period of FDA clinical hold (2013-2017) and involving 4 separate commercial partners over more than a decade. FDA approval includes a “black box warning” in the prescribing information with advice regarding the risk of serious and fatal encephalopathy, including Wernicke encephalopathy (WE), which, although rare and ultimately determined not to be caused by fedratinib, was the reason for the FDA clinical hold. This review describes the evolution of fedratinib from its initial development as a rationally designed JAK2 inhibitor through preclinical and clinical development (including the pivotal JAKARTA clinical trials), concluding with future directions encompassing the ongoing FREEDOM (trial for additional safety data) and FREEDOM2 trials (second-line trial in ruxolitinib-intolerant/resistant patients). After such a long road, the approval of fedratinib is welcome, providing a much-needed additional treatment option for patients with MF and revitalizing therapeutic development in myeloproliferative neoplasms (MPNs) more broadly.
Basic science/drug development
Discovery of JAK2V617F and development of JAK2 inhibitors in MPNs
The discovery of the JAK2V617F mutation in 2005 launched the era of rationally designed targeted therapy in MPNs.1-4 The JAK2V617F mutation, which activates JAK2 signaling, is the most common somatic mutation in MPNs and, importantly, is disease-initiating and central to MPN pathogenesis.5,6 The therapeutic potential of an oncogenic kinase allele was immediately apparent, and JAK2 inhibitors were rapidly developed and tested in the clinic. In 2011, ruxolitinib became the first JAK2 inhibitor approved by the FDA for the treatment of MF, followed by approval of fedratinib in 2019. Additional JAK2 inhibitors (eg, pacritinib and momelotinib) are currently in late-phase clinical trials for MF. Because the currently available JAK2 inhibitors are not JAK2V617F-mutant specific and because the other disease-initiating MPN-phenotypic driver mutations (ie, mutant calreticulin [CALR] and MPLW515L/K) also activate JAK-STAT signaling, the use of JAK2 inhibitors is not restricted to JAK2 mutant–induced MPNs. Although the clinical development of JAK2 inhibitors has focused primarily on MF, ruxolitinib is also FDA approved for the treatment of polycythemia vera in cases in which hydroxyurea resistance or intolerance has been demonstrated.7 FDA approval of fedratinib is currently restricted to MF.
Development of fedratinib, chemical structure, and spectrum of activity
Fedratinib was identified using rational structure-based techniques to design and optimize a novel series of pyrimidine-based inhibitors to target JAK2. Initially, a hit with a 50% inhibitory concentration (IC50) of ∼5 μM was identified from a kinase-inhibitor library, and 2 crystal structures available in the literature (2B7A.pdb for JAK2 and 1YVJ.pdb for JAK3)8,9 were used to design molecules guided by molecular modeling. With this approach, the series was rapidly optimized to yield low-nanomolar IC50 concentration inhibitors with drug-like properties, of which fedratinib was chosen for clinical development based on its IC50 of 3 nM against JAK2, target selectivity, metabolic stability, and promising drug properties. The chemical structure of the small molecule inhibitor fedratinib is shown in Figure 1.10
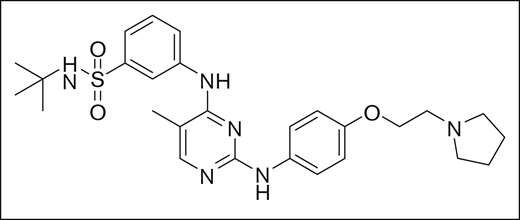
Fedratinib was identified to be an agent selective for JAK2 relative to other kinases in general and relative to other Janus family kinases in particular10 (Table 1). This distinct profile could be advantageous because JAK1, JAK3, and TYK2, the other members of the Janus family of kinases, are critical for proper immune function.11 Agents with activity against JAK1, JAK3, or TYK2, including ruxolitinib, have been shown to have activity as immunosuppressive agents in indications such as graft-versus-host disease,12 psoriasis,13 and rheumatoid arthritis.14 Because mortality associated with infections has been reported to be elevated in MF patients,15 further immune suppression may be problematic. The specificity of fedratinib was tested against purified kinase proteins from JAK family members and a wide range of kinases outside of the Janus family. The only kinase found to have an IC50 within 10× of the JAK2 IC50 was Flt3 with an IC50 of 15 nM, or 5× the JAK2 IC50. Within the Janus family of kinases, fedratinib was found to be 35-, 135-, and 334-fold less active against JAK1, JAK3, and Tyk2, respectively (Table 1).10 In comparison, ruxolitinib is equipotent against both JAK1 and JAK2, 6× less potent against JAK3 and 140× less potent against TYK2.16 Pacritinib and momelotinib are the most advanced other JAK inhibitors currently under development. Pacritinib is reported to be equipotent against JAK2 and FLT3, twofold less potent against TYK2, and significantly less active against other tested kinases.17 Momelotinib is reported to have roughly equal potency against JAK1, JAK2, and TYK2.18 As such, fedratinib is the most selective JAK2 inhibitor currently available.
Kinase profiles of current JAK2 inhibitors
| Kinase | Enzyme IC50, nM | |||
|---|---|---|---|---|
| Fedratinib | Ruxolitinib | Pacritinib | Momelotinib | |
| JAK1 | 105 | 3.3 | 1280 | 11 |
| JAK2 | 3 | 2.8 | 23 | 18 |
| JAK3 | 1002 | 428 | 520 | 155 |
| TYK2 | 405 | 19 | 50 | 17 |
| Kinase | Enzyme IC50, nM | |||
|---|---|---|---|---|
| Fedratinib | Ruxolitinib | Pacritinib | Momelotinib | |
| JAK1 | 105 | 3.3 | 1280 | 11 |
| JAK2 | 3 | 2.8 | 23 | 18 |
| JAK3 | 1002 | 428 | 520 | 155 |
| TYK2 | 405 | 19 | 50 | 17 |
Preclinical studies of fedratinib
Fedratinib (then TG101348), tested initially in preclinical MPN mouse models, was quickly shown to be safe and efficacious. In a retroviral JAK2V617F-driven MPN mouse model, fedratinib reduced blood counts and splenomegaly without toxicity.10 Similar efficacy was shown in an immunocompromised mouse xenotransplantation model in which human cord blood progenitors virally transduced with JAK2V617F were found to have diminished erythroid engraftment following fedratinib treatment.19 Antiproliferative effects of fedratinib were also demonstrated in MPN cell lines and in colony-forming unit assays using primary human MPN cells.10,19,20 Later, using a genetic Jak2V617F knockin mouse, it was demonstrated that, although fedratinib was effective at reducing blood counts and splenomegaly, it did not eradicate disease-propagating MPN stem cells,21 a finding that has been replicated in clinical trials in which the JAK2 inhibitors have not shown strong clonal selectivity for JAK2-mutant cells.6
Phase 1 study of fedratinib
Building on these promising preclinical studies, a multicenter phase 1 trial of fedratinib (then TG101348) was performed in patients with intermediate- or high-risk MF.22 Patients were assigned to 1 of 8 cohorts ranging from 30 mg to 800 mg of fedratinib once daily, according to a standard 3+3 cohort design. Fifty-nine patients were treated, including 28 in the dose-escalation phase. Fedratinib was administered continuously in 28-day cycles for 24 weeks. The maximum tolerated dose (MTD) was 680 mg per day and the dose-limiting toxicity was an asymptomatic and reversible increase in serum amylase levels. Dose-dependent gastrointestinal (GI) side effects (nausea, vomiting, diarrhea) were generally low grade with grade 3 events seen almost exclusively in the MTD cohort. On-target toxicity in the form of grade 3 or 4 cytopenias were highest for anemia (35% patients), followed by thrombocytopenia (24% patients), and/or neutropenia (10% patients). By 6 weeks, 39% of patients achieved a spleen response; and, although there was no significant change in cytokine levels, more than one-half of the patients treated reported improvement in constitutional MF-related symptoms. Given that fedratinib was generally well tolerated and showed evidence of clinical activity, it moved forward in clinical development.
Commercial development of fedratinib: TargeGen, Sanofi, Impact, Celgene
Fedratinib development took a tortuous relay path requiring multiple partners. Fedratinib was initially identified, entered into clinical development, and sponsored in phase 1 and 2 clinical trials by TargeGen, a San Diego–based biotech company. Based on promising clinical activity for fedratinib, in 2010 TargeGen was acquired by Sanofi. Sanofi then sponsored additional clinical studies including a pivotal 289-patient phase 3 trial (JAKARTA) in intermediate-2/high-risk MF patients,23 a phase 2 study (JAKARTA-2) in ruxolitinib-refractory or -intolerant patients,24 and a host of supporting pharmacokinetic and safety studies to support regulatory filings. In total, 877 subjects were treated with fedratinib in 18 clinical trials sponsored by TargeGen or Sanofi. The phase 3 study was completed and regulatory meetings were held with US and European authorities in anticipation of new drug application and marketing authorization application filings. Some patients enrolled in the phase 1 trials were on-drug for >5 years at this point. In November 2013, Sanofi notified the FDA of 8 subjects across all studies who had been treated with fedratinib who experienced neurological symptoms suggestive of WE, a neurological condition caused by thiamine malnutrition. In response, the FDA placed the drug on clinical hold. The following week, Sanofi terminated all clinical development of fedratinib. In 2016, Impact Biomedicines acquired all rights to fedratinib from Sanofi. In 2018, after clinical review finding that most of the subjects in question were experiencing cachexia or other systemic nutritional challenge, the FDA agreed to lift the clinical hold. In January 2018, Celgene acquired all rights to fedratinib through acquisition of Impact Biomedicines. Regulatory filings were made in the United States in late 2018 and fedratinib was ultimately approved by the FDA in August 2019.25
Pharmacokinetics, dosing considerations
Fedratinib is rapidly absorbed following single oral doses (10-680 mg)26 ; the time to maximum serum concentration is 2 to 4 hours following the administration of fedratinib 400 mg once daily.25 Fedratinib is metabolized by cytochrome P450 enzymes (CYP3A4, CYP2C19) and, as a result, dose reductions are required; in some cases, avoiding coadministration with cytochrome P450 inhibitors is required (potential drug-drug interactions should be checked prior to treatment initiation).25 Fedratinib is mainly excreted in feces, and the effective half-life is 41 hours.25 Age (20-95 years), sex, race (white, Asian), body weight (40-135 kg), mild to moderate hepatic impairment, or mild renal impairment do not significantly affect the pharmacokinetics of fedratinib.25 However, moderate (creatinine clearance, 30-59 mL/min) or severe (creatinine clearance, 15-29 mL/min) renal impairment results in increased fedratinib exposure; consequently, dose reductions are required for severe renal impairment.25
Clinical applications
Landscape of MF therapy prefedratinib
MF is characterized by clonal proliferation, progressive splenomegaly, marrow fibrosis, and constitutional symptoms, typically cytopenia and shortened life expectancy.27 The condition may be primary or secondary to a preexisting other MPN. Until the introduction of JAK inhibitors, and apart from rare instances of patients being able to undergo curative allogeneic stem cell transplantation, MF treatment focused upon individual facets of disease (ie, anemia). Ruxolitinib (Novartis Pharmaceuticals, Basel, Switzerland), the first-in-class JAK1/2 inhibitor, when compared with either placebo or best available therapy, significantly alleviated disease-related symptoms and reduced splenic volume, and later was also demonstrated to prolong life.28,29 However, although the majority of patients respond to ruxolitinib, some patients are resistant and others will become resistant/intolerant over time; the median duration of response in COMFORT trials being ∼3 years. Furthermore, a retrospective analysis of a phase 1/2 study of patients with MF treated with ruxolitinib found that outcomes after ruxolitinib discontinuation were poor, with a median overall survival of 14 months (survival after ruxolitinib discontinuation was ∼6 months in patients with clonal evolution).30 There are, therefore, gaps in the MF therapeutic algorithm, and, importantly for those patients with MF who have failed or become intolerant to ruxolitinib, the overall prognosis is often poor.31
Fedratinib in JAK inhibitor–naive patients
Given that the MTD of fedratinib was found to be 680 mg daily from the phase 1 trial22 (dose-limiting toxicity hyperamylasemia) and that there was favorable impact on splenomegaly and MF symptoms, the definitive randomized phase 3 trial was initiated. The JAKARTA study23 (Table 2) was launched at 94 centers worldwide, with 3 equal arms (400 mg daily, 500 mg daily, or placebo [whether 400 mg or 500 mg per day was optimal was unclear]) for 24 weeks with crossover from placebo after that time. The primary end point was reduction in spleen volume (SVR) by at least 35%, with confirmation 4 weeks later. The secondary end point was 50% reduction in symptom burden by the Myelofibrosis Symptom Assessment Form (MFSAF). Between 2011 and 2012, 289 patients were enrolled (96 at 400 mg of fedratinib, 97 at 500 mg of fedratinib, and 96 placebo). The SVR (all confirmed 4 weeks later) observed at week 24 was 35 in the 400-mg group (36%), 39 in the 500-mg group (40%), and 1 in the placebo group (1%). The MFSAF symptom response (noted by week 4, durable until week 24) was 33 of 91 for 400 mg (36%), 31 of 91 for 500 mg (34%), and 6 of 85 for placebo (7%). Responses were not correlated with MF disease subtype, prognostic risk, or JAK2-V617F mutation status (nb, this study preceded the discovery of the CALR mutation in MF). Anemia was the most common hematological toxicity, with an initial nadir but usually improvement. GI toxicities of nausea and diarrhea were the most common nonhematological toxicity, were usually low grade, responded to supportive-care medications (ie, loperamide and/or ondansetron), and diminished over trial conduct. It is particularly noteworthy that 4 cases of an ill-defined encephalopathy were noted in patients in the 500-mg fedratinib arm that were at least suspected of being WE, which led to an abrupt FDA-mandated clinical hold and closure of the JAKARTA study.
Comparison of JAKARTA (frontline) and JAKARTA-2 (second-line) trials
| JAKARTA1 (frontline)23 | JAKARTA2 (second line)24 | |
|---|---|---|
| Design | Phase 3/randomized PB controlled | Single arm |
| Dosing/arms | Placebo FEDR 400 mg FEDR 500 mg | FEDR 400 mg |
| Inclusion | Disease: primary, post-ET/PV MF Risk: DIPSS INT-2, high risk Prior RX: JAK-inhibitor naive | Disease: primary, post-ET/PV MF Risk: DIPSS INT-1 (symptomatic), INT-2, high risk Prior RX: ruxolitinib intolerant/refractory |
| Primary end point | >35% SVR | >35% SVR |
| Key secondary end point | ≥50% reduction in MFSAF-TSS | ≥50% reduction in MFSAF-TSS |
| Enrollment | N = 289 | N = 97 |
| Initial published response rates | ||
| Spleen volume response (>35% volume reduction) | FEDR 400 mg (36%) FEDR 500 mg (40%) Placebo (1%) | FEDR 400 mg (55% of 83 evaluable) |
| MFSAF-TSS (>50% reduction) | FEDR 400 mg (36%) FEDR 500 mg (34%) Placebo (7%) | FEDR 400 mg (26% of 90 evaluable) |
| Toxicity | Grade 1-2 GI toxicities Grade 3-4 cytopenias Suspected WE (more so in 500-mg arm) led to trial hold | Consistent with JAKARTA study toxicity • Low-grade GI TOX • Grade 3-4 anemia/thrombocytopenia |
| JAKARTA1 (frontline)23 | JAKARTA2 (second line)24 | |
|---|---|---|
| Design | Phase 3/randomized PB controlled | Single arm |
| Dosing/arms | Placebo FEDR 400 mg FEDR 500 mg | FEDR 400 mg |
| Inclusion | Disease: primary, post-ET/PV MF Risk: DIPSS INT-2, high risk Prior RX: JAK-inhibitor naive | Disease: primary, post-ET/PV MF Risk: DIPSS INT-1 (symptomatic), INT-2, high risk Prior RX: ruxolitinib intolerant/refractory |
| Primary end point | >35% SVR | >35% SVR |
| Key secondary end point | ≥50% reduction in MFSAF-TSS | ≥50% reduction in MFSAF-TSS |
| Enrollment | N = 289 | N = 97 |
| Initial published response rates | ||
| Spleen volume response (>35% volume reduction) | FEDR 400 mg (36%) FEDR 500 mg (40%) Placebo (1%) | FEDR 400 mg (55% of 83 evaluable) |
| MFSAF-TSS (>50% reduction) | FEDR 400 mg (36%) FEDR 500 mg (34%) Placebo (7%) | FEDR 400 mg (26% of 90 evaluable) |
| Toxicity | Grade 1-2 GI toxicities Grade 3-4 cytopenias Suspected WE (more so in 500-mg arm) led to trial hold | Consistent with JAKARTA study toxicity • Low-grade GI TOX • Grade 3-4 anemia/thrombocytopenia |
DIPSS INT-1/-2, Dynamic International Prognostic Scoring System–Intermediate-1/–Intermediate-2; ET, essential thrombocythemia; FEDR, fedratinib; MFSAF-TSS, Myelofibrosis Symptom Assessment Form–total symptom score; PB, peripheral blood; PV, polycythemia vera; RX, prescription; TOX, toxicity.
Fedratinib in JAK inhibitor–treated patients
JAKARTA-2 was a single-arm, open-label, nonrandomized, phase 2, multicenter study conducted to evaluate the utility of fedratinib in intermediate-1, intermediate-2, or high-risk PMF or post essential thrombocythemia–MF/post polycythemia vera–MF patients who had previously been treated with ruxolitinib, conducted temporally in parallel with the JAKARTA study24 (Table 2). Patients were deemed resistant or intolerant to ruxolitinib according to the individual investigator as no standard definition was available at that time. Patients received fedratinib at a starting dose of 400 mg daily. Dose escalation up to 600 mg was permitted if there was <50% reduction in palpable spleen size by the end of cycles 2 and 4. This study was also halted early due to the clinical hold placed by the FDA concerning WE. A recent reappraisal of the JAKARTA-2 study data used more stringent criteria for resistance/intolerance: relapse/refractory was defined as ruxolitinib therapy ≥3 months with initial response followed by spleen regrowth or a suboptimal response (defined as <10% SVR or <30% decrease in spleen size from baseline); intolerant was defined as ruxolitinib ≥28 days complicated by development of a red blood cell transfusion requirement (≥2 units per month for 2 months) or grade ≥3 thrombocytopenia, anemia, hematoma, and/or hemorrhage while receiving ruxolitinib.32 For the entire intention-to-treat cohort, with a median age of 67 years, SVR of ≥35% after 6 cycles was met by 31% (95% confidence interval, 22, 41). Of the 79 patients (81%) who met more stringent criteria for ruxolitinib resistance or intolerance, 30% (95% confidence interval, 21, 42) achieved at least 35% SVR following 6 cycles of treatment. Similar reductions in total symptom score of >50% were observed in 27% of both the intention-to-treat group and stringent criteria analyses groups. Clinically meaningful and statistically significant improvements from baseline were recorded across both total and individual symptoms and visits, though it is important to remember that this was an open-label study. Most common grade 3/4 hematological adverse events were anemia (46%) and thrombocytopenia (24%) and most common treatment-emergent nonhematological events were GI as for JAKARTA. Longer-term follow-up data are not available due to the early cessation of the study. No biological response data (ie, somatic mutation analysis, MPN-phenotypic driver mutation variant allele fraction change, fibrosis grade change, or durability of response) are available.
Resolving the issue of suspected WE with fedratinib
The suspicion that fedratinib might cause WE, a neurodegenerative condition characterized by a classical triad of oculomotor dysfunction, cerebellar dysfunction, and confusion, led to permanent suspension of the JAKARTA trials. Following central review of the original 8 reported potential cases of WE, only one definitive WE case (clinical and confirmatory magnetic resonance imaging) was identified33 ; the subject had >10% weight loss, poor performance status, and ataxia preenrolment suggesting prior neurodegeneration. During the study, the subject had uncontrolled GI toxicity without supplementary nutrition, illustrating clear present risk factors for WE. A further 2 cases had unconfirmed WE with suggestive symptoms with magnetic resonance imaging findings but confounding abnormalities (cerebral infarction; and suspected vertebra-basilar stroke and GI dysfunction). It was concluded that there was no evidence that fedratinib induces WE but a high index of suspicion is required. In August 2017, the FDA lifted the clinical hold that had been placed on the fedratinib development program (see Figure 2 for summary of key milestones in fedratinib development). Both new FREEDOM1 and FREEDOM2 studies include proactive management of GI symptoms to ensure adequate nutrition, measurement of thiamine, and also thiamine replacement as indicated with the “black box warning.”

Key milestones in the development of fedratinib. P, phosphorylation; Ph 1, phase 1; Ph 2, phase 2; Ph 3, phase 3; PI3K, phosphatidylinositol 3-kinase.
Key milestones in the development of fedratinib. P, phosphorylation; Ph 1, phase 1; Ph 2, phase 2; Ph 3, phase 3; PI3K, phosphatidylinositol 3-kinase.
Thiamine (vitamin B1) is an essential micronutrient, which cannot be made in the body. Although thiamine deficiency is rare in MPNs,34,35 thiamine levels should be checked prior to initiating fedratinib, periodically during treatment, and as clinically indicated. If thiamine deficiency is identified, thiamine should be repleted prior to starting fedratinib. If encephalopathy is suspected, fedratinib should be discontinued immediately and parenteral thiamine should be initiated until symptoms resolve or improve and thiamine levels normalize.
Fedratinib approval and clinical utilization implications
The approval of fedratinib will facilitate collection of real-world data as well as information from the current FREEDOM studies (NCT03755518 and the second-line NCT03952039). This will be important to understand duration of responses to fedratinib and whether thiamine deficiency and WE occur outside of the setting of the malnourished cachectic patient or one with excessive GI toxicity. Data from the JAKARTA and COMFORT studies do not guide clinicians concerning which drug (ruxolitinib or fedratinib) might be used first line, and a head-to-head study is not likely to be completed. Practical issues might include reimbursement and cost but worse GI toxicity; the suggestion that spleen responses may possibly be more pronounced with fedratinib is apparent from cross-trial comparisons and may influence drug choice. Whether there is a benefit to the lack of JAK1 inhibition with fedratinib, or wider kinome effects of this drug, remains uncertain. Further useful data might be to compare whether patients with a particular pattern of somatic mutations might respond differently. The issues surrounding when to choose to consider a second-line agent are of key importance because criteria used in clinical trials for ruxolitinib failure or intolerance are not fully applicable to real-world practice. Of note, a small retrospective case series (13 patients) found that patients who lose their response or have an inadequate response to ruxolitinib can be rechallenged with ruxolitinib following a period of treatment cessation and can achieve splenic and symptom responses on retreatment.36 Another issue is how to safely transition patients from ruxolitinib to fedratinib (and potentially vice versa). Here, there are concerns regarding the potential for a proinflammatory state and acute deterioration due to JAK inhibitor withdrawal, which can occur after patients substantially reduce the dose or stop ruxolitinib (it is unclear whether this occurs with fedratinib). In clinical practice, most patients will switch directly from 1 drug to another without “washing out” the first drug (Figure 3). Guidance is likely to be required for clinicians, patients, and their families.

Fedratinib in updated MF treatment guidelines
In 2017, the National Comprehensive Cancer Network (NCCN) developed and published the inaugural MPN guidelines for US-based MF as well as other MPNs.37 At the time of those inaugural guidelines, the main options available for MF patients were (1) observation, (2) medical therapy with ruxolitinib, (3) consideration of supportive cytoreduction with hydroxyurea or interferon, and (4) additional recommendations for agents to aid anemia. Ruxolitinib was recommended for consideration for (1) symptomatic low-risk MF patients, (2) intermediate-1–risk MF patients (especially if symptomatic, significant splenomegaly, and also not a candidate at that time for allogeneic stem cell transplant), and (3) intermediate-2 and high-risk MF with a platelet count ≥50 × 109/L (note with recommended dose reduction for those with a platelet count <100 × 109/L). Ruxolitinib did not play a role in the therapy of MF-associated anemia, given that on-target anemia derived from JAK2 inhibition, and could be used in accelerated or blast-phase MF but usually with a hypomethylating agent.
The approval of fedratinib in August 2019 led to an update of the NCCN MF guidelines (nccn.org), and took into consideration the data from JAKARTA, JAKARTA-2, and the FDA label for intermediate-2 and high-risk MF. On the basis of these aggregated data, as well as the prescribing and safety information, the guidelines included fedratinib as an option for initial therapy of intermediate-2 and high-risk MF for those with a platelet count ≥50 × 109/L. The safety and efficacy of using fedratinib for patients between a baseline platelet count of 50 × 109/L to 99 × 109/L was recently clarified by a pooled analysis of enrolled patients meeting these criteria from both JAKARTA and JAKARTA-2.38 Additionally, fedratinib was included as a second-line therapy for an MF patient who had failed frontline therapy with ruxolitinib. In addition to treatment recommendations, which frequently include >1 option given the nuances of individual patient management, the NCCN presents evidence blocks along 5 parameters of evaluation including efficacy, safety (ie, toxicity), quality of the evidence, consistency of the evidence, and affordability. Along these parameters, they did make a distinction between ruxolitinib on quality of the evidence (in second-line fedratinib in a single-arm trial), and safety, favoring ruxolitinib due to low-grade GI toxicity and low risk of WE (and resulting black box warning) with fedratinib. The NCCN panel did not feel that there was evidence yet for including fedratinib in the treatment guidelines for intermediate-1 (in the frontline) or symptomatic low-risk MF, although in the evidence blocks the panel acknowledged that the quality of the data for those situations with ruxolitinib is less than for intermediate-2 or high-risk patients. Additionally, these new data and clinical experiences will likely further impact the “NCCN Evidence Blocks.”
Future MF therapy pipeline
The pipeline for therapeutic options for MF remains very robust going into 2020 (Table 3). First, we highlight 2 additional JAK inhibitors seeking approval to join ruxolitinib and fedratinib as MF treatment options. Pacritinib, a JAK2/FLT3/IRAK1 inhibitor, which in addition to improvements in spleen volume and MF symptoms has, as a differentiator, the ability to be dosed fully in patients irrespective of thrombocytopenia and with safety and efficacy supporting this use in 2 large phase 3 trials (PERSIST 139 and 240 ), with another phase 3 study ongoing.41 Second, we highlight momelotinib, a JAK1/JAK2/activin A receptor type 1 (ACVR1) inhibitor that has, in addition to improvement in spleen volume and symptoms, also improved MF-associated anemia as proven in phase 3 trials (SIMPLIFY 142 and 243). Finally, we also include NS-108, a JAK2-selective inhibitor, currently enrolling patients intolerant or refractory/relapsed from prior JAK2 inhibitor therapy into the phase 2 portion of a phase 1/2 study.44 Next, multiple agents currently demonstrating benefit in combination with ruxolitinib that might similarly benefit from combination approaches with fedratinib include transforming growth factor (TGF) β ligand trap (luspatercept),45 bromodomain and extraterminal domain inhibitor (CPI-0610),46 B-cell lymphoma 2 (Bcl-2) inhibitor (navitoclax),47 and immunomodulatory (pomalidomide48 or thalidomide49 ). A phase 1b study of a heat shock protein 90 (HSP90) inhibitor (PU-H71) for patients on a stable dose of ruxolitinib also opened recently (NCT03935555). Additionally, even single-agent pathways show activity including telomerase inhibition (imetelstat),50 lysine-specific demethylase 1 (LSD1) inhibition,51 TGF β inhibition (AVID 200), second mitochondrial-derived activator of caspases mimetics (LCL161),52 and CD123 targeting (tagraxofusp),53 making this an exciting time for therapy development in MF. The approval of fedratinib further invigorates therapeutic development for MF. We anticipate that fedratinib data will be further augmented by the ongoing FREEDOM (NCT03755518) and FREEDOM 2 (NCT03952039) trials. FREEDOM, an efficacy and safety trial of fedratinib in myelofibrosis previously treated with ruxolitinib, includes an arm with luspatercept for anemia in combination; FREEDOM 2 is a second-line trial with a focus on ruxolitinib-intolerant/resistant patients. Additionally, we will learn further safety data regarding WE, and obtain data on intermediate-1 patients and additional data on use in ruxolitinib-intolerant/resistant patients that may expand the depth and breadth of recommendations for use of fedratinib in MF. Fedratinib will significantly and favorably impact patients with MF in the frontline and second line. Interesting potential combination approaches are also on the horizon for this drug.
MF drug-development pipeline
| Drug (class) | Efficacy | Comments | Ref. |
|---|---|---|---|
| JAK inhibitors | |||
| Momelotinib (JAK1/JAK2 inhibitor) | SIMPLIFY 1 • Spleen response 26.5% • Symptom response 29% • Greater rate of transfusion independence as compared with ruxolitinib | Ongoing phase 3 momentum study in second line for MF and symptoms/anemia (NCT04173494) | 42 |
| Pacritinib (JAK2/FLT3 inhibitor) | PERSIST 1 • Spleen response 25% (including 33% response in those with platelets <50 × 109/L) • Symptom response 36% rate | Ongoing phase 3 study PACIFICA41 (NCT03165734) | 39 |
| NS-018 (JAK2 inhibitor) | Phase 1 • Dose escalation completed | Ongoing phase 2 study for prior JAK2 inhibitor failures (NCT01423851) | 44 |
| Ruxolitinib combination studies | |||
| Luspatercept* (receptor type IIb and IgG1Fc domain) | Greater efficacy for anemia in combination with ruxolitinib than single agent (57% in nontransfusion dependent/53% in transfusion dependent) | Ongoing trial NCT03194542 | 45 |
| CPI-0610* (BET inhibitor) | Ongoing study active alone or in combination with ruxolitinib as second line; active in combination with ruxolitinib in front line | Ongoing trial NCT02158858 | 46 |
| Navitoclax* (Bcl-2 inhibitor) | Ongoing study, when added to suboptimal ruxolitinib responders’ responses seen for splenomegaly and symptoms | Ongoing trial NCT03222609 | 47 |
| Pomalidomide (IMID) | Ongoing study anemia responses seen when added to ruxolitinib | Ongoing trial NCT01644110 | 48 |
| Thalidomide (IMID) | Ongoing study responses seen in anemia and/or thrombocytopenia when added to ruxolitinib | Ongoing trial NCT03069326 | 49 |
| PU-H71 (HSP90 inhibitor) | Ongoing phase 1b study in MF patients on stable dose of ruxolitinib | Ongoing trial NCT03935555 | N/A |
| Single-agent alternative pathway inhibitors | |||
| AVID 200 (inhibits TGFβ 1/3) | Ongoing phase 1 | Ongoing trial NCT03895112 | N/A |
| Bomedemstat (IMG-7289; LSD1 inhibitor) | Ongoing phase 1 | Ongoing trial NCT03136185 | 51 |
| Imetelstat (telomerase inhibitor) | 27% response rate Responses more likely if JAK2-V617F mutated or ASXL1 mutated | Completed trial NCT02426086 | 50 |
| LCL161 (SMAC mimetic) | Ongoing phase 2 | Ongoing trial NCT02098161 | 52 |
| Tagraxofusp (SL-401; CD123 targeting) | Ongoing phase 1/2 | Ongoing trial NCT02268253 | 53 |
| Drug (class) | Efficacy | Comments | Ref. |
|---|---|---|---|
| JAK inhibitors | |||
| Momelotinib (JAK1/JAK2 inhibitor) | SIMPLIFY 1 • Spleen response 26.5% • Symptom response 29% • Greater rate of transfusion independence as compared with ruxolitinib | Ongoing phase 3 momentum study in second line for MF and symptoms/anemia (NCT04173494) | 42 |
| Pacritinib (JAK2/FLT3 inhibitor) | PERSIST 1 • Spleen response 25% (including 33% response in those with platelets <50 × 109/L) • Symptom response 36% rate | Ongoing phase 3 study PACIFICA41 (NCT03165734) | 39 |
| NS-018 (JAK2 inhibitor) | Phase 1 • Dose escalation completed | Ongoing phase 2 study for prior JAK2 inhibitor failures (NCT01423851) | 44 |
| Ruxolitinib combination studies | |||
| Luspatercept* (receptor type IIb and IgG1Fc domain) | Greater efficacy for anemia in combination with ruxolitinib than single agent (57% in nontransfusion dependent/53% in transfusion dependent) | Ongoing trial NCT03194542 | 45 |
| CPI-0610* (BET inhibitor) | Ongoing study active alone or in combination with ruxolitinib as second line; active in combination with ruxolitinib in front line | Ongoing trial NCT02158858 | 46 |
| Navitoclax* (Bcl-2 inhibitor) | Ongoing study, when added to suboptimal ruxolitinib responders’ responses seen for splenomegaly and symptoms | Ongoing trial NCT03222609 | 47 |
| Pomalidomide (IMID) | Ongoing study anemia responses seen when added to ruxolitinib | Ongoing trial NCT01644110 | 48 |
| Thalidomide (IMID) | Ongoing study responses seen in anemia and/or thrombocytopenia when added to ruxolitinib | Ongoing trial NCT03069326 | 49 |
| PU-H71 (HSP90 inhibitor) | Ongoing phase 1b study in MF patients on stable dose of ruxolitinib | Ongoing trial NCT03935555 | N/A |
| Single-agent alternative pathway inhibitors | |||
| AVID 200 (inhibits TGFβ 1/3) | Ongoing phase 1 | Ongoing trial NCT03895112 | N/A |
| Bomedemstat (IMG-7289; LSD1 inhibitor) | Ongoing phase 1 | Ongoing trial NCT03136185 | 51 |
| Imetelstat (telomerase inhibitor) | 27% response rate Responses more likely if JAK2-V617F mutated or ASXL1 mutated | Completed trial NCT02426086 | 50 |
| LCL161 (SMAC mimetic) | Ongoing phase 2 | Ongoing trial NCT02098161 | 52 |
| Tagraxofusp (SL-401; CD123 targeting) | Ongoing phase 1/2 | Ongoing trial NCT02268253 | 53 |
Spleen responses: 35% volume reduction. Symptom responses: 50% reduction in MFSAF or Myeloproliferative Neoplasms Symptom Assessment Form (MPNSAF) total symptoms.
Bcl-2, B-cell lymphoma 2; BET, bromodomain and extraterminal domain; HSP90, heat shock protein 90; IgG1, immunoglobulin G1; IMID, immunomodulatory imide; N/A, not applicable; Ref., reference; SMAC, second mitochondrial activator of caspases.
These trials have both single-agent and combination arms (with ruxolitinib).
Data-sharing requests may be e-mailed to the corresponding author, Ann Mullally (amullally@partners.org).
Acknowledgments
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL131835) (A.M.), the MPN Research Foundation (A.M.), and the Gabrielle’s Angel Foundation for Cancer Research (A.M.).
A.M. is a Scholar of The Leukemia & Lymphoma Society.
Authorship
Contribution: A.M. and R.M. designed the outline for the manuscript; and A.M., J.H., C.H., and R.M. drafted, edited, and approved the manuscript.
Conflict-of-interest disclosure: A.M. has received honoraria from Blueprint Medicines, Roche, and Incyte, and receives research support from Janssen. J.H. was an equity holder in Impact Biomedicines prior to its acquisition by Celgene. C.H. has received institutional research support from Celgene and Novartis, and honoraria from Celgene, Roche, Janssen, AOP Pharma, Novartis, and Incyte. R.M. receives consulting honoraria from Novartis, La Jolla Pharmaceutical Company, and Sierra Oncology, and research support from Celgene, Bristol-Myers Squibb, Incyte, Promedior, CTI BioPharma, AbbVie, and Genentech.
Correspondence: Ann Mullally, Harvard Medical School, Harvard Institutes of Medicine Building, Room 738, 77 Ave Louis Pasteur, Boston, MA 02115; e-mail: amullally@partners.org; and Ruben Mesa, Mays Cancer Center, UT Health San Antonio MD Anderson, 7979 Wurzbach Rd, San Antonio, TX 78006; e-mail: mesar@uthscsa.edu.

