

Key Points
Display of agonistic single-chain variable fragments on lentiviral vector particles activates T cells and induces their proliferation.
Such particles allow gene transfer in unprocessed blood and generate CAR T cells directly in vivo.

Abstract
Genetic modification of T lymphocytes is a key issue in research and therapy. Conventional lentiviral vectors (LVs) are neither selective for T cells nor do they modify resting or minimally stimulated cells, which is crucial for applications, such as efficient in vivo modification of T lymphocytes. Here, we introduce novel CD3-targeted LVs (CD3-LVs) capable of genetically modifying human T lymphocytes without prior activation. For CD3 attachment, agonistic CD3-specific single-chain variable fragments were chosen. Activation, proliferation, and expansion mediated by CD3-LVs were less rapid compared with conventional antibody-mediated activation owing to lack of T-cell receptor costimulation. CD3-LVs delivered genes not only selectively into T cells but also under nonactivating conditions, clearly outperforming the benchmark vector vesicular stomatitis-LV glycoproteins under these conditions. Remarkably, CD3-LVs were properly active in gene delivery even when added to whole human blood in absence of any further stimuli. Upon administration of CD3-LV into NSG mice transplanted with human peripheral blood mononuclear cells, efficient and exclusive transduction of CD3+ T cells in all analyzed organs was achieved. Finally, the most promising CD3-LV successfully delivered a CD19-specific chimeric antigen receptor (CAR) into T lymphocytes in vivo in humanized NSG mice. Generation of CAR T cells was accompanied by elimination of human CD19+ cells from blood. Taken together, the data strongly support implementation of T-cell–activating properties within T-cell–targeted vector particles. These particles may be ideally suited for T-cell–specific in vivo gene delivery.
Introduction
Because of their crucial role in adaptive immunity, T lymphocytes have always been important targets for gene therapy approaches. Their potential has been further underscored by the recent approval of 2 CD19-specific chimeric antigen receptor (CAR) T-cell therapies for treatment of hematological diseases in Europe and the United States.1,2 Several hundred clinical studies are ongoing assessing CAR T-cell therapies for various types of cancers and other indications.3-6
For genetic engineering, T lymphocytes are isolated from the patient’s blood, ex vivo activated by stimulation with recombinant antibodies against CD3 and CD28 (soluble, plate-, or bead-bound) in combination with cytokines such as interleukin (IL)-2, IL-7, and IL-15 followed by gene transfer and subsequent expansion before infusion.7,8 Genetic modification is most frequently accomplished by transduction with stably integrating γ-retroviral or lentiviral vectors (LVs) pseudotyped with the vesicular stomatitis (VSV) glycoprotein G. These vectors have a broad tropism and can be produced at high titers under good manufacturing practice conditions, but they also harbor essential drawbacks. First, their broad tropism confers transduction of many cell types including malignant B cells. Accidental transfer of the CD19-CAR into a single leukemic cell during manufacture has led to relapse and death of a patient.9 Second, T cells have to be activated before genetic engineering because resting T lymphocytes are not susceptible toward transduction with VSV-LVs.10 Optimizing gene delivery through engineering of vector particles is a valuable strategy to improve and simplify genetic modification of T cells. Receptor-targeted LVs (RT-LVs) use a cell surface protein of choice as entry receptor. This is achieved through retargeted glycoproteins that can be combined with any type of lentiviral capsid and genetic elements regulating expression of the gene of interest.11,12 Attachment to the targeted receptor is achieved by displaying a targeting domain, such as a single-chain antibody fragment (scFv). In particular, selective gene transfer is mediated by employing engineered glycoproteins from paramyxoviruses.13 Initially established with measles virus glycoproteins, those of the zoonotic Nipah virus (NiV) are superior with respect to particle yields and absence of immunity in large parts of the population.14 For CAR T-cell generation, RT-LVs recognizing CD4 or CD8 have been described.15 Both were recently shown to mediate the generation of CAR T cells directly in vivo in humanized mouse models.16-19
However, the most obvious cell surface marker for targeting T lymphocytes is CD3. As part of the T-cell receptor (TCR)–CD3 complex, it is exclusively expressed on T lymphocytes. The receptor complex is formed by the TCR, the 2 heterodimers CD3εγ and CD3εδ as well as CD3ζζ homodimer. All CD3 subunits possess activation motifs in their intracellular tails mediating signal transduction following antigen binding. Importantly, cross-linking of the extracellular domains by agonistic CD3-specific antibodies is sufficient to induce major histocompatibility complex-independent T-cell activation.20
Here, we show that T-cell activation and targeted gene delivery can be combined by displaying CD3-specific scFvs on NiV-based RT-LVs. These CD3-LVs are capable of activating T cells during the transduction process, mediating efficient gene delivery into nonactivated T lymphocytes in vitro, even in human whole blood in absence of any additional external stimuli. The most promising CD3-LV candidate generated functional CD19-specific CAR T cells directly in vivo in humanized mice, emphasizing the relevance of these novel LVs for therapeutic applications.
Materials and methods
Primary cells
Human peripheral blood mononuclear cells (PBMCs) were isolated from blood of healthy anonymous donors who had given informed consent, or from buffy coats purchased from the German Red Cross blood donation center (DRK Blutspendedienst Baden-Württemberg-Hessen), as previously described.21 PBMCs were cultured in T-cell medium (TCM; RPMI 1640 [Biowest] containing 10% fetal bovine serum [Biochrom], 2 mM l-glutamine [Thermo Fisher Scientific], 0.5% penicillin/streptomycin and 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [Sigma-Aldrich]) supplemented with 50 IU IL-2, or 25 IU/mL IL-7 and 50 IU/mL IL-15 (all Miltenyi Biotec). Antibody-based T-cell activation was performed in all experiments throughout the manuscript with the clone OKT3 (αCD3) and clone 15E8 (αCD28; both Miltenyi Biotec) as described.21 For assessment of unstimulated and cytokine-stimulated cells, freshly isolated PBMCs were cultured in TCM containing 50 U/mL IL-2, or 25 U/mL IL-7 and 50 U/mL IL-15, or without addition of cytokines, for 16 to 24 hours before incubation with vectors or vehicle. Cell lines are described in the supplemental Methods.
LV production and characterization
LVs were produced in HEK-293T (CRL-11268; ATCC) or Lenti-X-293T (Takara Bio) cells via polyethyleneimine transfection as described.22 Their activity was verified by transduction of Jurkat cells. LV particle yields were determined by nanoparticle tracking analysis or p24-specific enzyme-linked immunosorbent assay (HIV type 1 p24 Antigen ELISA; ZeptoMetrix Corporation) according to the manufacturer’s instructions and calculated as described.14,21
Vector copy number (VCN) was calculated as ratio of woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and human albumin quantified in genomic DNA of human cells carrying the WPRE as described previously.16
Ex vivo gene transfer
Jurkat cells were seeded at 2 to 4 × 104 cells per well and incubated with serial dilutions of vector stocks. Transgene expression was analyzed 72 to 96 hours later by flow cytometry as described in supplemental Methods. αCD3/αCD28-activated, IL-2-, or IL-7/IL-15-stimulated or unstimulated primary human PBMCs were seeded at 4 × 104 or 8 × 104 cells per 96-well, respectively, in medium supplemented with IL-2 or IL-7/IL-15, or in the absence of cytokines before 2 to 5 × 1010 CD3-LV particles were added. Unless otherwise indicated, CD3-LV transduction of PBMCs was carried out in presence of Vectofusin-1 (Miltenyi Biotec) as described previously.21 VSV-LV was used under optimal conditions (ie, the maximal tolerated dose of 1 × 109 particles in absence of Vectofusin-1). Cells were centrifuged at 850g, 32°C for 90 minutes, followed by addition of TCM containing appropriate cytokines. Medium was replenished every 2 to 3 days. Transgene expression was assessed by flow cytometry as described in supplemental Methods 6 and 12 days posttransduction.
Transduction in whole blood
Whole blood from healthy anonymous donors was collected in BD Vacutainer CPT tubes (Becton Dickinson) containing 0.1 M sodium citrate and Ficoll. Whole blood was transferred into 24-wells at 1 mL/well, and subsequently mixed with 4 to 8 × 1010 CD3-LV or 3 × 109 VSV-LV particles without transduction enhancers. In the VSV-LV setting, 1 μg/mL αCD3 and 3 μg/mL αCD28 antibodies were added along with vector particles. The mixture of blood and vector particles was incubated in a cell culture incubator on a tumbling table (120 rpm) for 6 hours before PBMCs were isolated and cultured further in TCM supplemented with IL-7 and IL-15.
Animal experiments
All animal experiments were conducted in accordance with the regulations of the German and French animal protection laws and the respective European guidelines. For in vivo gene transfer, 6-week-old NSG mice (NOD.Cg.PrkdcscidIL2rgtmWjl/Szj; Charles River Deutschland GmbH) were intraperitoneally injected with 5 × 106 αCD3-activated human PBMCs followed by intraperitoneal administration of 1.3 × 1012 TR66-LV or CD8-LV particles, or vehicle control. After 7 days, mice were euthanized, and blood, spleen, and peritoneal cells were collected, and single-cell suspensions were prepared and analyzed by flow cytometry as detailed in supplemental Methods.
For in vivo CAR delivery, NSG mice were humanized as described in supplemental Methods. Mice were randomly distributed into 3 groups 4 days before vector application. Group 1 and group 2 received 200 ng human IL-7 (Miltenyi Biotec) by subcutaneous injection 4 days and 1 day before vector application. Group 3 received phosphate-buffered saline. At day 0, group 2 and group 3 were IV injected with a single dose of 2 × 1011 TR66.opt-LV particles; mice of group 1 received vehicle as control. Mice were monitored regularly for weight and general wellbeing. When humane endpoints were reached, blood, spleen, and long bones were collected, single-cell suspensions were prepared and analyzed by flow cytometry.
Statistical analysis
All data were analyzed with GraphPad Prism 8 (GraphPad Software). Statistical differences in experiments were considered significant at P < .05.
Results
Generation of CD3-LVs
To generate CD3-LVs, we selected the agonistic CD3-specific antibodies TR66 and HuM291.23,24 A TR66 scFv had been previously described as part of a bispecific T-cell engager.25 Seven critical residues in the VL region were converted to the immunoglobulin G1 antibody consensus, yielding TR66.opt (supplemental Figure 1). TR66, TR66.opt, and HuM291 scFvs were C-terminally fused to the engineered NiV glycoprotein Gmut. Incorporation of the modified Gmut proteins into LV particles was confirmed by quantitative immunoblot analysis, with TR66-LV exhibiting the lowest and HuM291-LV the highest glycoprotein incorporation rate (supplemental Figure 2). In mixed cocultures of CD3+ Jurkat and CD3− Molt4.8 cells, CD3-LVs transduced Jurkat, but not Molt4.8 cells in contrast to VSV-LV (supplemental Figure 3A). Selectivity was further confirmed by absence of gene delivery into primary human umbilical vein endothelial cells (HUVECs), Nalm-6 tumor cells, and HepG2 liver cells, which were highly susceptible to VSV-LV gene transfer (Table 1).
Gene transfer activity of CD3-LVs on various cell types
| Vector | % Positive cells, mean ± SD | ||
|---|---|---|---|
| HUVEC* | HepG2* | Nalm-6† | |
| — | ND | ND | 0.02 ± 0.01 |
| TR66-LV | 0.29 ± 0.20 | 0.08 ± 0.11 | 0.05 ± 0.02 |
| TR66.opt-LV | 0.03 ± 0.04 | 0.09 ± 0.06 | 0.05 ± 0.03 |
| HuM291-LV | 0.23 ± 0.08 | 0.09 ± 0.13 | — |
| VSV-LV | 86.74 ± 3.73 | 32.53 ± 4.89 | 97.50 ± 0.37 |
| Vector | % Positive cells, mean ± SD | ||
|---|---|---|---|
| HUVEC* | HepG2* | Nalm-6† | |
| — | ND | ND | 0.02 ± 0.01 |
| TR66-LV | 0.29 ± 0.20 | 0.08 ± 0.11 | 0.05 ± 0.02 |
| TR66.opt-LV | 0.03 ± 0.04 | 0.09 ± 0.06 | 0.05 ± 0.03 |
| HuM291-LV | 0.23 ± 0.08 | 0.09 ± 0.13 | — |
| VSV-LV | 86.74 ± 3.73 | 32.53 ± 4.89 | 97.50 ± 0.37 |
ND, not detectable.
HUVEC and HepG2 cells (n = 2 technical replicates per condition) were transduced with 0.8 to 1 × 1010 CD3-LV particles or 9 × 106 VSV-LV particles and analyzed for GFP expression 4 days after vector incubation.
Nalm-6 cells (n = 3 different vector stocks per condition) were transduced with 5 to 8 × 109 CD3-LV particles or 4 × 108 VSV-LV particles and analyzed for CAR expression 6 days after vector incubation.
Next, gene delivery into αCD3/αCD28-activated primary human T lymphocytes was assessed. GFP expression was detectable for all CD3-LVs with TR66.opt-LV being most efficient (Figure 1A-B). CD3-LV-mediated gene transfer remained stable upon 12 days of cell culture (supplemental Figure 3B). Reproducibly more GFP+ cells were CD4+ than CD8+ with CD3-LVs and VSV-LV. CD4+ T cells contained a higher CD3 receptor density compared with CD8+ T cells under the applied culture conditions (supplemental Figure 4A). This, together with an enhanced proliferation of the CD4+ T cell subset, likely caused the observed skewing.
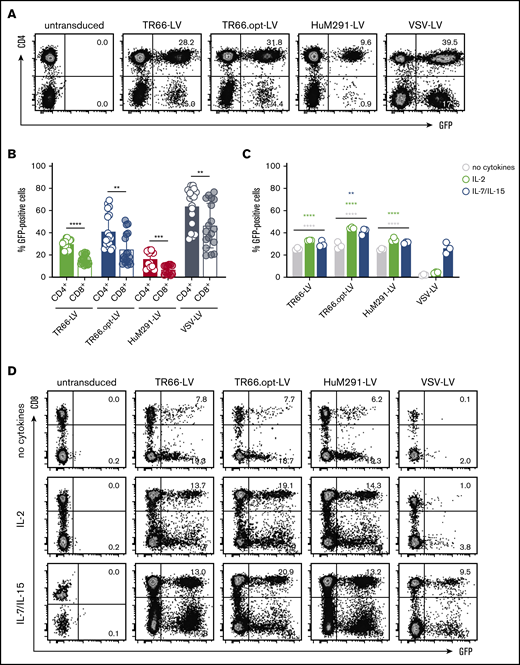
CD3-LVs transduce T cells independently of activation or cytokine treatment. (A-B) Primary human PBMCs isolated from different blood donations were activated with αCD3 and αCD28 antibodies for 3 days in the presence of IL-2 before they were incubated with CD3-LVs or VSV-LV. Green fluorescent protein (GFP) expression was determined 6 days later by flow cytometry. (A) Representative dot plots show GFP expression in CD4+ and CD4− populations gated for viable cells. (B) Scatter bar diagrams summarize the percentages of GFP+ cells as mean ± standard deviation (SD) of 3 independent experiments with 2-3 donors and 2-3 technical replicates. (C-D) Freshly isolated PBMCs were cultured overnight in presence of IL-2 or IL-7/IL-15, or in absence of cytokines, before they were transduced with CD3-LVs or VSV-LV. GFP expression was determined 6 days later by flow cytometry. (C) Scatter bar diagrams show the percentage of GFP+ cells in all viable single cells. (D) Representative dot plots show GFP expression in the CD8+ and CD8− populations gated for viable cells. Data are mean ± SD from 1 experiment with n = 3 technical replicates; biological replicates from additional experiments with IL-2- and IL-7/IL-15-stimulated cells are shown in supplemental Figure 6C-F. **P < .01, ***P < .001, ****P < .0001 by 2-way analysis of variance (ANOVA) with Dunnett's correction (comparison with VSV-LV for each condition), or unpaired Student t test.
CD3-LVs transduce T cells independently of activation or cytokine treatment. (A-B) Primary human PBMCs isolated from different blood donations were activated with αCD3 and αCD28 antibodies for 3 days in the presence of IL-2 before they were incubated with CD3-LVs or VSV-LV. Green fluorescent protein (GFP) expression was determined 6 days later by flow cytometry. (A) Representative dot plots show GFP expression in CD4+ and CD4− populations gated for viable cells. (B) Scatter bar diagrams summarize the percentages of GFP+ cells as mean ± standard deviation (SD) of 3 independent experiments with 2-3 donors and 2-3 technical replicates. (C-D) Freshly isolated PBMCs were cultured overnight in presence of IL-2 or IL-7/IL-15, or in absence of cytokines, before they were transduced with CD3-LVs or VSV-LV. GFP expression was determined 6 days later by flow cytometry. (C) Scatter bar diagrams show the percentage of GFP+ cells in all viable single cells. (D) Representative dot plots show GFP expression in the CD8+ and CD8− populations gated for viable cells. Data are mean ± SD from 1 experiment with n = 3 technical replicates; biological replicates from additional experiments with IL-2- and IL-7/IL-15-stimulated cells are shown in supplemental Figure 6C-F. **P < .01, ***P < .001, ****P < .0001 by 2-way analysis of variance (ANOVA) with Dunnett's correction (comparison with VSV-LV for each condition), or unpaired Student t test.
CD3-LVs outperform VSV-LV in transducing human T cells in absence of antibody activation
Both TR66 and HuM291 induce T-cell activation following CD3 cross-linking.23,24 We therefore hypothesized that the scFvs displayed on CD3-LV particles activate human T cells, allowing transduction without prior activation. For this purpose, gene delivery into freshly isolated PBMCs cultured either in absence of cytokines, or in presence of IL-2 or a combination of IL-7 and IL-15 was assessed. Notably, in contrast to CD3-LVs, VSV-LV exerted substantial cytotoxicity under these conditions (supplemental Figure 5A-E), resulting in a lower applicable particle dose. Strikingly, all CD3-LVs were capable of transducing T cells at similar efficiencies as observed at peak activation in all treatment conditions (Figure 1C-D). In contrast, VSV-LV mediated gene transfer into IL-2-activated and unstimulated T cells was substantially reduced (Figure 1C-D; supplemental Figure 6). Again, more GFP+CD4+ than GFP+CD8+ cells were obtained (Figure 1D; supplemental Figure 6B,D,F).
Binding of CD3-LVs to CD3/TCR activates T cells and induces proliferation
The agonistic function of CD3-specific scFvs displayed on the CD3-LVs may explain their transduction capacity in absence of activating antibodies. Indeed, incubation with CD3-LVs resulted in rapid upregulation of CD69 and CD25, whereas VSV-LV particles did not induce expression of either marker (Figure 2A-B). Expression kinetics of CD69 closely resembled that of αCD3/αCD28 stimulated cells: An initial strong increase was followed by return to baseline levels (Figure 2A). Although the percentage of CD69+ cells was basically identical between vector- and antibody-treated cells, CD69 expression levels were about twofold higher after antibody treatment (supplemental Figure 7A). The overall kinetic of CD25 expression levels were similar for CD3-LV stimulation and antibody treatment; however, upregulation following CD3-LV incubation was less pronounced and delayed (Figure 2B; supplemental Figure 7B).
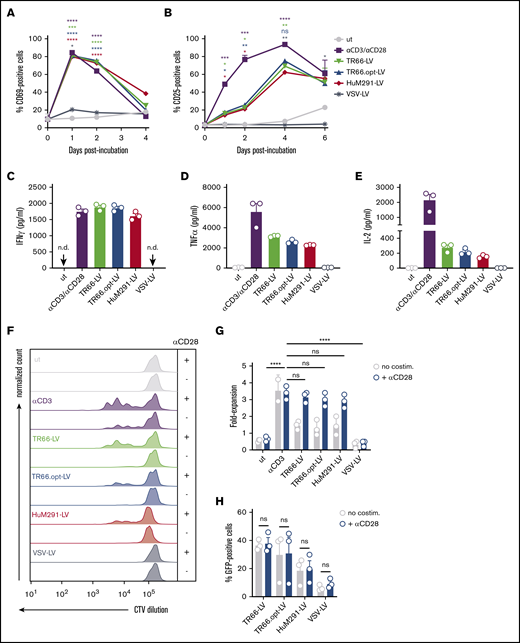
CD3-LVs activate cytokine-cultured T cells and induce cytokine production and proliferation. PBMCs isolated from adult blood were cultured overnight in the presence of IL-2 only and then transduced with the indicated CD3-LVs or with VSV-LV. Controls were left untransduced (ut) or activated with αCD3/αCD28 antibodies (1 µg/mL αCD3, 3 µg/ml αCD28) until first medium exchange. (A-B) Expression of the activation markers CD69 (A) and CD25 (B) on all viable cells was followed for 6 days by flow cytometry. The number of activation marker positive cells are shown. N = 3 donors, mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001 by 2-way ANOVA with Dunnett's correction. (C-E) One day postincubation with vector particles or recombinant antibodies, cytokines secreted into the cell culture supernatant were quantified. Scatter bar diagrams show the concentration of interferon-γ (C), tumor necrosis factor-α (D), and IL-2 (E) for each condition. Mean ± SD from 1 experiment with n = 3 triplicates each are shown. (F) PBMCs were stained with CellTrace Violet (CTV) before transduction ± costimulation to follow cell proliferation over time. Histograms show the fluorescence of CTV at day 5 posttransduction. Data are representative of 3 different donors. (G) Bar diagrams display T-cell expansion after 6 days compared with day 0 ± costimulation. N = 3 donors, mean ± SD. ns, nonsignificant; *P < .05 by 1-way ANOVA with Dunnett's correction. (H) Bar diagrams show percentages of GFP+ cells gated from all viable single cells ± costimulation at day 6 posttransduction. N = 3 donors. Mean ± SD. ns, nonsignificant by 2-way ANOVA with Sidak's correction.
CD3-LVs activate cytokine-cultured T cells and induce cytokine production and proliferation. PBMCs isolated from adult blood were cultured overnight in the presence of IL-2 only and then transduced with the indicated CD3-LVs or with VSV-LV. Controls were left untransduced (ut) or activated with αCD3/αCD28 antibodies (1 µg/mL αCD3, 3 µg/ml αCD28) until first medium exchange. (A-B) Expression of the activation markers CD69 (A) and CD25 (B) on all viable cells was followed for 6 days by flow cytometry. The number of activation marker positive cells are shown. N = 3 donors, mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001 by 2-way ANOVA with Dunnett's correction. (C-E) One day postincubation with vector particles or recombinant antibodies, cytokines secreted into the cell culture supernatant were quantified. Scatter bar diagrams show the concentration of interferon-γ (C), tumor necrosis factor-α (D), and IL-2 (E) for each condition. Mean ± SD from 1 experiment with n = 3 triplicates each are shown. (F) PBMCs were stained with CellTrace Violet (CTV) before transduction ± costimulation to follow cell proliferation over time. Histograms show the fluorescence of CTV at day 5 posttransduction. Data are representative of 3 different donors. (G) Bar diagrams display T-cell expansion after 6 days compared with day 0 ± costimulation. N = 3 donors, mean ± SD. ns, nonsignificant; *P < .05 by 1-way ANOVA with Dunnett's correction. (H) Bar diagrams show percentages of GFP+ cells gated from all viable single cells ± costimulation at day 6 posttransduction. N = 3 donors. Mean ± SD. ns, nonsignificant by 2-way ANOVA with Sidak's correction.
Activation by CD3-LVs resulted in secretion of the pro-inflammatory cytokines interferon-γ, tumor necrosis factor-α, and IL-2, whereas VSV-LV did not have this effect (Figure 2C-E). Furthermore, CD3-LVs induced T-cell proliferation, as evidenced by transition of most cells from the resting status into cell division (Figure 2F). CD3-LV-treated cells proliferated with doubling rates of about 5 days, whereas untreated and VSV-LV-treated T cells did not proliferate. Compared with antibody stimulation, entry into the cell cycle was delayed and significantly lower T-cell numbers were obtained (Figure 2G). However, CD3-LV-exposed T cells costimulated with αCD28 exhibited similar proliferation as αCD3/αCD28-treated cells (Figure 2F-G). Interestingly, costimulation did neither further enhance CD3-LV-mediated gene transfer (Figure 2H) nor improve cellular viability (supplemental Figure 5F). T cells exposed to CD3-LVs contained a higher percentage of TEFF (CD45RA+CD62L−) and TEM (CD45RA−CD62L−) cells compared with αCD3/αCD28-stimulated and CD3-LV/αCD28-treated cells, which somewhat differentiated into TCM (CD45RA−CD62L+) cells (supplemental Figure 7C). Thus, while CD3-LV-induced activation sufficed for efficient gene delivery, the vectors enabled generation of highly proliferative T cells in combination with αCD28 treatment.
CD3-LVs downmodulate the CD3/TCR complex
TCR triggering or CD3 crosslinking induces transient downmodulation of the TCR/CD3 complex from the T-cell surface.20 Accordingly, CD3-LV-treated and αCD3/αCD28 antibody-stimulated T cells showed strongly reduced CD3 and TCR surface expression within the first 2 days (Figure 3A-B). In contrast, VSV-LV treatment did not modulate CD3/TCR surface expression. Downregulation of CD3/TCR in response to antibody treatment was transient, returning to baseline expression after 6 days. Likewise, CD3/TCR expression returned to baseline for HuM291-LV after 6 days, whereas only a 50% recovery had occurred after TR66-LV and TR66.opt-LV treatment even after day 12 posttransduction (Figure 3A-B). Thus, duration of CD3 and TCR surface modulation differed depending on the scFv used.
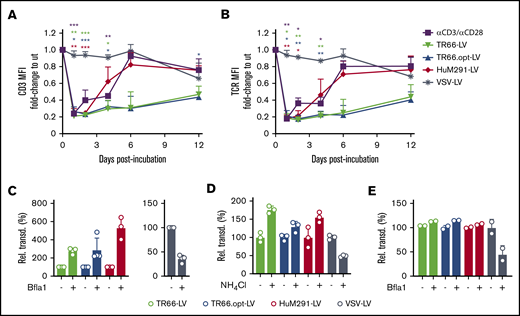
Endocytosis of the TCR/CD3 complex upon transduction. (A-B) Downmodulation of CD3 (A) and TCR (B) on PBMC stimulated only with IL-2 and transduced with CD3-LVs (in absence of Vectofusin-1), VSV-LV, or left untransduced but activated (αCD3/αCD28). The mean fluorescence intensities measured were normalized to those of untreated nonactivated cells. N = 3 donors, mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001 by 2-way ANOVA with Dunnett's correction. (C-D) Inhibition of endocytosis with Bafilomycin A1 (Bfla1) (C) or NH4Cl (D) increases transduction efficiency on Jurkat cells with CD3-LVs. Mean ± SD of 3-4 independent experiments (E), or 1 experiment with 3 technical replicates (D). *P < .05, **P < .01, ****P < .0001 by unpaired Student t test. (E) Transduction of primary PBMC cultured in IL-2 by CD3-LVs upon treatment with Bfla1. N = 2 technical replicates from 1 donor, mean ± SD.
Endocytosis of the TCR/CD3 complex upon transduction. (A-B) Downmodulation of CD3 (A) and TCR (B) on PBMC stimulated only with IL-2 and transduced with CD3-LVs (in absence of Vectofusin-1), VSV-LV, or left untransduced but activated (αCD3/αCD28). The mean fluorescence intensities measured were normalized to those of untreated nonactivated cells. N = 3 donors, mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001 by 2-way ANOVA with Dunnett's correction. (C-D) Inhibition of endocytosis with Bafilomycin A1 (Bfla1) (C) or NH4Cl (D) increases transduction efficiency on Jurkat cells with CD3-LVs. Mean ± SD of 3-4 independent experiments (E), or 1 experiment with 3 technical replicates (D). *P < .05, **P < .01, ****P < .0001 by unpaired Student t test. (E) Transduction of primary PBMC cultured in IL-2 by CD3-LVs upon treatment with Bfla1. N = 2 technical replicates from 1 donor, mean ± SD.
The observed endocytosis of CD3 could be a potential hurdle for CD3-LVs because RT-LVs enter at the cell membrane at neutral pH.14,15 To assess whether inhibition of endocytosis affects gene delivery, we incubated Jurkat cells with NH4Cl or Bafilomycin A1 (Bfla1) before vector particle addition. Transduction increased upon pretreatment with Bfla1, up to sixfold for HuM291-LV and threefold for the TR66-LVs (Figure 3C). Similar results were obtained with NH4Cl (Figure 3D), whereas VSV-LV-mediated gene transfer was substantially decreased by both inhibitors (Figure 3C-D). On primary cells, only a slight increase for CD3-LV-mediated gene delivery after endocytosis inhibition was observed, whereas the already low VSV-LV transduction rate had decreased even further (Figure 3E).
CD3-LVs transduce human T cells in whole blood
Vector particles were mixed into blood without addition of cytokines (Figure 4A). Remarkably, CD3-LVs selectively transduced CD3+ cells at rates ranging between 2% and 5% (Figure 4B-C). In contrast, VSV-LV achieved only low-level transduction and did not discriminate between CD3+ and CD3− cells. Only in presence of activating antibodies, comparable amounts of transduced T cells were generated with VSV-LV (Figure 4B-C). Analysis of vector integration by quantitative polymerase chain reaction confirmed genomic integration for all vectors (Table 2). In accordance with flow cytometry data, VCNs were higher for CD3-LVs. Examination of T-cell subsets showed that, contrary to our observations with isolated PBMCs, CD4+ and CD8+ T cells were equally well transduced by all vectors under these conditions (Figure 4D).

CD3-LVs transduce human T cells in whole blood. CD3-LVs and VSV-LV were tested for their ability to transduce T cells in whole blood of healthy donors in absence of additional stimuli and transduction enhancer. (A) Experimental procedure. (B) Representative dot plots show the percentages of GFP+ cells gated from all viable single cells at day 5 posttransduction in isolated PBMC. (C-D) Bar diagrams show the percentages of GFP+ cells of all viable single cells (C) and in CD4+ and CD8+ cell subsets (D) at day 5 posttranduction. *P < .05, **P < .01 by mixed-effect analysis with Sidak's correction. Mean ± SD of 3 independent experiments with 5 different donors. *P < .05, **P < .01 by mixed-effect analysis with Dunnett's correction.
CD3-LVs transduce human T cells in whole blood. CD3-LVs and VSV-LV were tested for their ability to transduce T cells in whole blood of healthy donors in absence of additional stimuli and transduction enhancer. (A) Experimental procedure. (B) Representative dot plots show the percentages of GFP+ cells gated from all viable single cells at day 5 posttransduction in isolated PBMC. (C-D) Bar diagrams show the percentages of GFP+ cells of all viable single cells (C) and in CD4+ and CD8+ cell subsets (D) at day 5 posttranduction. *P < .05, **P < .01 by mixed-effect analysis with Sidak's correction. Mean ± SD of 3 independent experiments with 5 different donors. *P < .05, **P < .01 by mixed-effect analysis with Dunnett's correction.
VCNs after transduction in whole blood
| Vector | VCNs, mean ± SD |
|---|---|
| TR66-LV | 1.17 ± 0.57 |
| TR66.opt-LV | 1.23 ± 0.05 |
| HuM291-LV | 1.03 ± 0.87 |
| VSV-LV | 0.16 ± 0.28 |
| VSV-LV + αCD3/αCD28* | 0.47 ± 0.59 |
| Vector | VCNs, mean ± SD |
|---|---|
| TR66-LV | 1.17 ± 0.57 |
| TR66.opt-LV | 1.23 ± 0.05 |
| HuM291-LV | 1.03 ± 0.87 |
| VSV-LV | 0.16 ± 0.28 |
| VSV-LV + αCD3/αCD28* | 0.47 ± 0.59 |
αCD3/αCD28 antibodies were added to the blood along with VSV-LV particles.
TR66-LV mediates efficient in vivo gene delivery into all T-cell subpopulations
To assess gene delivery directly in vivo, we intraperitoneally administered TR66-LV or CD8-LV encoding GFP into NSG mice transplanted with αCD3/αCD28-activated human PBMCs 1 day earlier (Figure 5A). Seven days after vector injection, GFP+ cells were detected in the peritoneal cavity of all TR66-LV-treated animals by flow cytometry (Figure 5B-C). Approximately 14% of all human CD3+ peritoneal cells were GFP+ in both vector-injected groups; similar amounts of transduced T cells were found in blood and spleen (Figure 5D-E). Gene transfer efficiency was comparable between TR66-LV and the established CD8-LV, as there was no significant difference in CD3+ GFP+ cells in all analyzed organs. However, TR66-LV transduced both CD4+ and CD8+ T cells at equal efficiency, whereas CD8-LV only modified the CD8 T-cell subset (Figure 5F-H). Closer assessment revealed an almost absolute selectivity of TR66-LV for its target cell population: <0.5% CD45− mouse cells and 0.1% CD45+ CD19+ cells were found within the GFP+ compartment (Figure 6A-D). This corresponded to less than 15 off-target cells within several thousand transduced cells.

TR66-LV mediates efficient in vivo gene delivery into all T-cell subpopulations. (A) Experimental outline: NSG mice were transplanted with αCD3/αCD28-activated human PBMC by intraperitoneal (IP) injection followed by vector administration 1 day later and analyzed for transgene expression by flow cytometry 7 days after vector application. (B) Representative dot plots show transduced T cells gated from all viable single CD3+ cells harvested from the peritoneal cavity at 7 days after vector injection. (C-E) Scatter plots summarize the percentages of GFP+ cells gated from all viable single CD3+ cells for peritoneum (C), blood (D), and spleen (E). (F-H) Respective scatter plots of GFP+ cells gated from CD4+ and CD8+ T cells isolated from peritoneum (F), blood (G), and spleen (H) at the day of analysis. N = 3 mice per group, mean ± SD. **P < .01, ***P < .001, ****P < .0001 by 1-way analysis of variance with Sidak's or Dunnett's correction.
TR66-LV mediates efficient in vivo gene delivery into all T-cell subpopulations. (A) Experimental outline: NSG mice were transplanted with αCD3/αCD28-activated human PBMC by intraperitoneal (IP) injection followed by vector administration 1 day later and analyzed for transgene expression by flow cytometry 7 days after vector application. (B) Representative dot plots show transduced T cells gated from all viable single CD3+ cells harvested from the peritoneal cavity at 7 days after vector injection. (C-E) Scatter plots summarize the percentages of GFP+ cells gated from all viable single CD3+ cells for peritoneum (C), blood (D), and spleen (E). (F-H) Respective scatter plots of GFP+ cells gated from CD4+ and CD8+ T cells isolated from peritoneum (F), blood (G), and spleen (H) at the day of analysis. N = 3 mice per group, mean ± SD. **P < .01, ***P < .001, ****P < .0001 by 1-way analysis of variance with Sidak's or Dunnett's correction.
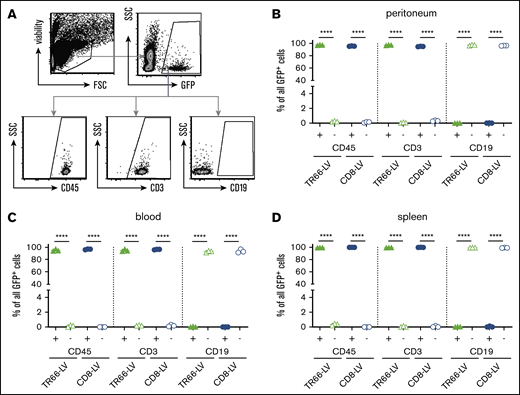
Selectivity of TR66-LV in vivo. (A) Strategy for backgating of GFP+ cells generated in PBMC-NSG mice of Figure 5. All viable single GFP+ cells were assessed for expression of human CD45, CD3, and CD19. (B-D) Scatter plots for the percentages of GFP+ cells in the indicated fractions of peritoneum (B), blood (C), and spleen (D). Data are mean ± SD from n = 3 animals per group. ****P < .0001 by 1-way ANOVA with Sidak's correction.
Selectivity of TR66-LV in vivo. (A) Strategy for backgating of GFP+ cells generated in PBMC-NSG mice of Figure 5. All viable single GFP+ cells were assessed for expression of human CD45, CD3, and CD19. (B-D) Scatter plots for the percentages of GFP+ cells in the indicated fractions of peritoneum (B), blood (C), and spleen (D). Data are mean ± SD from n = 3 animals per group. ****P < .0001 by 1-way ANOVA with Sidak's correction.
CAR delivery by CD3-LVs
Finally, we investigated CAR gene delivery into human T cells as a potential application of CD3-LVs. We packaged a second-generation CD19-CAR with a CD28 costimulatory domain and an extracellular myc tag for detection17 into TR66-LV and TR66.opt-LV particles (supplemental Figure 8A). Upon incubation with αCD3/αCD28-activated PBMCs, both CD3-LVs delivered the CAR into all T-cell subsets (supplemental Figure 8B). VCNs obtained with TR66-LV (0.8) and for TR66.opt-LV (2.4) were comparable to those obtained with VSV-LV (2.9). The generated CAR T cells were functionally active, eliminating the CD19+ B cells in the PBMC culture (supplemental Figure 8C). The majority of CAR+ and CAR− T cells exhibited a TCM phenotype, whereas untransduced cells constituted a mix of TEM and TCM cells (supplemental Figure 8D). In comparison with VSV-LV, fewer CAR T cells were generated, but these were enriched by more than 40-fold upon repeated addition of CD19+ Nalm-6 target cells (supplemental Figure 9A). CD4+ CAR T cells contributed more to this expansion than their CD8+ counterparts (supplemental Figure 9B). Both vectors also allowed generation of CAR T cells with IL-2-activated cells, but expansion in response to CD19+ target cells was only observed with TR66.opt-LV (supplemental Figure 9D-E). Interestingly, CD3-LV-generated CAR T cells appeared to be less exhausted than their VSV-LV-generated counterparts under both culture conditions (supplemental Figure 9C,F).
For assessment of in vivo CAR gene delivery, TR66.opt-LV was selected. To mimic in vivo preclinical settings, we used humanized NSG (huNSG) mice transplanted with human CD34+ human stem and progenitor cells. To assess if prestimulation by IL-7 is required, one group of animals received human IL-7 before vector administration, whereas the other received only TR66.opt-LV (Figure 7A). Random distribution of human lymphocyte levels between groups was verified before vector injection (supplemental Figure 10A-C). Three weeks after vector administration, CAR+ T cells were detected in the blood of most vector-treated animals (Figure 7B-D). Five of 6 mice receiving cytokine and vector had detectable CAR T cells in the blood, whereas in absence of IL-7, 3 of 5 animals clearly showed CAR signals in CD3+ cells (supplemental Figure 11). Importantly, CAR+ cells were only detected in the CD3+/CD45+ T-cell compartment (Figure 7D). CAR expression peaked between week 3 and 4 after injection in both groups and declined back to baseline after 5 weeks. Interestingly, CAR T-cell levels were significantly higher in mice having received IL-7 pretreatment (Figure 7C-D; supplemental Figure 11). Both the CD4+ and CD8+ subset expressed the CAR, with significantly more CAR+ cells in the CD8+ T-cell compartment (supplemental Figure 10D-E), in spite of no difference in CD3 density between the subsets at the day of injection (supplemental Figure 4B).
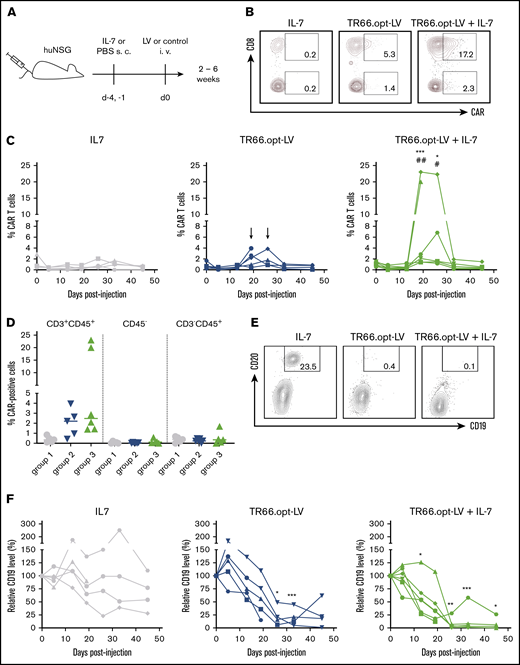
CD3-targeted LVs mediate functional CAR T-cell generation in vivo. For in vivo CAR delivery, huNSG mice were subcutaneously (SC) injected with human IL-7 or phosphate-buffered saline 4 days and 1 day before IV injection of TR66.opt-LV harboring the CD19-CAR or vehicle as control. (A) Experimental outline. (B) Representative plots show CAR+ cells in CD8+ and CD8− T cells at day 26 postinjection of vector. (C) Line diagrams show CAR+ cells in CD3+ cells in the peripheral blood of individual mice in group 1 (IL-7), group 2 (TR66.opt-LV), and group 3 (TR66.opt-LV + IL-7). *,#P < .05, **,##P < .01, ***,###P < .001 comparing group 3 to group 1 (*) and group 2 (#) by 2-way ANOVA with Tukey's correction. Arrows point to tendencies for CAR T-cell detection in group 2, which were not statistically significant. (D) Scatter dot plots compare the percentages of CAR+ cells within the CD3+CD45+ T-cell compartment, CD45− mouse cells and CD3−CD45+ non-T cells. (E-F) B-cell levels were determined in peripheral blood of the animals to assess CD19-CAR T-cell functionality. (E) Representative dot plots show CD19+CD20+ B cells at day 26 postinjection. (F) Line diagrams summarize relative levels of CD19+ cells compared with day 0 in the peripheral blood of individual mice in group 1 (IL-7), group 2 (TR66.opt-LV), and group 3 (TR66.opt-LV + IL-7). All data from n = 5-6 mice per group. *P < .05, **P < .01, ***P < .001 comparing groups 2 and 3 to group 1 by 2-way ANOVA with Turkey's correction.
CD3-targeted LVs mediate functional CAR T-cell generation in vivo. For in vivo CAR delivery, huNSG mice were subcutaneously (SC) injected with human IL-7 or phosphate-buffered saline 4 days and 1 day before IV injection of TR66.opt-LV harboring the CD19-CAR or vehicle as control. (A) Experimental outline. (B) Representative plots show CAR+ cells in CD8+ and CD8− T cells at day 26 postinjection of vector. (C) Line diagrams show CAR+ cells in CD3+ cells in the peripheral blood of individual mice in group 1 (IL-7), group 2 (TR66.opt-LV), and group 3 (TR66.opt-LV + IL-7). *,#P < .05, **,##P < .01, ***,###P < .001 comparing group 3 to group 1 (*) and group 2 (#) by 2-way ANOVA with Tukey's correction. Arrows point to tendencies for CAR T-cell detection in group 2, which were not statistically significant. (D) Scatter dot plots compare the percentages of CAR+ cells within the CD3+CD45+ T-cell compartment, CD45− mouse cells and CD3−CD45+ non-T cells. (E-F) B-cell levels were determined in peripheral blood of the animals to assess CD19-CAR T-cell functionality. (E) Representative dot plots show CD19+CD20+ B cells at day 26 postinjection. (F) Line diagrams summarize relative levels of CD19+ cells compared with day 0 in the peripheral blood of individual mice in group 1 (IL-7), group 2 (TR66.opt-LV), and group 3 (TR66.opt-LV + IL-7). All data from n = 5-6 mice per group. *P < .05, **P < .01, ***P < .001 comparing groups 2 and 3 to group 1 by 2-way ANOVA with Turkey's correction.
Examination of CD19+ B cells in the blood of the animals confirmed CAR T-cell generation by TR66.opt-LV because CD19+ cell levels dropped significantly concomitant to the emergence of CAR T cells (Figure 7E-F). By day 26, B cells were almost completely eliminated in 5 of 6 mice injected with IL-7 and TR66.opt-LV and 4 of the TR66.opt-LV-treated animals. Curiously, CD19+ cell depletion was durable only in IL-7-pretreated mice. In animals injected with vector only, CD19+ cells reappeared 6 weeks postinjection.
Discussion
We describe here a new class of LVs combining cell type–specific gene delivery with targeted activation of specific cells. Key for this achievement was the display of agonistic CD3-specific scFvs, which activate human T lymphocytes upon binding, inducing cytokine release and proliferation. Notably, this activation occurs in absence of a costimulatory signal. although this can to lead to activation-induced cell death, especially after long-term cultivation,26 the viability of CD3-LV-treated T cells was equivalent if not better than that of cells treated with VSV-LV. In vivo, the activity of CAR T cells generated with CD3-LV was detectable over the whole observation period of up to 6 weeks suggesting that AICD may not be an issue in this particular experimental setting.
The agonistic nature of CD3-LVs allows T-cell transduction in unstimulated, cytokine-only-stimulated PBMCs or even untouched human blood. Under these conditions (ie, in absence of activating antibodies), only about 1% of T cells reside in the G2/M phase of the cell cycle and are thus amenable to gene delivery with conventional LVs.27 This value is well in agreement with the transduction rates we observed with VSV-LV. Transduction rates mediated by CD3-LVs were substantially higher, thus opening up new options for genetic engineering of human T lymphocytes, both ex vivo and in vivo. TR66-derived LVs performed slightly better than HuM291-LVas despite lower glycoprotein incorporation rates. Differences in scFv affinity may play a role here, although parental antibody affinities are similar.23,28
Because of its T-cell–restricted expression, attempts have been undertaken before to target vector particles to CD3. These included LVs codisplaying engineered Sindbis virus glycoproteins and a membrane-anchored version of the CD3-specific antibody OKT3,29 as well as pseudotyping of LV particles with VSV G and a retroviral glycoprotein fused to an OKT3-derived antibody fragment.30 Although the latter study showed preferential transduction of human T cells, both systems were limited in gene delivery efficiency and selectivity. They have therefore never been further evaluated for their in vivo performance. TR66-LVs in contrast did not show any significant off-target activity, neither ex vivo with cell lines including tumor cells (Table 1), nor after injection into humanized mice, where mouse cells and CD3− human cells were unaffected by the applied CD3-LV particles. This precise delivery to T cells may enable in vivo transfer of genes inducing pathogenicity when expressed in nontarget cells. The recently described fatal consequence of CAR gene delivery into tumor cells may then be avoidable.9
More recently, synthetic nanoparticles displaying a fragment of the mouse CD3-specific antibody 145-2C11 have been described.31 Upon infusion into leukemia-bearing mice, the nanoparticles mediated in vivo CAR T-cell generation and subsequent tumor elimination after 5 daily administrations of 3 × 1011 particles. This dose is more than 7 times higher than the single injection required with TR66.opt-LV particles to generate CAR T cells in vivo, indicating superior gene delivery with CD3-LVs. This is remarkable because CD3 is rapidly internalized following crosslinking and therefore an ideal target receptor for nanoparticles, whose cell entry depends completely on endocytosis. In contrast, CD3-LVs rely on fusion with the cell membrane at neutral pH for cell entry, which we confirmed by the increased gene transfer after blocking endocytosis. Although T-cell activation in response to CD3-targeted nanoparticles was not assessed, it is likely that they induce similar activation of murine as CD3-LVs of human T cells because 145-2C11 is known to strongly activate T cells and is frequently employed for murine T-cell activation and expansion.32
Most circulating T cells are in a quiescent, noncycling state in which pre- and postentry blocks restrict transduction by HIV-1–derived LVs.10,33,34 Prolonged IL-7 exposure results in transition into the G1b phase of the cell cycle,27 subsequent inactivation of host restriction factors such as SAMHD135 and, ultimately, permissiveness toward HIV-1 infection in absence of TCR stimulation.36 We have therefore previously used IL-7 to minimally stimulate circulating T cells to facilitate gene delivery before injection of CD8-LV into humanized mice.16,19 Here, we show that in vivo CAR gene delivery with CD3-LV is possible in absence of IL-7 pretreatment. However, IL-7–stimulated animals contained higher numbers of CAR T cells. Moreover, B-cell depletion as consequence of the in vivo delivered CD19-CAR was more sustained. IL-7 particularly stimulates naïve and memory T cells, inducing homeostatic proliferation, while preserving their less differentiated phenotypes.37 Less differentiated CAR T cells, because of their greater proliferative potential, lower degree of exhaustion, and prolonged in vivo persistence, have been associated with durable clinical responses.38-40 It is likely that in presence of IL-7, CD3-LV particles hit more naïve and memory T cells, which then mediated permanent CAR T-cell activity resulting in durable B-cell depletion. Furthermore, expression of the late activation marker CD71 was upregulated in IL-7-treated huNSG mice just before vector administration (supplemental Figure 12). This preactivation may have resulted in more robust and homogenous transduction as previously observed ex vivo,41 leading to CAR T-cell persistence after elimination of target cells.
Among many potential fields of application, adoptive T-cell therapies, in particular genetic engineering of T cells with TCRs or CARs, appear especially relevant for CD3-LVs. Current manufacturing procedures are not only very complex, time-consuming, and costly, but the extensive manipulations before infusion may also dampen efficacy.42 CD3-LVs could contribute to overcome these limitations. Strategies may include direct injection in vivo as well as ex vivo CAR T-cell generation with abbreviated cultivation procedures.43 In each case, this will not only require safety, biodistribution, and efficacy studies in large immunocompetent animal models, but also establishment of good manufacturing practice production for this novel vector type.
Send data sharing requests via e-mail to the corresponding author, C.J.B., at christian.buchholz@pei.de.
Acknowledgments
The authors thank Gundula Braun, Julia Brynza, Manuela Gallet, and Tatjana Weidner for excellent technical assistance in production of vector particles and quantitative polymerase chain reaction analyses; Julia D. S. Hanauer for support during the NSG mouse experiment; Jessica Hartmann (all Paul-Ehrlich-Institut, Langen, Germany) for helpful discussions and support in data analysis; Jean-François Henry, Nadine Aguilera, and Tiphaine Dorel (Plateau de Biologie Experimental de la Souris, ENS de Lyon, Lyon, France) for taking care of the animals; and Véronique Pierre and Emilie Laurent (Center for Research in Infectious Diseases [CIRI], ENS de Lyon) for technical assistance during the huNSG mouse experiment and flow cytometry analysis.
This work was supported by grants from the LOEWE Center Frankfurt Cancer Institute funded by the Hessen State Ministry for Higher Education, Research and the Arts (III L 5 - 519/03/03.001 - (0015)) and the European Union (Horizon 2020 Framework Programme, CARAT [667980]) (C.J.B.).
Authorship
Contribution: A.M.F. planned and performed the experiments, analyzed data, supervised work and drafted the manuscript; A.H.B., L.S., and S.A. planned and performed in vitro experiments and analyzed data; I.C.S. designed the TR66.opt scFv; U.S. initiated TR66 and HuM291 antibody selection and provided the sequences; F.B.T. contributed to planning and performance of experiments and supported data analysis and interpretation; E.V. provided the huNSG mice and planned the corresponding experiment together with A.M.F., S.A., and F.B.T.; E.V., F.F., and S.P. performed and analyzed the huNSG mouse experiment and discussed the data together with A.M.F., F.B.T., and C.J.B.; and C.J.B. initiated and supervised the study, acquired grants, and revised the manuscript.
Conflict-of-interest disclosure: E.V., I.C.S., and C.J.B are listed as inventors on patents on receptor-targeted LVs that have been licensed to the patent commercializing agencies ipal GmbH and Pulsalys. The remaining authors declare no competing financial interests.
Correspondence: Christian J. Buchholz, Molecular Biotechnology and Gene Therapy, Paul-Ehrlich-Institut, Paul-Ehrlich-Straße 51-59, 60528 Langen, Germany; christian.buchholz@pei.de.

