

Key Points
Pacritinib demonstrated clinical benefit and was well tolerated in advanced myelofibrosis patients, including those with severe thrombocytopenia.
Pharmacokinetic and pharmacodynamic data support testing pacritinib 200 mg twice per day in a pivotal randomized phase 3 trial.

Abstract
PAC203 is a randomized dose-finding study of pacritinib, an oral JAK2/IRAK1 inhibitor, in patients with advanced myelofibrosis who are intolerant of or resistant to ruxolitinib. Patients were randomized 1:1:1 to pacritinib 100 mg once per day, 100 mg twice per day, or 200 mg twice per day. Enhanced eligibility criteria, monitoring, and dose modifications were implemented to mitigate risk of cardiac and hemorrhagic events. Efficacy was based on ≥35% spleen volume response (SVR) and ≥50% reduction in the 7-component total symptom score (TSS) through week 24. Of 161 patients, 73% were intolerant of and 76% had become resistant to ruxolitinib; 50% met criteria for both. Severe thrombocytopenia (platelet count <50 × 103/μL) was present in 44%. SVR rates were highest with 200 mg twice per day (100 mg once per day, 0%; 100 mg twice per day, 1.8%; 200 mg twice per day, 9.3%), particularly among patients with baseline platelet counts <50 × 103/μL (17%; 4 of 24). Although TSS response rate was similar across doses (100 mg once per day, 7.7%; 100 mg twice per day, 7.3%; 200 mg twice per day, 7.4%), median percent reduction in TSS suggested a dose-response relationship (–3%, −16%, and −27%, respectively). Pharmacokinetic and pharmacodynamic modeling based on all available data showed greatest SVR and TSS reduction at 200 mg twice per day compared with lower doses. Common adverse events were gastrointestinal events, thrombocytopenia, and anemia. There was no excess of grade ≥3 hemorrhagic or cardiac events at 200 mg twice per day. Pacritinib 200 mg twice per day demonstrated clinical activity and an acceptable safety profile and was selected as the recommended dose for a pivotal phase 3 study in patients with myelofibrosis and severe thrombocytopenia. This trial was registered at www.clinicaltrials.gov as #NCT03165734.
Introduction
Pacritinib is a novel inhibitor of JAK2, interleukin-1 receptor-associated kinase 1 (IRAK1), FLT3, and CSF-1R that has demonstrated clinical benefit in patients with myelofibrosis compared with best available therapy in PERSIST-1 and PERSIST-2 phase 3 studies.1,2 Unlike the JAK inhibitors that are currently approved in the United States for myelofibrosis (ruxolitinib and fedratinib), pacritinib has been studied in patients presenting with severe thrombocytopenia (platelet counts <50 × 103/μL). In the PERSIST studies, pacritinib was associated with clinically meaningful spleen volume responses (SVRs) and improvements in disease-associated symptoms in a substantial proportion of patients, including those with severe baseline thrombocytopenia and those who had previously received ruxolitinib. Severe thrombocytopenia and failure of initial treatment represent poor prognostic factors for patients with myelofibrosis. Severe thrombocytopenia is associated with advanced disease, a higher risk of bleeding, increased risk of leukemic transformation,3 and a shorter overall survival (median of ∼15 months).4-6 After discontinuation of first-line ruxolitinib therapy, median survival is only 13 to 14 months, and it is ∼8 months for patients with platelet counts <100 × 103/μL.7-9
The PERSIST studies indicate that pacritinib may address a significant unmet need in patients with severe thrombocytopenia, including those who are intolerant of or resistant to ruxolitinib. Concerns over high-grade cardiac and bleeding events in these studies, however, prompted development of the PAC203 phase 2 study, which was designed to identify the recommended pacritinib dosage and to establish risk minimization measures, such as exclusion criteria for patients with preexisting bleeding episodes or cardiac disease, increased cardiac monitoring, and detailed dose modification guidelines. Here, we present the results of the phase 2 PAC203 randomized dose-finding study in patients with advanced myelofibrosis who were intolerant of or had become resistant to ruxolitinib.
Methods
Study design and objective
The phase 2 PAC203 study was an open-label, randomized, dose-finding study of pacritinib in patients with myelofibrosis previously treated with ruxolitinib. The primary objective was to determine the recommended dose of pacritinib. Secondary objectives were to examine the dose-response relationship for efficacy and safety and to further characterize pharmacokinetics (PK) and pharmacodynamics (PD) of pacritinib. The selection of a recommended dose was to be based on PAC203 efficacy and safety data through week 24 as well as dose- and exposure-response analyses using all available data for pacritinib-treated patients from PAC203 and previous studies. The study was conducted at 62 sites (supplemental Methods). The study was approved by the institutional review boards at each institution and conducted in accordance with the principles outlined in the Declaration of Helsinki.
Patients
Adult patients with primary or secondary myelofibrosis were eligible if they had intermediate-1, intermediate-2, or high-risk disease according to the Dynamic International Prognostic Scoring System10 and were intolerant of ruxolitinib, which was defined as treatment for 28 days or more, with treatment complicated by a requirement for red blood cell (RBC) transfusion, grade ≥3 anemia, thrombocytopenia, hematoma, and/or hemorrhage while being treated with a dosage of <20 mg twice per day or were resistant to ruxolitinib. Ruxolitinib failure was defined as treatment for 3 or more months with <10% SVR or <30% decrease in spleen length or regrowth to these parameters. Additional eligibility criteria included splenomegaly ≥5 cm below the left costal margin, total symptom score (TSS) ≥10 on the Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS 2.0, 7-component version11 ) or single symptom score ≥5 or 2 scores ≥3, including only the symptoms of left upper quadrant pain, bone pain, itching, or night sweats; Eastern Cooperative Oncology Group performance status of 0 to 2; peripheral blast count <10%; absolute neutrophil count >0.5 × 109/L; adequate liver, renal, and coagulation parameters; left ventricular ejection fraction ≥45%; and life expectancy ≥6 months. Patients with grade ≥2 bleeding events within the previous 3 months and those treated with anticoagulation or antiplatelet agents (except for aspirin at dosages ≤100 mg per day) within the past 14 days were excluded. Patients with New York Heart Association Class ≥2 heart failure, grade ≥2 cardiac conditions within the previous 6 months, QTc prolongation >450 msec during screening, and those treated with agents that prolong the QT interval within the past 14 days were excluded. Additional eligibility criteria are described in supplemental Methods. All patients provided written informed consent.
Randomization and treatment
Patients were randomized 1:1:1 using an interactive Web response system to receive pacritinib at 1 of 3 doses: 100 mg once per day, 100 mg twice per day, or 200 mg twice per day. Randomization was stratified by baseline platelet count (≤50 × 103/μL, >50 × 103/μL to ≤100 × 103/μL, and >100 × 103/μL) and geographic region (North America vs Europe vs rest of world). PAC203 was an open-label study, and therefore treatment assignments were known.
Patients started treatment at their randomly assigned dose. Dose escalation was not permitted; dose modification guidelines were provided for treatment-related events, as described in supplemental Methods. Patients in all arms were treated through week 24 or until progressive disease (increase in spleen volume ≥25%, splenic irradiation, splenectomy, or leukemic transformation), unacceptable toxicity, or withdrawal from study. Patients who experienced clinical benefit at 24 weeks had the option of continuing to receive pacritinib until study completion, at which point they could transition to an expanded access (individual patient/named patient basis) program.
Study assessments
Spleen volume (by magnetic resonance imaging or computed tomography) was reviewed centrally by a blinded, independent radiology facility at baseline, every 12 weeks on study, and at the end of treatment. Patient-reported symptoms were recorded once per day using the MPN-SAF TSS 2.0. TSS was calculated as the sum of 7 scores (tiredness, satiety, abdominal discomfort, night sweats, pruritis, bone pain, and left rib pain), in contrast to the 6-component version (excluding tiredness), which supported the approval of other JAK inhibitors.12,13 Scores for Patient Global Impression of Change (PGIC)14 were assessed every 12 weeks until the end of treatment. Myeloid mutations were assessed using an International Organization for Standardization (ISO 15189:2012)–accredited Illumina TruSeq Custom Amplicon Panel that assessed 32 genes and reported variants with allele frequency ≥1%.15
Safety monitoring included study visits, laboratory assessments, QTc monitoring (reviewed centrally by an independent cardiologist blinded to treatment assignment) at baseline and at the end of weeks 4, 12, and 24 and every 12 weeks on study treatment, as well as monitoring of left ventricular ejection fraction at the end of weeks 4, 12, and 24 and every 24 weeks on study treatment. Dose modification guidelines, including those for ≥2 grade reduction in platelet counts, grade ≥2 hemorrhage, and grade ≥2 cardiac toxicity, were more stringent than those used in the previous PERSIST-1 and PERSIST-2 studies. Adverse events were classified and graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events v4.03. Cardiac and hemorrhagic events were classified by Standardized Medical Dictionary for Regulatory Activities (MedDRA) Query (SMQ). See supplemental Methods for additional details.
Population PK and PD modeling
Population PK modeling was conducted to describe pacritinib PK and to identify intrinsic and extrinsic factors that could potentially influence pacritinib PK among individuals. Population PK/PD modeling was conducted to assess the relationship between pacritinib plasma concentration and change from baseline spleen volume and TSS score. Population PK modeling was based on all available data from 16 studies (11 phase 1 and 5 phase 2 or 3 studies), including healthy volunteers and patients with myelofibrosis and other hematologic malignancies. Population PK/PD modeling was based on PERSIST-1, PERSIST-2, and PAC203 trial data and analyzed SVR and TSS reduction as continuous variables. Associations between exposure and efficacy outcomes were evaluated on binomial end points (responder or nonresponder based on ≥35% SVR or ≥50% reduction in TSS) using logistic regression. All modeling work was performed using NONMEM version 7.3 (ICON Development Solutions, Dublin, Ireland), and exposure-efficacy evaluations were conducted using R Statistical Software (version 3.5.3; https://www.R-project.org/). Further details on modeling are outlined in supplemental Methods.
Statistical analysis
A sample size of 150 patients was selected to ensure adequate precision to evaluate efficacy and safety. With 50 patients per arm, if the true incidence rate for a specified safety event is 5%, then the chance of observing at least 1 event among 50 patients would be 92%. Primary efficacy and safety analyses were performed in patients who received at least 1 dose of pacritinib. As an exploratory analysis, SVR and TSS end points were analyzed among patients who received at least 1 dose of pacritinib and had evaluable baseline and follow-up assessments relevant for that end point (evaluable population). The number of patients with ≥50% reduction in TSS and those with a ≥35% reduction in SVR were summarized using counts and percent. PGIC scores and safety analyses were summarized by treatment arm using descriptive statistics. Although no formal hypothesis testing was planned, an ad hoc Cochran-Armitage trend test was performed to evaluate efficacy end points.
Results
Patient disposition and exposure
From July 2017 to January 2019, 165 patients were randomly assigned, and 161 received treatment with pacritinib 100 mg once per day (n = 52), 100 mg twice per day (n = 55), or 200 mg twice per day (n = 54; Figure 1). Baseline demographic and disease characteristics were balanced across arms (Table 1). Median platelet count was 55× 103/μL, and 44.1% of patients (n = 71) had a platelet count <50 × 103/μL. Hemoglobin was <10 g/dL in 70.8% of patients (n = 114). Median duration of previous ruxolitinib exposure was 1.7 years; 76% met study-defined criteria for ruxolitinib failure, 73% for ruxolitinib intolerance, and 50% for both. The majority of patients (59%) had ≥1% peripheral blasts, with a median of 2.0% peripheral blasts on all arms. Molecular profiling performed on a subset of patients (n = 110) revealed a mean of 2.5 mutations (range, 1-11 mutations) per patient. High molecular risk mutations (ASXL1, SRSF2, EZH2, IDH1/2, U2AF1Q157)16 were detected in 40% of patients, and 7.3% had TP53 mutations (associated with poor prognosis and leukemic transformation17 ).

Patient disposition. Outcomes for all screened and randomized patients are shown. BID, twice per day; QD, once per day.
Patient disposition. Outcomes for all screened and randomized patients are shown. BID, twice per day; QD, once per day.
Baseline patient and disease characteristics by treatment arm
| Characteristic | Pacritinib | ||
|---|---|---|---|
| 100 mg once per d (n = 52) | 100 mg twice per d (n = 55) | 200 mg twice per d (n = 54) | |
| Median age (IQR), y | 69.5 (64.0-74.0) | 69.0 (66.0-73.0) | 68.5 (62.0-73.0) |
| Male sex | 31 (59.6) | 29 (52.7) | 32 (59.3) |
| Myelofibrosis diagnosis | |||
| Primary | 28 (53.8) | 28 (50.9) | 37 (68.5) |
| Post polycythemia vera | 16 (30.8) | 18 (32.7) | 10 (18.5) |
| Post essential thrombocythemia | 8 (15.4) | 9 (16.4) | 7 (13.0) |
| Previous ruxolitinib treatment | |||
| Failure | 40 (76.9) | 41 (74.5) | 42 (77.8) |
| Intolerance | 38 (73.1) | 41 (74.5) | 38 (70.4) |
| Both | 26 (50.0) | 28 (50.9) | 26 (48.1) |
| Median duration of previous ruxolitinib treatment (IQR), y | 1.7 (0.7-3.0) | 1.8 (0.8-3.5) | 1.6 (0.4-3.3) |
| DIPSS risk score* | |||
| Intermediate-1 | 9 (17.3) | 14 (25.5) | 12 (22.2) |
| Intermediate-2 | 25 (48.1) | 27 (49.1) | 28 (51.9) |
| High | 18 (34.6) | 14 (25.5) | 14 (25.9) |
| ECOG PS | |||
| 0 | 11 (21.2) | 14 (25.5) | 17 (31.5) |
| 1 | 32 (61.5) | 29 (52.7) | 29 (53.7) |
| 2 | 9 (17.3) | 12 (21.8) | 8 (14.8) |
| Platelet count, × 103/μL | |||
| Median (IQR) | 59 (41 -111 ) | 53 (33 -116 ) | 59 (29 -91 ) |
| <50 | 23 (44.2) | 24 (43.6) | 24 (44.4) |
| Hemoglobin <10g/dL | 35 (67.3) | 38 (69.1) | 41 (75.9) |
| RBC transfusion status† | |||
| Dependent | 15 (28.8) | 15 (27.3) | 21 (38.9) |
| Independent | 24 (46.2) | 23 (41.8) | 20 (37.0) |
| Intermediate | 13 (25.0) | 16 (29.1) | 13 (24.1) |
| Platelet transfusion dependent‡ | 6 (11.5) | 5 (9.1) | 6 (11.1) |
| Peripheral blasts ≥1% | 32 (61.5) | 30 (54.5) | 32 (59.3) |
| Median peripheral blasts (IQR), % | 2.0 (1.0-3.0) | 2.0 (0.9-4.0) | 2.0 (1.0-2.0) |
| Median white blood cell (IQR), ×109/L | 10.9 (4.9-34.9) | 7.8 (3.5-18.6) | 7.1 (4.2-12.6) |
| Median spleen length (IQR), cm | 12.0 (9.5-19.5) | 15.0 (11.0-20.0) | 14.0 (10.0-20.0) |
| Median spleen volume (IQR), cm3 | 2434 (1694-3041) | 2169 (1651-2839) | 2655 (1630-4069) |
| Characteristic | Pacritinib | ||
|---|---|---|---|
| 100 mg once per d (n = 52) | 100 mg twice per d (n = 55) | 200 mg twice per d (n = 54) | |
| Median age (IQR), y | 69.5 (64.0-74.0) | 69.0 (66.0-73.0) | 68.5 (62.0-73.0) |
| Male sex | 31 (59.6) | 29 (52.7) | 32 (59.3) |
| Myelofibrosis diagnosis | |||
| Primary | 28 (53.8) | 28 (50.9) | 37 (68.5) |
| Post polycythemia vera | 16 (30.8) | 18 (32.7) | 10 (18.5) |
| Post essential thrombocythemia | 8 (15.4) | 9 (16.4) | 7 (13.0) |
| Previous ruxolitinib treatment | |||
| Failure | 40 (76.9) | 41 (74.5) | 42 (77.8) |
| Intolerance | 38 (73.1) | 41 (74.5) | 38 (70.4) |
| Both | 26 (50.0) | 28 (50.9) | 26 (48.1) |
| Median duration of previous ruxolitinib treatment (IQR), y | 1.7 (0.7-3.0) | 1.8 (0.8-3.5) | 1.6 (0.4-3.3) |
| DIPSS risk score* | |||
| Intermediate-1 | 9 (17.3) | 14 (25.5) | 12 (22.2) |
| Intermediate-2 | 25 (48.1) | 27 (49.1) | 28 (51.9) |
| High | 18 (34.6) | 14 (25.5) | 14 (25.9) |
| ECOG PS | |||
| 0 | 11 (21.2) | 14 (25.5) | 17 (31.5) |
| 1 | 32 (61.5) | 29 (52.7) | 29 (53.7) |
| 2 | 9 (17.3) | 12 (21.8) | 8 (14.8) |
| Platelet count, × 103/μL | |||
| Median (IQR) | 59 (41 -111 ) | 53 (33 -116 ) | 59 (29 -91 ) |
| <50 | 23 (44.2) | 24 (43.6) | 24 (44.4) |
| Hemoglobin <10g/dL | 35 (67.3) | 38 (69.1) | 41 (75.9) |
| RBC transfusion status† | |||
| Dependent | 15 (28.8) | 15 (27.3) | 21 (38.9) |
| Independent | 24 (46.2) | 23 (41.8) | 20 (37.0) |
| Intermediate | 13 (25.0) | 16 (29.1) | 13 (24.1) |
| Platelet transfusion dependent‡ | 6 (11.5) | 5 (9.1) | 6 (11.1) |
| Peripheral blasts ≥1% | 32 (61.5) | 30 (54.5) | 32 (59.3) |
| Median peripheral blasts (IQR), % | 2.0 (1.0-3.0) | 2.0 (0.9-4.0) | 2.0 (1.0-2.0) |
| Median white blood cell (IQR), ×109/L | 10.9 (4.9-34.9) | 7.8 (3.5-18.6) | 7.1 (4.2-12.6) |
| Median spleen length (IQR), cm | 12.0 (9.5-19.5) | 15.0 (11.0-20.0) | 14.0 (10.0-20.0) |
| Median spleen volume (IQR), cm3 | 2434 (1694-3041) | 2169 (1651-2839) | 2655 (1630-4069) |
All data are no. (%) unless otherwise specified.
DIPSS, Dynamic International Prognostic Scoring System; ECOG PS, Eastern Cooperative Oncology Group performance status; IQR, interquartile range.
High molecular risk as defined by Tefferi et al.16
RBC transfusion status as defined by Gale criteria.28
Platelet transfusion dependence defined by any platelet transfusion required during the past month.
At study completion, 40 patients (24.8%) were still receiving treatment; 34 of these patients continued therapy off-study under expanded access. Discontinuation as a result of adverse events (AEs) was more common at 100 mg twice per day (20.0%) and 200 mg twice per day (18.5%) than at 100 mg once per day (11.5%). Other reasons for treatment discontinuation included physician decision (39.1%) and withdrawal by the patient (14.9%). Protocol-defined progression of disease occurred in all dosing arms, including splenic progression (100 mg once per day, n = 13; 100 mg twice per day, n = 10; 200 mg twice per day, n = 16) and leukemic transformation (100 mg once per day, n = 2; 100 mg twice per day, n = 0; 200 mg twice per day, n = 2, including 1 with concomitant splenic progression). Patients reached week 24 and had evaluable efficacy data at similar rates across dosing arms for SVR (100 mg once per day, 50.0% [26 of 52]; 100 mg twice per day, 47.3% [26 of 55]; 200 mg twice per day, 50% [27 of 54]) and TSS (100 mg once per day, 44.2% [23 of 52]; 100 mg twice per day, 49.1% [27 of 55]; 200 mg twice per day, 44.4% [24 of 54]).
Median duration of pacritinib treatment on study was 23 weeks (100 mg once per day), 20 weeks (100 mg twice per day), and 21 weeks (200 mg twice per day). Patients treated at 200 mg twice per day had higher rates of dose interruptions and reductions (35.2% and 20.4%, respectively) than those at 100 mg twice per day (20.0% and 9.1%) or 100 mg once per day (19.2% and 0%). Dose intensity (average daily ratio of actual vs planned dose intensity) was high across arms: 100 mg once per day, 99.1%; 100 mg twice per day, 94.7%; 200 mg twice per day, 90.7%.
SVR
At week 24, the SVR response rate was highest in the 200 mg twice per day arm (9.3%) compared with lower dose arms (100 mg once per day, 0%; 100 mg twice per day, 1.8%; Ptrend = .012; Table 2). The median percent reduction in spleen volume was greatest on the 200 mg twice per day arm (–10.1%; interquartile range, −23.8% to 2.5%) compared with lower doses (Figure 2A). Across all arms, baseline cytopenias were common in patients with an SVR response: 6 of 6 responders had baseline hemoglobin <10g/dL, and 4 of 6 responders had baseline platelet counts <50 × 103/μL. Notably, of the 5 SVR responders at 200 mg twice per day, 4 had baseline platelet counts <50 × 103/μL; thus, the SVR rate among patients with severe thrombocytopenia was 16.7% (4 of 24). In the evaluable population with spleen volume data at week 24, the SVR rate at 200 mg twice per day was 18.5%, and it was 30.8% overall and among patients with severe baseline thrombocytopenia.
Summary of spleen volume and symptom reduction from baseline to week 24, by dosing arm
| Pacritinib | ||||||
|---|---|---|---|---|---|---|
| 100 mg once per d | 100 mg twice per d | 200 mg twice per d | ||||
| n/N | % | n/N | % | n/N | % | |
| SVR rate | ||||||
| Overall | 0/52 | 0 | 1/55 | 1.8 | 5/54 | 9.3 |
| Evaluable | 0/26 | 0 | 1/26 | 3.8 | 5/27 | 18.5 |
| Patients with platelet count <50 × 103/μL | ||||||
| Overall | 0/23 | 0 | 0/24 | 0 | 4/24 | 16.7 |
| Evaluable | 0/9 | 0 | 0/10 | 0 | 4/13 | 30.8 |
| Median percent spleen volume change from baseline (IQR) | −6.7 (–14.5 to 11.3) | −0.1 (–14.2 to 9.2) | −10.1 (–23.8 to 2.5) | |||
| TSS response rate | ||||||
| Overall | 4/52 | 7.7 | 4/55 | 7.3 | 4/54 | 7.4 |
| Evaluable | 4/23 | 17.4 | 4/27 | 14.8 | 4/24 | 16.7 |
| Patients with platelet count <50 × 103/μL | ||||||
| Overall | 2/23 | 8.7 | 0/24 | 0 | 2/24 | 8.3 |
| Evaluable | 2/9 | 22.2 | 0/10 | 0 | 2/13 | 15.4 |
| Median percent TSS change from baseline (IQR) | −3.1 (–30.4 to 29.3) | −16.0 (–43.5 to 0.8) | −27.3 (–39.2 to 1.2) | |||
| Pacritinib | ||||||
|---|---|---|---|---|---|---|
| 100 mg once per d | 100 mg twice per d | 200 mg twice per d | ||||
| n/N | % | n/N | % | n/N | % | |
| SVR rate | ||||||
| Overall | 0/52 | 0 | 1/55 | 1.8 | 5/54 | 9.3 |
| Evaluable | 0/26 | 0 | 1/26 | 3.8 | 5/27 | 18.5 |
| Patients with platelet count <50 × 103/μL | ||||||
| Overall | 0/23 | 0 | 0/24 | 0 | 4/24 | 16.7 |
| Evaluable | 0/9 | 0 | 0/10 | 0 | 4/13 | 30.8 |
| Median percent spleen volume change from baseline (IQR) | −6.7 (–14.5 to 11.3) | −0.1 (–14.2 to 9.2) | −10.1 (–23.8 to 2.5) | |||
| TSS response rate | ||||||
| Overall | 4/52 | 7.7 | 4/55 | 7.3 | 4/54 | 7.4 |
| Evaluable | 4/23 | 17.4 | 4/27 | 14.8 | 4/24 | 16.7 |
| Patients with platelet count <50 × 103/μL | ||||||
| Overall | 2/23 | 8.7 | 0/24 | 0 | 2/24 | 8.3 |
| Evaluable | 2/9 | 22.2 | 0/10 | 0 | 2/13 | 15.4 |
| Median percent TSS change from baseline (IQR) | −3.1 (–30.4 to 29.3) | −16.0 (–43.5 to 0.8) | −27.3 (–39.2 to 1.2) | |||
SVR is defined as ≥35% reduction; TSS (7-component version) response is defined as ≥50% reduction. Response rates are provided for the overall population (all treated patients) and for the evaluable population (patients with week 24 data).
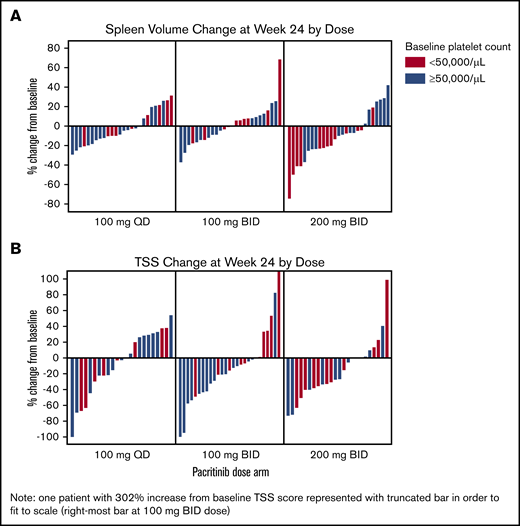
Spleen volume and TSS reduction in evaluable patients. Waterfall plots for SVR (A) and TSS (B) reduction from baseline to week 24. TSS is the 7-component version of the Total Symptom Score (inclusive of “tiredness”).
Spleen volume and TSS reduction in evaluable patients. Waterfall plots for SVR (A) and TSS (B) reduction from baseline to week 24. TSS is the 7-component version of the Total Symptom Score (inclusive of “tiredness”).
Symptom reduction
At week 24, the TSS response rate was similar across all arms (100 mg once per day, 7.7%; 100 mg twice per day, 7.3%; 200 mg twice per day, 7.4%; Table 2). Among the evaluable population with TSS data at week 24, the TSS response rate was also similar across arms (17.4%, 14.8%, 16.7%; Ptrend > .05). Baseline cytopenias were common among patients with a TSS –50% response: 8 of 12 responders had hemoglobin <10 g/dL, and 4 of 12 responders had platelet counts <50 × 103/μL. Although response rates were similar across arms, when analyzed as a continuous variable, the percent decrease in TSS showed deeper reductions with escalating doses, with the greatest median reduction at 200 mg twice per day (–27.3%; interquartile range, −39.2% to 1.2%; Figure 2B). TSS reduction occurred in both spleen-related symptoms (satiety, abdominal discomfort, and left rib pain) and cytokine-related symptoms (night sweats, itching, and bone pain), particularly on the 200 mg twice per day arm (Figure 3). The subset of TSS responders hardly overlapped with SVR responders; the proportion of patients who achieved either response was highest at 200 mg twice per day (14.8%) compared with 100 mg twice per day (7.3%) or 100 mg once per day (7.7%).
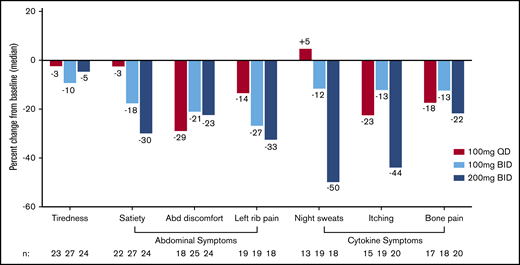
Change in individual symptom scores per MPN-SAF TSS 2.0 between baseline and week 24 by dosing arm. Median percent change in individual symptom scores show greatest improvement on the 200 mg twice per day dose for the majority of abdominal- and cytokine-related symptoms. Abd, abdominal; MPN-SAF TSS, Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score.
Change in individual symptom scores per MPN-SAF TSS 2.0 between baseline and week 24 by dosing arm. Median percent change in individual symptom scores show greatest improvement on the 200 mg twice per day dose for the majority of abdominal- and cytokine-related symptoms. Abd, abdominal; MPN-SAF TSS, Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score.
The percentage of patients reporting at least minimal improvement on the PGIC score at week 24 was greatest in the 200 mg twice per day arm (33.3% [18 of 54]) compared with 100 mg twice per day (23.6% [13 of 55]) or 100 mg once per day (19.2% [10 of 52]) doses. Among evaluable patients, more than one third of patients on all arms reported that their symptoms were much or very much improved by week 24 (Figure 4).
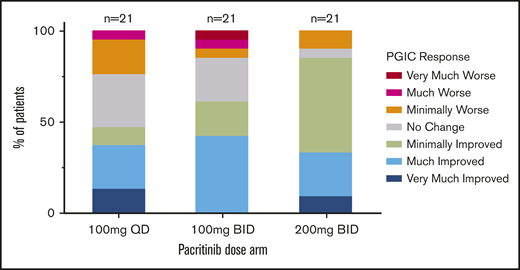
PGIC assessment (evaluable population) at week 24 by dosing arm. Number of patients with any improvement in disease symptoms was greatest at 200 mg twice per day (n = 18) compared with lower doses (100 mg once per day, n = 10; 100 mg twice per day, n = 13).
PGIC assessment (evaluable population) at week 24 by dosing arm. Number of patients with any improvement in disease symptoms was greatest at 200 mg twice per day (n = 18) compared with lower doses (100 mg once per day, n = 10; 100 mg twice per day, n = 13).
Changes in hematologic parameters
Platelet counts remained stable for the majority of patients on study, including those with severe thrombocytopenia at baseline (Figure 5A), although some patients required dose modifications or discontinuations because of thrombocytopenia (supplemental Table 1). Among patients who had received RBC transfusions within 90 days of enrollment or during screening, the median rate of RBC transfusions (units per month) was stable or decreasing through week 24 (Figure 5B), and reductions in transfusion burden by 50% or greater were observed in all dosing arms: 100 mg once per day: 17.9% (5 of 28); 100 mg twice per day: 35.5% (11 of 31); 200 mg twice per day: 14.7% (5 of 34). Among patients with baseline hemoglobin levels ≤10 g/dL, a larger proportion treated at 200 mg twice per day had hemoglobin increase by ≥1 g/dL (100 mg once per day: 2.8% [n = 1]; 100 mg twice per day: 2.8% [n = 1]; 200 mg twice per day: 9.5% [n = 4]) and by ≥2 g/dL (100 mg once per day, 0%; 100 mg twice per day, 0%; 200 mg twice per day, 4.8% [n = 2]) in the absence of transfusion in the 8 weeks leading up to week 24. Among patients who were dependent on RBC transfusions at baseline, a subset of patients in all arms achieved transfusion independence by week 24: 100 mg once per day: 13% (2 of 15); 100 mg twice per day: 13% (2 of 15); 200 mg twice per day: 9.5% (2 of 21).
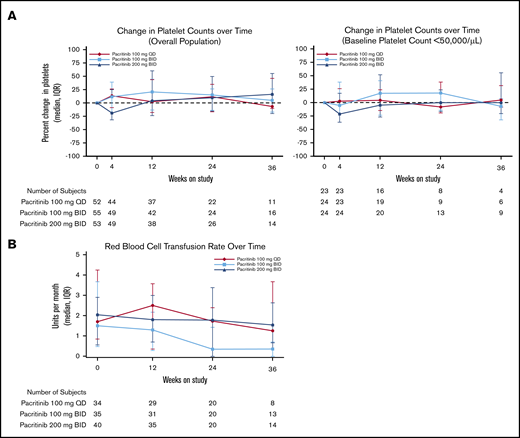
Changes in hematologic parameters. (A) Median percent change in platelet counts on study treatment by dosing arm. (B) Median number of RBC transfusions per month among patients who received RBC transfusions before study treatment by dosing arm.
Changes in hematologic parameters. (A) Median percent change in platelet counts on study treatment by dosing arm. (B) Median number of RBC transfusions per month among patients who received RBC transfusions before study treatment by dosing arm.
Safety
The most commonly reported treatment-emergent AEs are listed in Table 3. The majority of common nonhematologic AEs were mild or moderate in severity, and rates were similar across dosing arms with the exception of gastrointestinal events, which occurred more commonly in patients treated at 200 mg twice per day (72.2% [39 of 54]) than at lower doses (100 mg once per day: 50.0% [26 of 52]; 100 mg twice per day: 54.5% [30 of 55]). These events were largely grade 1 or 2 (81 of 95, or 85.3% of patients with events) and usually occurred within the first 8 weeks of treatment. Diarrhea was common but generally manageable with standard antidiarrheal agents and tended to be short-lived: the median duration of pacritinib-related diarrhea at 200 mg twice per day was 2 weeks. Neoplasms were reported in 5 patients (3.1%); all reports were skin cancers, and no cases of neoplasms were reported at 200 mg twice per day.
Summary of most common treatment-emergent AEs
| Pacritinib, no. (%) | ||||||
|---|---|---|---|---|---|---|
| 100 mg once per d (n = 52) | 100 mg twice per d (n = 55) | 200 mg twice per d (n = 54) | ||||
| Any grade | Grade 3 to 4 | Any grade | Grade 3 to 4 | Any grade | Grade 3 to 4 | |
| Nonhematologic AEs | ||||||
| Diarrhea | 10 (19.2) | 1 (1.9) | 12 (21.8) | 2 (3.6) | 16 (29.6) | 3 (5.6) |
| Nausea | 12 (23.1) | 0 | 11 (20.0) | 0 | 15 (27.8) | 0 |
| Fatigue | 9 (17.3) | 3 (5.8) | 13 (23.6) | 2 (3.6) | 13 (24.1) | 2 (3.7) |
| Abdominal pain | 9 (17.3) | 0 | 6 (10.9) | 2 (3.6) | 13 (24.1) | 3 (5.6) |
| Fever | 8 (15.4) | 1 (1.9) | 9 (16.4) | 1 (1.8) | 7 (13.0) | 1 (1.9) |
| Peripheral edema | 7 (13.5) | 0 | 5 (9.1) | 0 | 9 (16.7) | 1 (1.9) |
| Decreased appetite | 6 (11.5) | 0 | 4 (7.3) | 0 | 10 (18.5) | 1 (1.9) |
| Pruritis | 2 (3.8) | 0 | 10 (18.2) | 1 (1.8) | 6 (11.1) | 0 |
| Constipation | 2 (3.8) | 0 | 1 (1.8) | 0 | 10 (18.5) | 0 |
| Pneumonia | 4 (7.7) | 2 (3.8) | 2 (3.6) | 2 (3.6) | 5 (9.3) | 5 (9.3) |
| Hyperuricemia | 1 (1.9) | 1 (1.9) | 2 (3.6) | 1 (1.8) | 3 (5.6) | 3 (5.6) |
| Hyponatremia | 0 | 0 | 3 (5.5) | 3 (5.5) | 3 (5.6) | 2 (3.7) |
| Dehydration | 0 | 0 | 3 (5.5) | 3 (5.5) | 4 (7.4) | 1 (1.9) |
| Hypertension | 0 | 0 | 3 (5.5) | 3 (5.5) | 2 (3.7) | 1 (1.9) |
| Hematologic AEs | ||||||
| Thrombocytopenia* | 11 (21.2) | 10 (19.2) | 12 (21.8) | 12 (21.8) | 22 (40.7) | 18 (33.3) |
| Anemia | 5 (9.6) | 5 (9.6) | 6 (10.9) | 4 (7.3) | 13 (24.1) | 11 (20.4) |
| Neutropenia† | 3 (5.8) | 3 (5.8) | 3 (5.5) | 3 (5.5) | 3 (5.6) | 3 (5.6) |
| Pacritinib, no. (%) | ||||||
|---|---|---|---|---|---|---|
| 100 mg once per d (n = 52) | 100 mg twice per d (n = 55) | 200 mg twice per d (n = 54) | ||||
| Any grade | Grade 3 to 4 | Any grade | Grade 3 to 4 | Any grade | Grade 3 to 4 | |
| Nonhematologic AEs | ||||||
| Diarrhea | 10 (19.2) | 1 (1.9) | 12 (21.8) | 2 (3.6) | 16 (29.6) | 3 (5.6) |
| Nausea | 12 (23.1) | 0 | 11 (20.0) | 0 | 15 (27.8) | 0 |
| Fatigue | 9 (17.3) | 3 (5.8) | 13 (23.6) | 2 (3.6) | 13 (24.1) | 2 (3.7) |
| Abdominal pain | 9 (17.3) | 0 | 6 (10.9) | 2 (3.6) | 13 (24.1) | 3 (5.6) |
| Fever | 8 (15.4) | 1 (1.9) | 9 (16.4) | 1 (1.8) | 7 (13.0) | 1 (1.9) |
| Peripheral edema | 7 (13.5) | 0 | 5 (9.1) | 0 | 9 (16.7) | 1 (1.9) |
| Decreased appetite | 6 (11.5) | 0 | 4 (7.3) | 0 | 10 (18.5) | 1 (1.9) |
| Pruritis | 2 (3.8) | 0 | 10 (18.2) | 1 (1.8) | 6 (11.1) | 0 |
| Constipation | 2 (3.8) | 0 | 1 (1.8) | 0 | 10 (18.5) | 0 |
| Pneumonia | 4 (7.7) | 2 (3.8) | 2 (3.6) | 2 (3.6) | 5 (9.3) | 5 (9.3) |
| Hyperuricemia | 1 (1.9) | 1 (1.9) | 2 (3.6) | 1 (1.8) | 3 (5.6) | 3 (5.6) |
| Hyponatremia | 0 | 0 | 3 (5.5) | 3 (5.5) | 3 (5.6) | 2 (3.7) |
| Dehydration | 0 | 0 | 3 (5.5) | 3 (5.5) | 4 (7.4) | 1 (1.9) |
| Hypertension | 0 | 0 | 3 (5.5) | 3 (5.5) | 2 (3.7) | 1 (1.9) |
| Hematologic AEs | ||||||
| Thrombocytopenia* | 11 (21.2) | 10 (19.2) | 12 (21.8) | 12 (21.8) | 22 (40.7) | 18 (33.3) |
| Anemia | 5 (9.6) | 5 (9.6) | 6 (10.9) | 4 (7.3) | 13 (24.1) | 11 (20.4) |
| Neutropenia† | 3 (5.8) | 3 (5.8) | 3 (5.5) | 3 (5.5) | 3 (5.6) | 3 (5.6) |
All events are reported regardless of relatedness and are those occurring as any grade in ≥15% or as grade 3 to 4 in ≥5% of patients in any arm.
Includes platelet count decrease.
Includes neutrophil count decrease.
The most common hematologic AEs were thrombocytopenia and anemia, both of which occurred more frequently in patients treated at 200 mg twice per day (Table 3). The majority of thrombocytopenia and anemia AEs were classified as grade 3 or 4, although it is important to note that a majority of patients had baseline grade ≥2 thrombocytopenia (101 of 161, or 62.7% with platelet count <75 × 103/μL) and anemia (114 of 161, or 70.8% with hemoglobin <10 g/dL), and thus development of higher-grade AEs required a relatively small decline in platelet counts, which were already depressed as a consequence of the disease. Dose modifications as a result of thrombocytopenia occurred in all arms, although the majority occurred on the 100 mg twice per day and 200 mg twice per day arms; among patients treated at 200 mg twice per day, 3 patients required dose reductions and 3 required drug discontinuation because of thrombocytopenia, as shown in supplemental Table 1.
Severe cardiac events (per SMQ) were uncommon. Grade 1 and 2 events, most commonly peripheral edema and QT prolongation, were more common at 200 mg twice per day (40.7% [n = 22]) than at lower doses (100 mg once per day: 21.2% [n = 11]; 100 mg twice per day: 21.8% [n = 12]) (supplemental Table 2). The most common higher-grade cardiac event was decreased left ventricular ejection fraction detected on per-protocol cardiac imaging: 2 events were described at each of the lower doses (100 mg once per day: 3.8% [2 of 52]; 100 mg twice per day: 3.6% [2 of 55]), and no such event was reported at 200 mg twice per day. There was no excess in grade 3 or 4 cardiac events at the highest dose (100 mg once per day: 5.8% [n = 3]; 100 mg twice per day: 5.5% [n = 3]; 200 mg twice per day: 3.7% [n = 2]; no grade 4 events). The mean change in QTc interval between baseline and week 24 was +10 to 11 msec on all arms; no patient had a QTc >500 msec (supplemental Table 3; supplemental Figure 1). There was one grade 5 cardiac event on study, which occurred in a patient receiving 100 mg twice per day who died of heart failure in the setting of progressive hyperleukocytosis.
Bleeding events (per SMQ) were more common at 200 mg twice per day (42.6% [n = 23]) than at lower doses (100 mg once per day: 36.5% [n = 19]; 100 mg twice per day: 25.5% [n = 14]) but were largely grade 1 or 2 in severity, with the most common being epistaxis and bruising (supplemental Table 2). There was no excess in grade 3 or 4 bleeding events at the highest dose (100 mg once per day: 7.7% [n = 4]; 100 mg twice per day: 0% [n = 0]; 200 mg twice per day: 5.6% [n = 3]; no grade 4 events). The most common high-grade bleeding event was epistaxis. Two grade 5 bleeding events occurred, 1 at 100 mg twice per day and 1 at 200 mg twice per day, both subdural hemorrhages. Although bleeding events of all grades were more common in patients with baseline platelet counts <50 × 103/μL (100 mg once per day: 60.9% [n = 14]; 100 mg twice per day: 33.3% [n = 8]; 200 mg twice per day: 75.0% [n = 18]), the magnitude of between-arm differences in bleeding rates in this subgroup was similar to that of the population as a whole.
The rate of fatal events was similar across all arms: 100 mg once per day: 7.7% (n = 4; sepsis, disease progression, tuberculosis, and general state deterioration); 100 mg twice per day: 5.5% (n = 3; disease progression, subdural hemorrhage, and heart failure); 200 mg twice per day: 5.6% (n = 3; sepsis, respiratory failure, subdural hematoma).
Population PK and PD modeling
Population PKs for pacritinib were established on the basis of pooled data from 16 studies that included 630 patients who received pacritinib at daily doses of 100 to 600 mg (supplemental Methods). Pacritinib population PKs were characterized by a 2-compartment disposition model with first-order absorption and elimination. Both absorption and bioavailability were influenced by the amount of drug administered. Systemic exposure (area under the concentration-time curve, maximum concentration, and minimum concentration) for the 100 mg once per day and 100 mg twice per day regimens was generally lower than that achieved with higher doses (200 mg twice per day and 400 mg once per day), although there was overlap (supplemental Table 4). Simulations indicate that pacritinib concentrations are expected to reach steady state within a week. Accumulation of drug after multiple doses seems to be low. Patient factors (age, sex, performance status, platelet count, hemoglobin, and hepatic function) were not significantly associated with pacritinib concentrations.
The population PK/PD modeling for pacritinib includes 2-compartment, first-order absorption and elimination with dose-dependent absorption and bioavailability. Population PK/PD modeling based on data from PAC203 and both of the phase 3 PERSIST studies showed that higher pacritinib doses were associated with greater SVR (n = 280) and TSS reduction (n = 282) (Figure 6). Exposure-response analysis showed that higher systemic pacritinib levels were significantly associated with higher SVR rates (based on quartiles for area under the time-concentration curve and minimum concentration in 182 patients; P < .05). Although there was a trend toward high TSS response rate with higher exposure to pacritinib, this was not statistically significant (n = 162, P < .1; supplemental Figure 2).

Dose-response modeling. Modeling based on efficacy data from previous phase 3 (PERSIST-1 and PERSIST-2) studies as well as the PAC203 phase 2 study. Increasing doses are associated with better responses for both spleen volume and symptom score reduction.
Dose-response modeling. Modeling based on efficacy data from previous phase 3 (PERSIST-1 and PERSIST-2) studies as well as the PAC203 phase 2 study. Increasing doses are associated with better responses for both spleen volume and symptom score reduction.
Discussion
Data from PAC203 and from dose- and exposure-response modeling demonstrate that pacritinib 200 mg twice per day provides greater efficacy compared with lower doses and has a manageable safety profile. The PAC203 study was conducted in a population with advanced myelofibrosis and prolonged previous exposure to ruxolitinib. A substantial number of patients had high-risk molecular mutations, including a higher proportion with TP53 mutations than has been reported in other myelofibrosis cohorts, including those with previous ruxolitinib exposure (7.3% on PAC203 vs 1% to 4% previously reported18-21 ). The majority of patients were anemic and thrombocytopenic, a profile characteristic of the myelodepletive myelofibrosis phenotype.22 The use of current first-line therapies for myelofibrosis, such as ruxolitinib and fedratinib, is limited in myelodepletive disease because these drugs can exacerbate cytopenias. Myelofibrosis patients with baseline platelet counts <50 × 103/μL were excluded from pivotal trials that led to approval of other JAK inhibitors,13,23 and studies of ruxolitinib in thrombocytopenic patients have focused exclusively on those with baseline platelet counts of 50 × 103/μL and above.24,25 In contrast, pacritinib has demonstrated clinical benefit across groups of myelofibrosis patients in multiple clinical studies,1,2 and most myelofibrosis patients with severe thrombocytopenia are able to tolerate pacritinib at 200 mg twice per day without requiring dose reductions or holds.
As observed in PERSIST-1 and PERSIST-2, patients with severe thrombocytopenia treated on PAC203 attained spleen and symptom responses despite previous exposure to ruxolitinib. Although the biologic basis of this well-documented observation is not fully understood, it is likely a function of both the kinase inhibition profile of pacritinib, which includes IRAK1, and differences in myelofibrosis biology in which thrombocytopenia and associated low JAK2 allele burden may represent a distinct myelofibrosis subgroup that is particularly responsive to pacritinib.26 The correlation between disease genotype and response to pacritinib therapy will be a correlative of interest in ongoing studies.
Pacritinib was generally well tolerated across all dose levels on PAC203, with manageable toxicities because of appropriate patient selection and the use of dose modification guidelines. Although the 200 mg twice per day dose was associated with higher rates of AEs, there was no excess in high-grade cardiac or hemorrhagic events compared with lower doses, and AE rates were lower than previously observed on the 200 mg twice per day arm of PERSIST-2. In fact, bleeding rates at 200 mg twice per day (42.6%) were comparable to those observed in ruxolitinib-treated patients enrolled in the COMFORT studies (32.6%)27 , which excluded patients with platelet counts <100 × 103/μL. These comparisons suggest that the PAC203 risk minimization measures were effective.
A limitation of the PAC203 study is that the dose modification guidelines required an open-label study. Although lack of blinding should have little impact on the evaluation of SVR and key safety measures, it is possible that patient-reported outcomes could be biased by knowledge of treatment assignment.
The results of this phase 2 study informed the dose selection for an ongoing phase 3 randomized study (PACIFICA) comparing pacritinib 200 mg twice per day with physician’s choice therapy (including low-dose ruxolitinib) for patients with myelofibrosis and severe thrombocytopenia (NCT03165734). PACIFICA will include the risk mitigation measures implemented in the PAC203 study. There is a lack of available treatment options for patients with severe thrombocytopenia, including those who are intolerant of or resistant to ruxolitinib. Pacritinib remains the only prospectively evaluated therapy supported by clinical data for treating patients with myelofibrosis and severe thrombocytopenia, an area of unmet need and the focus of the current phase 3 trial.
Send data sharing requests via e-mail to the corresponding author, Aaron T. Gerds, at gerdsa@ccf.org.
Acknowledgments
Medical editing assistance was provided by Janis Leonoudakis, and was funded by CTI BioPharma Corp.
This work was supported by CTI BioPharma Corp.
Authorship
Contribution: J.O.M. and A.R.C. devised the trial; A.R.C. wrote the protocol; A.T.G., M.R.S., B.L.S., M.T., M.E., C.N.H., A.Y., A.V., A.J.M., J.-J.K., V.G.-G., P.B., R.K.R., C.B.M., J.P., S.T.O., and J.O.M. enrolled patients; A.T.G., M.R.S., J.O.M., S.A.B., D.R.M., K.I., S.T., K.R.-T., J.A.S., and A.R.C. analyzed the data; J.A.S. and K.R.-T. performed statistical analysis; A.J.M. and J.O. performed correlative analysis of genomic samples; D.R.M. and S.T. performed PK/PD modeling; S.A.B. and S.D. provided administrative and technical support; A.R.C. and J.O.M. supervised the conduct of the study; S.A.B. drafted the manuscript; and all authors critically reviewed the manuscript for intellectual content and approved the final manuscript.
Conflict-of-interest disclosure: A.T.G. served on advisory boards for Kartos Therapeutics, CTI BioPharma, Promedior, Galecto, and PharmaEssentia. M.R.S. received research funding from Astex, Incyte, Takeda, and TG Therapeutics, has equity in Karyopharm, served on advisory boards and DSMBs for AbbVie, Bristol Myers Squibb, Celgene, Karyopharm, Ryvu, Sierra Oncology Takeda, and TG Therapeutics, and has consulted for Karyopharm and Ryvu. B.L.S. has consulted for Incyte, Celgene/Bristol Myers Squibb, and CTI BioPharma, has received funding from Celgene/Bristol Myers Squibb, and Incyte, and has served on the speaker’s bureau for Alexion and Agios. M.T. served on advisory boards for Bristol Myers Squibb, CTI BioPharma, Imago, Novartis, and Takeda. M.E. served on the board for AOP. C.N.H. has consulted for Novartis, AstraZeneca, CTI BioPharma, Roche, AOP, Sierra Oncology, Promedior, Constellation, Celgene/Bristol Myers Squibb, and Imago and is on the speaker’s bureau for Novartis and Janssen. A.Y. received honoraria from Incyte, Novartis, and Agios. A.V. served on the speaker’s bureau for Novartis, Bristol Myers Squibb-Celgene, Takeda, and AOP and on the advisory board for Novartis, Bristol Myers Squibb/Celgene, Incyte, CTI BioPharma, and AbbVie. A.J.M. has consulted for Novartis, Bristol Myers Squibb/Celgene, and AbbVie, has received research funding from Novartis, Bristol Myers Squibb/Celgene, and CTI BioPharma, has received honoraria from Novartis and CTI BioPharma, and has served on the speaker’s bureau for Novartis. J.-J.K. has served on advisory boards for Novartis, CTI BioPharma, AOP, Celgene, and AbbVie. V.G.-G. has received honoraria from and served on advisory committees for Novartis, Pfizer, and Celgene/Bristol Myers Squibb. P.B. has received honoraria from Incyte, Celgene/Bristol Myers Squibb, CTI BioPharma, and Kartos Therapeutics, and research funding from Incyte, Celgene/Bristol Myers Squibb, CTI BioPharma, Kartos, Constellation, Blueprint Medicines, Astellas, Pfizer, NS Pharma, and Promedior. R.K.R. has consulted for Constellation, Incyte, Celgene, Promedior, CTI, Jazz Pharmaceuticals, Blueprint, Stemline, and Galecto and received research funding from Incyte, Stemline, and Constellation. C.B.M. has received honoraria from Incyte and Bristol Myers Squibb, has served on the speaker’s bureau for Incyte and Bristol Myers Squibb, has served on advisory boards for Incyte and CTI BioPharma, and has received institutional research support from Incyte, Bristol Myers Squibb, and CTI BioPharma. S.T.O. has served as a consultant for and served on advisory boards for Incyte, Gilead Sciences, Novartis, Celgene/Bristol Myers Squibb, Blueprint Medicines, Kartos Therapeutics, Disc Medicine, PharmaEssentia, and CTI BioPharma. D.R.M. and K.I. have consulted for CTI BioPharma. J.O.M. has received institutional research funding from Incyte, CTI BioPharma, Janssen, PharmaEssentia, Novartis, Merck, Arog, Merus, Promedior, Kartos, Forbius and Roche and has received consulting fees from Incyte, Geron, Kartos, Celgene, Promedior, Prelude, and Galecto. The remaining authors declare no competing financial interests.
Correspondence: Aaron T. Gerds, Cleveland Clinic, 9500 Euclid Ave, CA60, Cleveland, OH 44195; e-mail: gerdsa@ccf.org.

