

Key Points
PNH in a family was caused by CN-LOH of a germline heterozygous PIGB mutation.
CN-LOH on chromosome 15 is related to a shared 70-kbp microdeletion including TM2D3 and TARSL2 in a patient and her mother.

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare hematopoietic stem cell (HSC) disorder characterized by defective synthesis of the glycosylphosphatidylinositol (GPI) anchors as a result of somatic mutations in the X-linked PIGA gene. The disease is acquired. No constitutional PNH has been described. Here, we report familial PNH associated with unusual inflammatory symptoms. Genetic analysis revealed a germline heterozygous PIGB mutation on chromosome 15 without mutations in PIGA or any of the other genes involved in GPI biosynthesis. In vitro data confirmed that transfection of the mutant PIGB could not restore the surface expression of GPI-anchored proteins (APs) in PIGB-deficient Chinese hamster ovary cells. Homozygosity was caused by copy number–neutral loss of heterozygosity (CN-LOH) of the germline PIGB mutation, leading to deficient expression of GPI-APs in the affected blood cells of the index patient and her mother. The somatic event leading to homozygosity of the germline mutant PIGB gene involved a 70-kbp microdeletion of chromosome 15q containing the TM2D3 and TARSL2 genes, which was implicated in chromosome 15q mosaicism. Interestingly, we detected the deletion in both the patient and her mother. A sister of the mother, who carried the same germline PIGB mutation but without this microdeletion involving TM2D3 and TARSL2, did not have a PNH clone or CN-LOH. In conclusion, we describe PNH caused by CN-LOH of a germline heterozygous PIGB mutation in a patient and her mother and hypothesize that the 70-kbp microdeletion may have contributed to the PNH clone in both.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired clonal disease characterized by intravascular hemolysis, thrombophilia, and a varying degree of bone marrow failure.1,2 In patients with PNH, the affected blood cells show either reduced expression or complete absence of glycosylphosphatidylinositol (GPI)-anchored proteins (APs), including the complement regulatory proteins decay accelerating factor (DAF or CD55) and membrane inhibitor of reactive lysis (CD59). Deficiency of these complement regulatory proteins renders erythrocytes sensitive to complement-mediated destruction and chronic intravascular hemolysis.3 PNH is caused by a somatic mutation of the PIGA gene in the hematopoietic stem cell (HSC) and subsequent clonal expansion (PIGA-PNH).4 PIGA is involved in the first step of GPI biosynthesis5 and is the only X-linked gene among the 27 genes involved in this process.6 A somatic mutation in PIGA on 1 allele is sufficient to render cells GPI deficient in women as well as men due to X-chromosome inactivation in women.5 Here, we report familial PNH (PIGB-PNH) caused by a homozygous mutation in the PIGB gene on chromosome 15q, which is also involved in the GPI biosynthesis pathway.7,8 The index patient is 57 years old and has unusual PNH. She was diagnosed with aplastic anemia at age 8 years and PNH at the age of 14 years. She has had recurrent autoinflammatory symptoms as well as hemolysis since early childhood. Her PNH clone size is nearly 100% in not only granulocytes and monocytes but also erythrocytes. We show that her PNH clone and the one in her mother were generated by homozygosity of a germline PIGB mutation caused by copy number–neutral loss of heterozygosity (CN-LOH). We also show that the regions of the CN-LOH in the patient and her mother contained PIGB and a 70-kbp microdeletion including the TM2D3 and TARSL2 genes on the same chromosome 15q, which was reported to be closely associated with CN-LOH–dependent mosaic hematopoiesis among healthy blood donors.9 Our data suggest that the 70-kbp microdeletion including the TM2D3 and TARSL2 genes is causal to generation of PNH clones in this family.
Methods
Patient samples
Peripheral blood samples from the patient and family members were taken after informed consent was obtained. Remaining material from a diagnostic cervical biopsy that had been obtained in recent years was used after obtaining informed consent. The study was approved by the independent ethics committee of the Radboudumc and Osaka University review board and was conducted in agreement with Good Clinical Practice guidelines and according to the Declaration of Helsinki.
Flow cytometrical analysis and cell sorting of PNH and non-PNH cells
For analysis of granulocytes and monocytes, cells were stained with CD45-KO, CD14-ECD, CD64-PECy7, CD15-PacificBlue, and CD24-APC monoclonal antibodies (Beckman Coulter). Within the CD64pos cell population, the PNH monocytes (CD14neg/dim and fluorescein-labeled proaerolysin [FLAER]neg/dim) and non-PNH monocytes (CD14pos/FLAERpos) were selected. Within the CD15pos cell population, the PNH granulocytes (CD24neg/dim and FLAERneg/dim) and non-PNH granulocytes (CD24pos/FLAERpos) were selected and sorted. For analysis of lymphocytes, cells were stained with CD3-APCA750 (Beckman Coulter), CD19-BUV395, and CD48-PE (BioLegend). Within the CD3pos cell population, PNH T cells (CD48neg/dim and FLAERneg/dim) and non-PNH T cells (CD48pos/ FLAERpos) were selected and sorted. The detection limit was 0.1%.
DNA isolation
DNA was isolated from whole blood, granulocytes isolated with Ficoll-Paque PLUS (GE Healthcare), and sorted PNH and non-PNH cells using the NucleoSpin Blood QuickPure (Machery-Nagel). DNA from paraffin-embedded cervical tissue was isolated using Chelex-100 (Bio-Rad).
Target exome sequencing
Target exome sequencing was performed for all 27 GPI pathway genes (PIGA, PIGQ, PIGY, PIGC, PIGP, PIGH, PIGL, PIGW, PIGM, PIGX, PIGV, PIGN, PIGB, PIGO, PIGG, PIGZ, PIGK, PIGS, PIGT, PIGU, GPAA1, PGAP1, PGAP5, PGAP2, PGAP3, PGAP4, and PGAP6) with genomic DNA (gDNA) from the patient’s granulocytes using the HaloPlex HS Target Enrichment kit (Agilent Technologies) and the MiSeq instrument (Illumina) according to the manufacturer’s instructions. Exome data processing, variant calling, and variant annotation were performed using Sure Call (3.5.1.46) software (Agilent Technologies). The PIGB mutation in the patient was validated by Sanger sequencing. The presence or absence of the same PIGB mutation in the patient’s mother, daughter, aunt, and uncle was studied by Sanger sequencing.
Functional analysis of mutant PIGB
Complementary DNAs (cDNAs) of hemagglutinin-tagged human PIGB and its mutant were subcloned into an SRα-driven pME vector.10 These plasmids were cotransfected with a luciferase-expressing plasmid (to determine the transfection efficiency) by electroporation into PIGB-deficient Chinese hamster ovary (CHO) cells11 that were permanently transfected with human CD59 and DAF cDNA. Cell-surface levels of GPI-APs were determined by flow cytometric analysis after staining either with anti-CD59 or anti-DAF monoclonal antibodies or with FLAER.
SNP array
Single-nucleotide polymorphism (SNP) array analysis of chromosome 15q was performed by LSI Medience using CytoScan HD Array(Thermo Fisher Scientific).
Digital PCR
TM2D3 gene copy number was determined using QuantStudio 3D Digital polymerase chain reaction (PCR) system (Thermo Fisher).
Measurement of cytokines and serum proteins
Interleukin-6 (IL-6), IL-18, and serum amyloid A in serum samples were measured by ELISA (enzyme-linked immunosorbent assay).
Results
Clinical findings and laboratory analysis
Our investigations focused on our patient (III-1), her mother (II-7), her daughter (IV-1), and 2 available siblings of her mother (II-10 and II-11) (Figure 1A).

Pedigree of the family and GPI-AP-deficiency of the patient and her mother. (A) Pedigree. Family members tested are indicated by an asterisk. The patient (III-1) and her mother (II-7) have a PNH clone. The aunt (II-10) has a heterozygous PIGB mutation c.695G>A but no PNH clone. The daughter (IV-1) and uncle (II-11) have no PIGB mutation. (B) Flow cytometry of blood cells from the patient (III-1) and her mother (II-7).
Pedigree of the family and GPI-AP-deficiency of the patient and her mother. (A) Pedigree. Family members tested are indicated by an asterisk. The patient (III-1) and her mother (II-7) have a PNH clone. The aunt (II-10) has a heterozygous PIGB mutation c.695G>A but no PNH clone. The daughter (IV-1) and uncle (II-11) have no PIGB mutation. (B) Flow cytometry of blood cells from the patient (III-1) and her mother (II-7).
Our patient (III-1) is a 57-year-old woman with a normal appearance and no dysmorphic features. She has a remarkable history of inflammatory symptoms with fever since early childhood. At age 8 years, aplastic anemia was diagnosed, which was attributed to repeated chloramphenicol courses. Treatment with oxymetholone and corticosteroids resulted in a partial response with persisting mild leukopenia and thrombocytopenia. At age 13 years, she suffered from acute unexplained pericarditis with fever, polyarthritis, exanthematous skin lesions, tonsillitis, and hemolytic anemia. At that time, she was screened for autoimmune diseases. The anti-nuclear antibody test result was negative. The lupus erythematosus cell test result was negative. A skin biopsy specimen showed no evidence of lupus erythematosus or any other immune-complex disease. At age 14 years, PNH was diagnosed (based on results of an acid Ham test and, later, a sucrose lysis test). Hemoglobin was 10.5 g/dL, and lactate dehydrogenase (LDH) was 2.1 times the upper limit of normal. Since that time, she suffered from fatigue and had frequent hemolytic crises, with typical PNH symptoms such as abdominal pain and dark urine but also atypical inflammatory symptoms, such as joint pain and urticaria. She had a deep venous thrombosis at age 19 years and a myocardial infarction at the age of 37 years. She denied treatment with the complement inhibitor eculizumab when this became available. More recently, the blood examination showed a hemoglobin level of 11.1 g/dL and an LDH level 3.6 times the upper limit of normal; during a hemolytic crisis, the hemoglobin level was 10.6 g/dL and the LDH level was 6.5 times the upper limit of normal (supplemental Table 1). IL-6, IL-18, and serum amyloid A were measured on one occasion after the patient (III-1) had experienced an episode of fever and urticaria. The symptoms had resolved the day before the blood sample was taken. IL-6 was not elevated, but IL-18 was elevated (1190 pg/mL [normal level, <211 pg/mL]), as was serum amyloid A (120 mg/L [normal level, <6.4 mg/L]). Serum amyloid A was mildly elevated (7.8 mg/L) 1 month after the occurrence of inflammatory symptoms (supplemental Table 2).
Remarkably, PNH clone size was consistently 99% to 100% not only in monocytes and granulocytes but also in erythrocytes (Figure 1B). A clone size of 82% was detected in the T-cell fraction. All PNH cells were type II cells with reduced rather than absent expression of GPI-APs. Analysis of peripheral blood of the patient’s clinically healthy mother (II-7) at age 80 years showed the presence of a small PNH clone in granulocytes (4%), monocytes (4%), and erythrocytes (0.9%) and a moderate PNH clone in T cells (15%) (Figure 1B). The mother’s PNH cells were also type II only. No PNH clone was detected in the peripheral blood of family members II-10, II-11, and IV-1 (Figure 1A). The mother and other family members never reported PNH symptoms.
Genetic analysis of the patient and family members
Targeted exome sequencing of GPI pathway genes using gDNA from the patient’s (III-1) granulocytes revealed a homozygous single-nucleotide substitution in PIGB, NM_004855: c.695G>A p.Arg232His (supplemental Figure 1), which was confirmed by Sanger sequencing (Figure 2A). Sanger sequencing of gDNA from cervical tissue from the patient (III-1) showed a heterozygous PIGB mutation (Figure 2A). Together these results suggest that the heterozygous mutation exists in the germline and a somatic event resulted in homozygosity in the PNH clone. The same heterozygous PIGB mutation was detected in the patient’s mother (II-7), indicating that the germline PIGB mutation was inherited from her. The mutation was also detected in the patient's aunt (II-10), but not in the patient’s uncle (II-11) or daughter (IV-1) (Figure 2A).
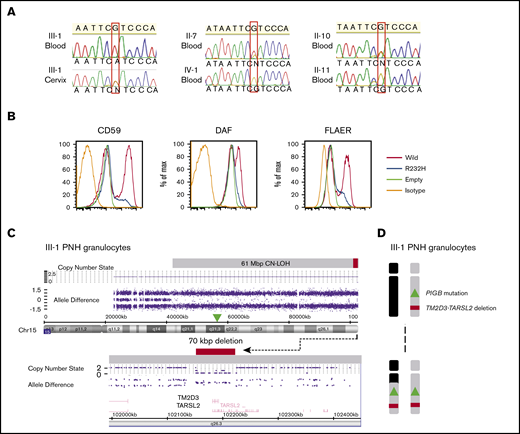
Genetic analyses of the affected patient (III-1) and family members and in vitro functional analysis of the mutant PIGB. (A) The inherited PIGB mutation confirmed by Sanger sequencing in blood cells (homozygous) and somatic cells derived from cervix (heterozygous) of III-1 and in blood cells (heterozygous) from the patient’s mother (II-7) and aunt (II-10). The mutation was not present in blood cells from the daughter (IV-1) or uncle (II-11). (B) Functional analysis of the mutant PIGB cDNA. Mutant PIGB cDNA could not rescue the surface expression of GPI-APs through transient transfection in PIGB-deficient CHO cells. (C) SNP array analysis of chromosome 15q (Chr15) in PNH-type granulocytes from III-1 using CytoScan HD Array. The PNH clone had a CN-LOH region spanning 61 Mbp with a small deletion spanning 70 kbp, including the TM2D3 and TARSL2 genes. (D) Outcome of the CN-LOH in the patient (III-I); the inherited PIGB mutation and microdeletion became homozygous by the somatic CN-LOH.
Genetic analyses of the affected patient (III-1) and family members and in vitro functional analysis of the mutant PIGB. (A) The inherited PIGB mutation confirmed by Sanger sequencing in blood cells (homozygous) and somatic cells derived from cervix (heterozygous) of III-1 and in blood cells (heterozygous) from the patient’s mother (II-7) and aunt (II-10). The mutation was not present in blood cells from the daughter (IV-1) or uncle (II-11). (B) Functional analysis of the mutant PIGB cDNA. Mutant PIGB cDNA could not rescue the surface expression of GPI-APs through transient transfection in PIGB-deficient CHO cells. (C) SNP array analysis of chromosome 15q (Chr15) in PNH-type granulocytes from III-1 using CytoScan HD Array. The PNH clone had a CN-LOH region spanning 61 Mbp with a small deletion spanning 70 kbp, including the TM2D3 and TARSL2 genes. (D) Outcome of the CN-LOH in the patient (III-I); the inherited PIGB mutation and microdeletion became homozygous by the somatic CN-LOH.
Functional analysis of the PIGB mutant
Flow cytometric analysis of various GPI-APs on PIGB-deficient CHO cells transfected with wild type or mutant PIGB cDNAs revealed that the 695G>A mutation greatly reduced PIGB activity (Figure 2B).
SNP array analysis showing CN-LOH in the PNH cells
SNP array analysis of chromosome 15q of gDNA from the patient’s (III-1) granulocytes demonstrated a 61 Mbp CN-LOH spanning ∼74% of the telomeric side of chromosome 15q (Figure 2C, top). The CN-LOH region included PIGB, accounting for the homozygosity of the PIGB mutation (Figure 2D). SNP array analysis also revealed a homozygous microdeletion of ∼70 kbp close to the telomere that included TM2D3 and part of the TARSL2 genes (Figure 2C, bottom). To investigate whether this microdeletion was also derived from the mother’s allele with the PIGB mutation, the copy number of the TM2D3 gene was determined by digital PCR in gDNA from the patient’s granulocytes and whole blood cells from family members. The normalized copy number was almost 0 in the patient (III-1), 0.5 in the mother (II-7), and 1 in her daughter (IV-1), aunt (II-10), uncle (II-11), and a healthy control, suggesting that the microdeletion was derived from the mother’s allele together with the PIGB mutation (Figure 3A-B). The aunt (II-10) has the same germline PIGB mutation as the patient (III-1) and the mother (II-7) but lacks the 70-kbp microdeletion and had no detectable PNH cells, suggesting an association between the 70-kbp microdeletion and PNH.
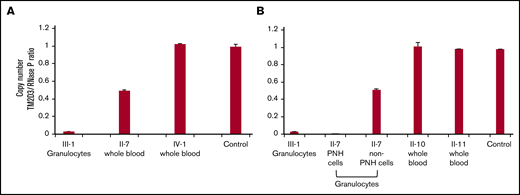
Detailed genetic analyses of the family members. (A-B) Copy-number analysis of the TM2D3 gene using the QuantStudio 3D Digital PCR system. Non-PNH cells from II-7 had heterozygous deletions, and PNH cells from II-7 and III-1 had homozygous deletions, whereas other family members showed normal copy number. Experiments shown in panels A and B were performed at the different time points using the same samples in case of III-1 and control. Standard deviation bars are shown from the independent triplicate analysis.
Detailed genetic analyses of the family members. (A-B) Copy-number analysis of the TM2D3 gene using the QuantStudio 3D Digital PCR system. Non-PNH cells from II-7 had heterozygous deletions, and PNH cells from II-7 and III-1 had homozygous deletions, whereas other family members showed normal copy number. Experiments shown in panels A and B were performed at the different time points using the same samples in case of III-1 and control. Standard deviation bars are shown from the independent triplicate analysis.
The PNH and non-PNH granulocytes and T cells from the mother were sorted and evaluated using digital PCR and SNP array to determine the involvement of CN-LOH in PNH cells. The normalized copy number of the TM2D3 gene, which was 0 in the PNH granulocytes (similar to the patient), was 0.5 in the non-PNH granulocytes (Figure 3B).
In addition, SNP array analysis revealed that PNH granulocytes and T cells of the mother (II-7) contained multiple clones with CN-LOH regions of different lengths (Figure 4A). Based on allele difference data (supplemental Figures 2 and 3; Figure 4B), 4 CN-LOH regions could be distinguished spanning 59-, 64-, 75-, and >80-Mbp telomeric regions, including PIGB and the 70-kbp deletion. The major clone, with a 59-Mbp CN-LOH, occupied ∼55% of PNH granulocytes and T cells as estimated from the pattern of allele differences. Three other clones in the mother bearing 64-, 75-, and >80-Mbp CN-LOH occupied ∼10%, 10%, and 25%, respectively, of both PNH granulocytes and T cells (Figure 4B).
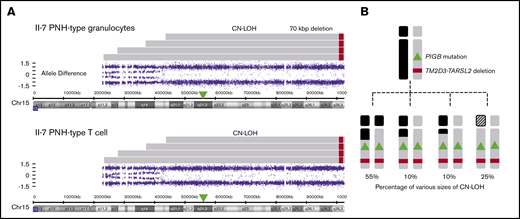
Various PNH clones with CN-LOH regions of different lengths from II-7 (mother). (A) SNP array analysis of chromosome 15q in PNH-type granulocytes and T cells from II-7 using CytoScan HD Array. There were at least 4 clones with different sizes of CN-LOH in PNH cells, and the sizes and number of CN-LOH clones in granulocytes and T cells were almost the same. As the probes for chromosome 15p were missing in the kit, the exact spanning size of the longest CN-LOH was unknown (shaded areas). (B) Percentage of each clone with different sizes of CN-LOH estimated from the pattern of allele differences.
Various PNH clones with CN-LOH regions of different lengths from II-7 (mother). (A) SNP array analysis of chromosome 15q in PNH-type granulocytes and T cells from II-7 using CytoScan HD Array. There were at least 4 clones with different sizes of CN-LOH in PNH cells, and the sizes and number of CN-LOH clones in granulocytes and T cells were almost the same. As the probes for chromosome 15p were missing in the kit, the exact spanning size of the longest CN-LOH was unknown (shaded areas). (B) Percentage of each clone with different sizes of CN-LOH estimated from the pattern of allele differences.
Discussion
We describe the first family with PNH and the first PNH resulting from a homozygous mutation in the PIGB gene
The clinical picture of PNH requires reduced or absent expression of GPI-APs on the surface of blood cells as a result of defective GPI synthesis, commonly because of a PIGA mutation in the HSC and expansion of the abnormal clone for which an additional event is required.12-14
PNH in the index patient was diagnosed at an early age, when flow cytometry, now used in routine diagnostics for PNH, was not yet available. When flow cytometry became available, the index patient had a remarkable presence of exclusively type II PNH cells, no type III cells, and almost no normal blood cells. Commonly, in untreated PNH patients, the PNH clone in erythrocytes is smaller than the clones in granulocytes and monocytes; moreover, erythrocyte clone size is not 100%. In the index patient (III-1), all erythrocytes, granulocytes, and monocytes were PNH cells. Even during episodes of more severe hemolysis (LDH >4× upper limit of normal), no normal red cells could be detected (data not shown). This implies that hematopoiesis in the patient was from PNH HSCs only.
The same PIGB mutation has been found in a family with inherited GPI deficiency caused by compound heterozygous germline mutations, with the main symptoms being intellectual disability and seizures, but these patients have no hemolysis.8 The index patient and her family have normal cognition, and there is no history of seizures.
In this PIGB-PNH case, the maternal allele of the patient had a pathogenic PIGB germline mutation and a 70-kbp microdeletion near the telomere, which included the TM2D3 and TARSL2 genes. The acquired CN-LOH spanning 61 Mbp, which includes PIGB and the 70-kbp microdeletion, renders both the PIGB mutation and the microdeletion homozygous. A similar CN-LOH was found in the mother’s PNH cells, but not in the aunt, who had the same germline PIGB mutation, but not the 70-kbp microdeletion. The aunt had no detectable PNH cells. This association of the specific microdeletion with the occurrence of CN-LOH strongly suggests that it is required for the development of PIGB-PNH.
Using computational techniques, it has been suggested that the presence of this 70-kbp deletion increased chromosome 15q mosaicism, such as CN-LOH and loss events, by a factor of 698.9 The loss of the 70-kbp region might enhance mitotic recombination, a major cause of CN-LOH, or may be responsible for clonal expansion after occurrence of CN-LOH.15 Further studies are needed for clarification of this mechanism.
For the onset of PNH, the GPI-defective cells must expand. In PIGA-PNH, the PIGA mutation itself is not sufficient to drive clonal expansion.12-14 It is still not completely understood what mechanism is responsible for outgrowth or sometimes vanishing of the mutated cells in PNH. Selective pressure by the immune system and/or the presence of secondary mutations have been hypothesized to play a role.16-19 Recently, mutations of the PIGT gene, another GPI pathway gene (on chromosome 20), have been reported to cause PNH (PIGT-PNH). These patients had a germline mutation in one allele of PIGT and an acquired 8- to 18-Mbp deletion including PIGT in the other allele rendering the mutation homozygous. In PIGT-PNH, loss of tumor suppressor genes in the deleted region of chromosome 20q likely plays a role in expansion of the PIGT-defective clone,20 As for PIGB-PNH, before the onset of PNH, the index patient suffered from aplastic anemia, which might have brought a strong stimulus for hematopoiesis, leading to generation of CN-LOH and clonal expansion at an early age. The patient’s mother, when first examined at age 80 years, had a small population of PNH cells that contained multiple clones generated by CN-LOH. It is not understood why the mother’s PNH clones are much smaller than her daughter’s PNH clone. The mother never had clinical PNH, but it cannot be ruled out that a larger PNH clone was present at some time in her life. This hypothesis is strengthened by the fact that the PNH clone in the T-cell fraction of the mother is 15%, whereas the T-cell clone size in other studied cases of PNH is small compared with granulocytes and monocytes, most likely due to thyroid regression at a young age, before full expansion of the PNH clone.21 It is well known that a PNH clone often changes size over the years.
The patient in this study had inflammatory symptoms partly similar to the few patients with PIGT-PNH described. However, the clinical histories are different; PIGT-PNH patients had experienced autoinflammation caused by the small PNH clone long before clinical symptoms of PNH appeared, whereas in the patient with PIGB-PNH, autoinflammation-like symptoms were noted not long before the diagnosis of PNH, and hemolytic attacks often co-occur with autoinflammation-like symptoms, such as urticaria and fever (supplemental Table 2). The co-occurrence of episodes of autoinflammatory symptoms and episodes of increased PNH-related hemolysis renders a causal relationship very likely. The observation that the mother did not have symptoms of PNH or autoinflammation could indicate that not only PNH symptoms but also inflammatory symptoms require a certain minimum clone size to occur.
In PIGT-defective cells, free, non-protein-linked GPI accumulates.22 Free GPIs are transported to the cell surface, which can be detected by T5 antibody. T5 recognizes the N-acetylgalactosamine side chain of free GPI (supplemental Figure 4). Inflammasome activation is associated with free GPI and complement activation in PIGT-PNH.20 PIGB is the enzyme that attaches the third mannose to GPI (supplemental Figure 4). It is known that a GPI biosynthetic intermediate bearing only 2 mannoses accumulates in PIGB-defective cells.7 We speculate that similar to free GPI in PIGT-PNH cells, these GPI intermediates are transported to the cell surface. At present, we cannot definitely conclude that PIGB-PNH shows autoinflammation, because this is only one patient’s symptom. Further study with accumulated cases is required to determine whether and how the GPI intermediate and complement activation might also be causally involved in inflammation in PIGB-PNH (supplemental Figure 4). In conclusion, we report a new form of PNH caused by PIGB mutations with CN-LOH.
Send data sharing requests via e-mail to the corresponding author, Yoshiko Murakami (yoshiko@biken.osaka-u.ac.jp).
Acknowledgments
The authors thank Konnie Hebeda, Paul Rombout (Department of Pathology, Radboud University Medical Center), and Junji Takeda (Osaka University) for discussion; Shota Nakamura and Daisuke Motooka (Osaka University) for running the next-generation sequencer; and Keiko Kinoshita, Saori Umeshita, Kae Imanishi (Osaka University), and Michiko Nakamura (Wakayama Medical University) for technical help.
This work was supported by the Japan Society for the Promotion of Science and Ministry of Education, Culture, Sports, Science and Technology KAKENHI grants JP16H04753 and JP17H06422 and a grant from the Japan Society of Complement Research.
Authorship
Contributions: S.L., P.M., N.I., T.K., and Y.M. designed the study; S.L., C.S., N.B., F.P., J.H.J., N.I., and Y.M. acquired the data and conducted experiments; and S.L., P.M., T.K., and Y.M. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yoshiko Murakami, Yabumoto Department of Intractable Disease Research, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamada-oka, Suita, Osaka 565-0871, Japan; e-mail: yoshiko@biken.osaka-u.ac.jp.

