

Key Points
CRISPR/Cas9 screen identified JMJD1C Jumonji domain as critical for MLLr leukemogenesis. H3K36me is a marker for JMJD1C activity.
JMJD1C loss leads to increased IL-3 signaling. This effect is blunted in leukemia cells with activating RAS mutations.

Abstract
JMJD1C, a member of the lysine demethylase 3 family, is aberrantly expressed in mixed lineage leukemia (MLL) gene-rearranged (MLLr) leukemias. We have shown previously that JMJD1C is required for self-renewal of acute myeloid leukemia (AML) leukemia stem cells (LSCs) but not normal hematopoietic stem cells. However, the domains within JMJD1C that promote LSC self-renewal are unknown. Here, we used clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease (Cas9) negative-selection screening and identified a requirement for the catalytic Jumonji (JmjC) domain and zinc finger domain for leukemia cell survival in vitro and in vivo. In addition, we found that histone H3 lysine 36 methylation (H3K36me) is a marker for JMJD1C activity at gene loci. Moreover, we performed single cell transcriptome analysis of mouse leukemia cells harboring a single guide RNA (sgRNA) against the JmjC domain and identified increased activation of RAS/MAPK and the JAK-STAT pathway in cells harboring the JmjC sgRNA. We discovered that upregulation of interleukin 3 (IL-3) receptor genes mediates increased activation of IL-3 signaling upon JMJD1C loss or mutation. Along these lines, we observed resistance to JMJD1C loss in MLLr AML bearing activating RAS mutations, suggesting that RAS pathway activation confers resistance to JMJD1C loss. Overall, we discovered the functional importance of the JMJD1C JmjC domain in AML leukemogenesis and a novel interplay between JMJD1C and the IL-3 signaling pathway as a potential resistance mechanism to targeting JMJD1C catalytic activity.
Introduction
Acute myeloid leukemia (AML) cells have been shown to follow a leukemia stem cell (LSC) model. Similar to hematopoietic stem cells (HSCs), AML LSCs are rare cells at the apex of AML hierarchy and have the ability to self-renew and partially differentiate into blasts, which represent the bulk of cells.1-3 The LSC model implies that long-term remission for patients with AML depends on the eradication of LSCs.4 Identifying the factors that are required for LSCs, but not HSCs, and understanding the molecular mechanism of their function may lead to novel targeted therapies in AML. One of the most common translocations found in AML involves the mixed lineage leukemia (MLL) gene. In MLL-rearranged (MLLr) leukemias, the N terminus of MLL1 is fused to 1 of >50 partners. MLLr leukemia accounts for 5% to 10% of adult leukemia and >70% of infant leukemia and carries an intermediate to poor prognosis. The most common MLL fusion in AML is MLL-AF9.5,6
We have recently shown that JMJD1C, a Jumonji domain–containing protein of the lysine demethylase 3 (KDM3) family, is aberrantly expressed in mouse MLL-AF9 LSCs and in human MLLr leukemias. JMJD1C is required for AML LSC self-renewal in MLL-AF9 and Hoxa9/Meis1 murine leukemia models, but it is dispensable for normal HSC function. JMJD1C is a member of the KDM3 family that includes KDM3A, KDM3B, and JMJD1C (official nomenclature). KDM3A and KDM3B have been shown to be histone H3 lysine 9 mono- and dimethylation (H3K9me1/2) demethylases.7-9 JMJD1C was first characterized in a yeast 2-hybrid assay as thyroid receptor-interacting protein 8.10 JMJD1C protein contains a catalytic Jumonji (JmjC) domain, the catalytic domain found in the Jumonji family of demethylase,11 and a zinc finger domain (ZFD). The ZFD in other members of the KDM3A family has been implicated in determining substrate specificity8,9 ; however, the precise mechanism is unknown. The enzymatic activity of JMJD1C is still under debate. JMJD1C was initially shown to be an H3K9me1/2 demethylase, and it acts as a coactivator for the androgen receptor through demethylating the repressive H3K9-methyl mark.12,13 However, subsequent studies using similar approaches to assess the enzymatic activity of JMJD1C drew conflicting conclusions on its H3K9me1/2 demethylase activity,9,14,15 with the latest study showing weak activity toward H3K9me1 but not H3K9me2.16 Collectively, this demonstrates that the substrate for JMJD1C is not definitively established. Functionally, Jmjd1c constitutive knockout mice exhibit preweaning lethality with incomplete penetrance, defects in male gametogenesis,14 mydriasis and homeotic transformation of the vertebrae.17 In humans, germline variants of JMJD1C are associated with an increased risk of developing intracranial germ cell tumors.18 Using a short hairpin RNA approach, JMJD1C has also been shown to repress neural differentiation of human embryonic stem cells by maintaining miR-302 expression,19 maintaining mouse embryonic stem cell self-renewal,20 and regulating MyoD expression in myogenesis.21 Consistent with our previous finding, a requirement for JMJD1C in MLL-AF9 and AML1-ETO leukemias has also been demonstrated by hairpin knockdown15,16 ; however, the molecular mechanism through which JMJD1C promotes LSC self-renewal is still unknown.
In this study, we used a clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease (Cas9) domain screening approach to identify functionally important domains within JMJD1C and examined their role and the underlying mechanism for their requirement in AML leukemogenesis.
Methods
A detailed description of mouse experiments, cell culture, viral production and transduction of cells, cell assay (including colony forming cell [CFC] assay, apoptosis, cell proliferation, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), inhibitor treatment) and cell sorting, selective enrichment and identification of adapter-tagged DNA ends by sequencing (SITE-seq), RNA sequencing (RNA-seq), chromatin immunoprecipitation sequencing (ChIP-seq), single-cell RNA-seq, and data analysis is included in supplemental Methods.
Results
CRISPR/Cas9 screen identifies JmjC domain and ZFD as important for JMJD1C function in AML
We have shown previously that JMJD1C is required for AML LSC function. However, the large size of the JMJD1C protein (∼280 kDa) and the resulting extreme inefficiency in gene delivery prevented us from performing rescue experiments in JMJD1C-null cells to determine whether the enzymatic activity of JMJD1C is required for its function. This is important because a number of the demethylases have been shown to exert crucial enzyme-independent functions.22-25 Alternatively, a recent study by Shi et al26 showed that the CRISPR/Cas9 system can be used in negative-selection screens to identify domains within a given protein that are functionally important for cancer cell survival. CRISPR/Cas9 typically generates one third of in-frame mutations and two thirds of out-of-frame mutations in a target cell population. Functionally important domains are much less tolerant of in-frame mutation in comparison with nonessential domains, resulting in the depletion of cells bearing the single guide RNAs (sgRNAs) targeting critical domains. To perform an unbiased screen against the entire JMJD1C protein, we tiled 234 sgRNAs to cover the coding region of the Jmjd1c gene at ∼30-bp intervals, cloned the sgRNAs into a lentiviral vector with a TdTomato marker gene, and transduced them into clonal Cas9-expressing mouse MLL-AF9 leukemia cells (MLL-AF9-Cas9) (Figure 1A; supplemental Figure 1A). The percentages of sgRNA+ cells were analyzed by flow cytometry at 2 and 14 days following transduction (Figure 1B). As a positive control, an sgRNA against Dot1l, a methyltransferase known to be essential for MLL-AF9 leukemia,27 was included. sgRNAs against the ZFD (26-aa) and JmjC domain (225-aa) and their immediate surrounding regions showed the most significant depletion, up to 17-fold (Figure 1B). To validate this result, we used 5 sgRNAs each against the ZFD and JmjC domain, the negative-control sgRNAs against Renilla and firefly luciferase, and positive controls against Rpa3 and Brd4.26 The sgRNA against both domains of JMJD1C showed significant negative selection, consistent with our screening result (Figure 1C-F). We corroborated on-target mutations generated by ZFD and JmjC sgRNAs by next-generation sequencing (supplemental Figure 1E). Lastly, we obtained similar results in an independent clone of MLL-AF9-Cas9 leukemia cells (supplemental Figure 1B-D), validating that the effect we observed is not clone specific.
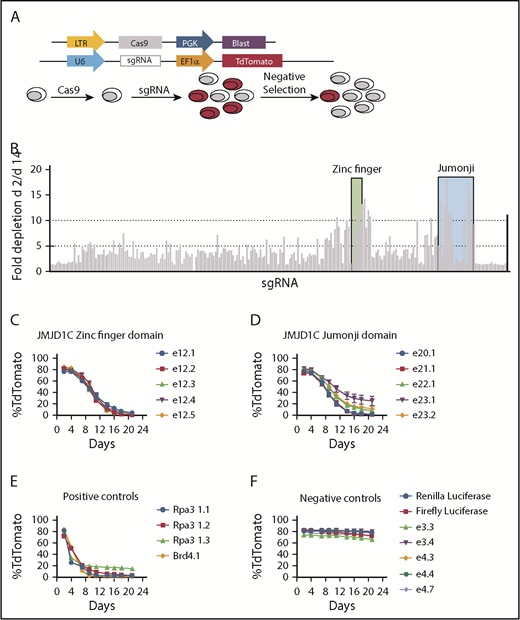
Negative-selection screen using CRISPR/Cas9. (A) Schematics of the screen. Vector used to establish a clonal Cas9-expressing MLL-AF9 leukemia cell line and vector used for sgRNA transduction (upper). Experimental scheme (lower). (B) Fold changes in TdTomato positivity (day 2/day 14) during 14 days in culture. Each bar represents an sgRNA targeting Jmjd1c. The black bar represents sgRNA against Dot1l. Shaded areas are sgRNAs targeting ZFD (green) and Jumonji domain (blue). (C-F) Time course of flow cytometry analysis of TdTomato level after lentiviral transduction of sgRNAs. (B-F) Data are mean ± standard deviation of independently transduced triplicate samples.
Negative-selection screen using CRISPR/Cas9. (A) Schematics of the screen. Vector used to establish a clonal Cas9-expressing MLL-AF9 leukemia cell line and vector used for sgRNA transduction (upper). Experimental scheme (lower). (B) Fold changes in TdTomato positivity (day 2/day 14) during 14 days in culture. Each bar represents an sgRNA targeting Jmjd1c. The black bar represents sgRNA against Dot1l. Shaded areas are sgRNAs targeting ZFD (green) and Jumonji domain (blue). (C-F) Time course of flow cytometry analysis of TdTomato level after lentiviral transduction of sgRNAs. (B-F) Data are mean ± standard deviation of independently transduced triplicate samples.
JMJD1C JmjC domain and ZFD are required for MLL-AF9–mediated leukemogenesis
Our sgRNA screening data suggest that the JmjC domain and ZFD are important for MLL-AF9 leukemia cell survival and proliferation. To investigate the role of these domains in MLL-AF9 leukemogenesis, we transduced MLL-AF9-Cas9 cells with sgRNAs against these domains, as well as the Renilla luciferase as control. We then sorted the transduced cells by the TdTomato marker gene and subjected the cells to a CFC assay. Leukemic cells harboring JmjC or ZFD sgRNAs showed impaired clonogenic potential, and the colonies appeared smaller and diffused, indicative of cell differentiation (Figure 2A-B). Consistently, we observed impaired proliferation (Figure 2C) and increased expression of the myeloid differentiation marker Mac1 upon mutating these domains (Figure 2E). Moreover, we observed a significant increase in apoptosis of leukemia cells harboring JmjC and ZFD sgRNAs (Figure 2D). As an additional control, we compared a mock-transduction control with that of Renilla sgRNA; we found no differences between the 2 in similar assays as above. The only exception was apoptosis, for which the Renilla control showed a small, but significant, increase (supplemental Figure 2A-F). Furthermore, we mapped genome-wide off-target sites of all the sgRNAs used by SITE-seq.28 Our results showed that all the identified off-target sites (≥20 cliff reads) are within intergenic region or introns (supplemental Table 8 presents all sites unfiltered). One exception is e.20.1 sgRNA in the St6galnac1 gene, where an off-target site was identified in the 3′ untranslated region close to the 3′ end of the last exon. No prominent role has been described for this gene in hematopoiesis or leukemia. The consistency between the 2 sgRNAs used for each domain, the result of the off-target assay, and the consistency of phenotypes observed between the CRISPR/Cas9 approach and the knockout mouse model29 demonstrate that the phenotype of cells harboring JMJD1C sgRNAs is due to on-target effects of the guide RNAs. Overall, these data suggest that the JmjC domain and ZFD of JMJD1C are required for cell proliferation, cell survival, and clonogenic activity in MLL-AF9 leukemia.
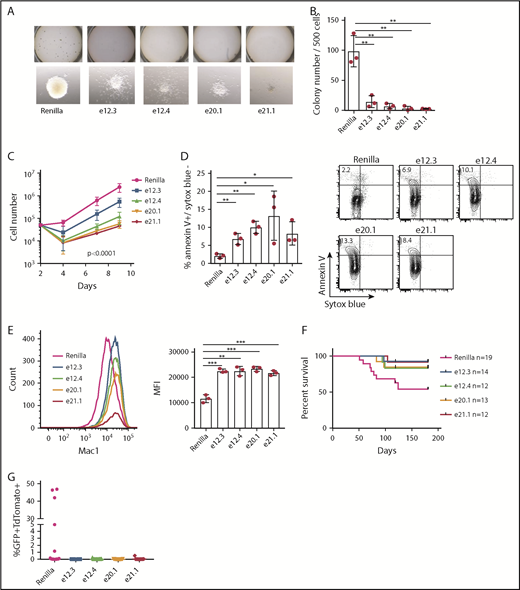
Effect of mutating JMJD1C ZFD and Jumonji domain on MLL-AF9 leukemogenesis. (A-B) CFC assay after transduction of MLL-AF9-Cas9 cells with sgRNAs against ZFD (e12.3, e12.4) and Jumonji domain (e20.1, e21.1). (A) Colonies in methylcellulose culture (upper panels). Individual colony morphology (original magnification ×80; lower panels). (B) Colony counts 1 week after plating. (C) Proliferation assay of sorted GFP+ (marks MLL-AF9) TdTomato+ (marks sgRNA) MLL-AF9-Cas9 leukemia cells after sgRNA transduction. Data are mean ± standard deviation (SD) of 3 independent experiments. The P value was determined by 2-way analysis of variance (ANOVA). For multiple comparisons, P < .05 for day 7 and P < .0001 for day 9 between Renilla and all other sgRNAs, with the exception of Renilla vs e12.3 on day 7 (not statistically significant). (D) Apoptosis assay (left panel). Data are mean ± SD from 3 independent experiments. Representative flow plot; numbers within the upper left quadrant indicate average percentage (right panel). (E) Representative flow cytometry analysis of Mac1 of MLL-AF9-Cas9 leukemia cells 7 days after transduction (left panel). Mean fluorescence intensity (MFI; geometric mean) of Mac1 expression (right panel). Data are mean ± SD from 3 independent experiments. (F) Survival curves of secondary recipient mice that received MLL-AF9-Cas9 cells after transduction with respective sgRNAs. P = .040 overall, log-rank test. Pairwise log-rank test P values between Renilla and JMJD1C sgRNAs: P = .025 for e12.3; P = .137 for e12.4; P = .096 for e20.1, and P = .048 for e21.1. (G) Peripheral blood GFP+TdTomato+ cell percentage 8 weeks after transplantation. P = .05, multiple comparison, 1-way ANOVA. Renilla vs e12.3, P = .06; Renilla vs e12.4, e20.1, and e21.1, P = .08 . *P < .05, **P < .01, ***P < .001, unpaired 2-tailed Student t test.
Effect of mutating JMJD1C ZFD and Jumonji domain on MLL-AF9 leukemogenesis. (A-B) CFC assay after transduction of MLL-AF9-Cas9 cells with sgRNAs against ZFD (e12.3, e12.4) and Jumonji domain (e20.1, e21.1). (A) Colonies in methylcellulose culture (upper panels). Individual colony morphology (original magnification ×80; lower panels). (B) Colony counts 1 week after plating. (C) Proliferation assay of sorted GFP+ (marks MLL-AF9) TdTomato+ (marks sgRNA) MLL-AF9-Cas9 leukemia cells after sgRNA transduction. Data are mean ± standard deviation (SD) of 3 independent experiments. The P value was determined by 2-way analysis of variance (ANOVA). For multiple comparisons, P < .05 for day 7 and P < .0001 for day 9 between Renilla and all other sgRNAs, with the exception of Renilla vs e12.3 on day 7 (not statistically significant). (D) Apoptosis assay (left panel). Data are mean ± SD from 3 independent experiments. Representative flow plot; numbers within the upper left quadrant indicate average percentage (right panel). (E) Representative flow cytometry analysis of Mac1 of MLL-AF9-Cas9 leukemia cells 7 days after transduction (left panel). Mean fluorescence intensity (MFI; geometric mean) of Mac1 expression (right panel). Data are mean ± SD from 3 independent experiments. (F) Survival curves of secondary recipient mice that received MLL-AF9-Cas9 cells after transduction with respective sgRNAs. P = .040 overall, log-rank test. Pairwise log-rank test P values between Renilla and JMJD1C sgRNAs: P = .025 for e12.3; P = .137 for e12.4; P = .096 for e20.1, and P = .048 for e21.1. (G) Peripheral blood GFP+TdTomato+ cell percentage 8 weeks after transplantation. P = .05, multiple comparison, 1-way ANOVA. Renilla vs e12.3, P = .06; Renilla vs e12.4, e20.1, and e21.1, P = .08 . *P < .05, **P < .01, ***P < .001, unpaired 2-tailed Student t test.
Our in vitro findings led us to examine whether the JmjC domain and ZFD are required for MLL-AF9 leukemogenesis in vivo. We transplanted sorted MLL-AF9-Cas9 cells harboring sgRNAs against these domains into recipient mice. Our result demonstrates that mutating the JmjC domain and ZFD with sgRNAs significantly prolonged the survival of the recipient mice compared with Renilla controls (Figure 2F). The percentage of leukemic cells in the peripheral blood was significantly higher in the Renilla control vs JMJD1C sgRNAs groups at 8 weeks after transplant (Figure 2G). We did not observe any differences between Renilla and mock-transduced groups in our transplant experiment (supplemental Figure 2G). Examination of bone marrow (BM), spleen, liver, and thymus from mice harboring sgRNAs against JMJD1C that succumbed to the disease showed leukemia infiltration in all of the organs (supplemental Figure 2I). Nonetheless, Sanger sequencing of amplicons encompassing exons 12 or 20 from the BM cells of these mice showed a mixture of mutated and wild-type allele or completely wild-type leukemia cells (supplemental Table 5). Collectively, the outgrowth of wild-type cells suggests that there is strong selection pressure against mutating these domains. Overall, these data indicate that the ZFD and JmjC domain are important for JMJD1C’s function in MLL-AF9 leukemia in vivo.
JMJD1C affects gene transcription by modulating H3K36 methylation
Although initially described as a H3K9me1/2 demethylase, the demethylase activity of JMJD1C is still under debate. Histone demethylases can have dual activity toward multiple histone marks.30 To explore this possibility, we performed genome-wide profiling of H3K27me and H3K36me, which have been shown to be substrates for some KDMs that bear activity toward H3K929 and H3K9me3/me1 by ChIP-seq. Genome-wide mapping of these marks has shown that H3K9me2/3 and K27me2/3 are associated with gene repression, whereas H3K9/K27me1 and K36me1/2/3 are associated with gene activation.31 We first performed ChIP-seq in JMJD1C-null cells29 vs control leukemia cells. Examination of the methylation levels in the top differentially expressed genes by RNA-seq, such as Gas7 and Abcg1,29 in these cells showed an increase in H3K36me3 levels (supplemental Figure 3A), which we validated by ChIP–quantitative polymerase chain reaction (supplemental Figure 3B). In contrast, we did not observe changes in H3K27me3 or H3K9me1/2/3 on these genes (supplemental Figure 3A). To examine whether there is a correlation between H3K36me3 and gene expression on a genome-wide scale, the ChIP-seq signal was quantified as the total number of reads per million from the transcription start to end sites and subjected to gene set enrichment analysis (GSEA). We found enrichment of JMJD1C-regulated genes (derived from RNA-seq comparing JMJD1C-null and control cells29 ) in H3K36me3 data sets (supplemental Figure 3C) but not in H3K27me3 or H3K9me3/2/1 data sets (supplemental Figure 3D-G). We then performed RNA-seq and ChIP-seq in MLL-AF9 cells harboring JMJD1C sgRNAs (Figure 3A-C). We identified 790 upregulated genes and 115 downregulated genes comparing MLL-AF9 cells harboring sgRNAs against Renilla vs those against JMJD1C (Figure 3B-C). GSEA analysis showed enrichment of the differentially regulated genes identified in Jmjd1c-knockout MLL-AF9 leukemia, as well as similar sets of genes that we reported previously, including those of myeloid differentiation genes and HOXA9-regulated gene transcription programs (Figure 3D-E). Lastly, to examine whether there is a correlation between gene expression and H3K36me3, we performed GSEA and found enrichment of differentially expressed genes between Renilla and JMJD1C sgRNAs in the H3K36me3 data set (Figure 3F). To summarize, changes in H3K36 methylation, but not that of H3K27me3 or K9me1/2/3, correlate with changes in gene expression upon JMJD1C loss. Therefore, methylated H3K36 at gene loci represents a marker for JMJD1C activity.
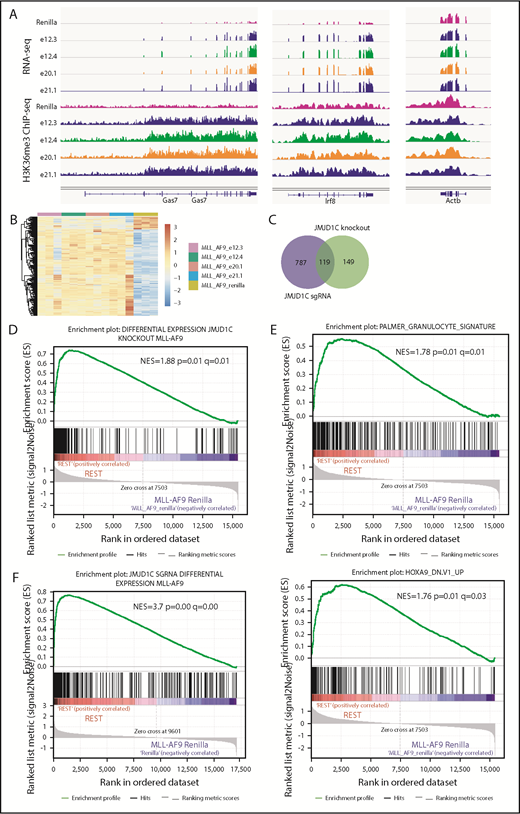
Changes in histone modifications upon loss of JMJD1C. (A) Representative snapshots of RNA-seq and H3K36me3 ChIP-seq (triplicate and duplicate biological replicates, respectively; 1 representative replicate is shown) results in Jmjd1c sgRNA vs Renilla sgRNA cells: Irf8 and Gas7 are genes repressed by JMJD1C, and Actb is a control gene. (B) Heat map of differentially expressed genes (adjusted P < .05 and fold change > 1.5) between MLL-AF9 cells harboring Renilla vs JMJD1C sgRNAs. (C) Venn diagram of overlapping differentially regulated genes in the knockout and sgRNA RNA-seq data set. P < 8.5−97, exact hypergeometric test. GSEA enrichment plots of differentially regulated genes between Jmjd1cf/f and Jmjd1c−/− cells29 (D), indicated gene sets (E, top and bottom) in MLL-AF9 cells harboring Renilla (Renilla) vs JMJD1C sgRNAs (REST) RNA-seq data. (F) GSEA enrichment plots of differentially regulated genes between MLL-AF9 cells harboring Renilla vs JMJD1C sgRNAs in H3K36me3 ChIP-seq data of Renilla vs JMJD1C sgRNAs (REST).
Changes in histone modifications upon loss of JMJD1C. (A) Representative snapshots of RNA-seq and H3K36me3 ChIP-seq (triplicate and duplicate biological replicates, respectively; 1 representative replicate is shown) results in Jmjd1c sgRNA vs Renilla sgRNA cells: Irf8 and Gas7 are genes repressed by JMJD1C, and Actb is a control gene. (B) Heat map of differentially expressed genes (adjusted P < .05 and fold change > 1.5) between MLL-AF9 cells harboring Renilla vs JMJD1C sgRNAs. (C) Venn diagram of overlapping differentially regulated genes in the knockout and sgRNA RNA-seq data set. P < 8.5−97, exact hypergeometric test. GSEA enrichment plots of differentially regulated genes between Jmjd1cf/f and Jmjd1c−/− cells29 (D), indicated gene sets (E, top and bottom) in MLL-AF9 cells harboring Renilla (Renilla) vs JMJD1C sgRNAs (REST) RNA-seq data. (F) GSEA enrichment plots of differentially regulated genes between MLL-AF9 cells harboring Renilla vs JMJD1C sgRNAs in H3K36me3 ChIP-seq data of Renilla vs JMJD1C sgRNAs (REST).
JMJD1C regulates the interleukin-3 signaling pathway
To further examine the molecular mechanism of JMJD1C mutations in MLL-AF9 leukemia, we performed single-cell gene-expression analysis. We focused on the JmjC domain because of its known enzymatic activity. We profiled ∼1000 mouse MLL-AF9-Cas9 cells harboring sgRNAs against Renilla or JmjC domain (e20.1) using 10× Genomics technology. The data analysis was done using the Seurat single cell analysis package.32 As expected, t-distributed stochastic neighbor embedding analysis33 revealed distinct clustering of control cells transduced with sgRNA against Renilla vs that of the JmjC domain (Jumonji; Figure 4A; supplemental Figure 4A,C). Further analysis uncovered 2 clusters within control cells, Renilla 1 and Renilla 2, and 3 clusters within mutated cells, Jumonji 1, Jumonji 2, and Jumonji 3 (Figure 4B). These clusters differ in their cell cycle status, with Renilla 1 having the highest percentage of cells in the S phase of the cell cycle (Figure 4C). A comparison of the enrichment of an MLL-AF9 LSC signature34 between these clusters demonstrated that Renilla clusters have the highest expression levels, whereas Jumonji 1, Jumonji 2, and Jumonji 3 have progressively less expression (Figure 4D). Single-cell trajectory analysis by Monocle35 showed that Renilla 2, Jumonji 2, and Jumonji 3 occupied 3 end states, with the Renilla 1 cluster progressing toward Renilla 2, and Jumonji 1 spreading on the paths to Jumonji 2 and Jumonji 3, suggesting that Jumonji 1 is in a transitional state (Figure 4E; supplemental Figure 4D) between Renilla 2 and Jumonji 2 and Jumonji 3. To further understand the differences between clusters, we performed SCENIC analysis36 to examine active regulons (transcription factors and their target genes) in each cluster. Whereas Renilla 1, Renilla 2, and Jumonji 1 have very few active regulons, Jumonji 3 has the most active regulons (Figure 4F; supplemental Figure 4B), including transcription factors important for monocyte/macrophage development: Mef2c,37,38 Klf2,39 Klf3,40,41 Klf6,42 and Irf8.43-45 Moreover, MAST46 analysis (GSEA adapted for single-cell RNA-seq data) comparing Jumonji 2 and Jumonji 3 using a collection of hematopoietic fingerprint gene sets47 showed that Jumonji 2 has the greatest enrichment of genes found to be highly expressed in granulocytes, whereas Jumonji 3 has the greatest enrichment of genes that are most highly expressed in monocytes (Figure 4H). Overall, these data suggest that a simple hierarchy exists in Renilla cells, with Renilla 1 bearing more LSC properties and Renilla 2 bearing more mature cell properties. Upon mutation of JMJD1C, cells can be driven into 2 distinct differentiated states, reflecting the plasticity in lineage determination of the leukemia cells.
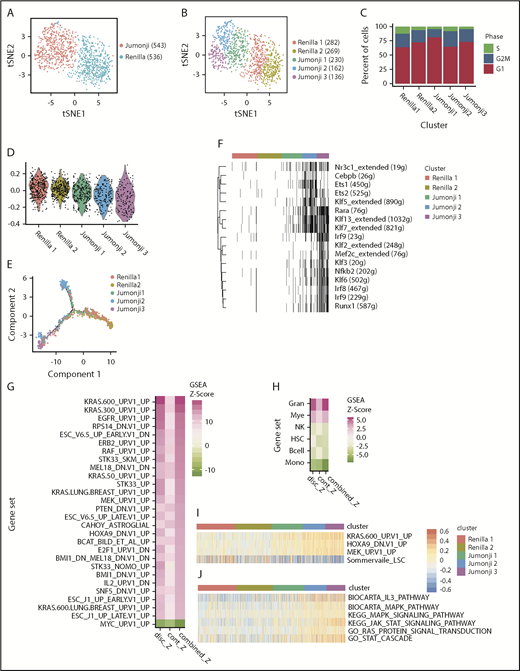
Single-cell transcriptome analysis of JMJD1C Jumonji domain–mutated MLL-AF9 leukemia cells. (A-B) T-distributed stochastic neighbor embedding plot of single-cell gene-expression data of mouse MLL-AF9-Cas9 leukemia cells 7 days after transduction with sgRNA against Renilla or JMJD1C JmjC domain (Jumonji). (A) Cell phenotype. (B) Cell clusters identified within phenotypes. The numbers of cells are indicated in parenthesis. (C) Cyclone cell cycle status. (D) Expression level of Somerville LSC34 signature. (E) Monocle single-cell trajectory analysis. (F) SCENIC regulon analysis. Black bars indicate that a regulon is activated within a cell, across all clusters identified in panel B. (G-H) Decomposed Z score of MAST analysis on C6 oncogenic signature from the Molecular Signature Database (all Renilla cells vs all Jumonji cells). (G) Pink: enrichment in Jumonji; green: enrichment in Renilla sample, and hematopoietic fingerprint gene sets (Jumonji 2 cells vs Jumonji 3 cells). (H) Pink: enrichment in Jumonji 2; green: enrichment in Jumonji 3. GSVA analysis of top KRAS pathway (I; identified in panel G) and RAS/MAPK and JAK-STAT pathways (J). P < .01 for all pairwise comparisons in panel D, with the exception of Renilla 1 vs Renilla 2 (not significant), unpaired 2-tailed Student t test with Bonferroni correction. Gran, granulocytes; Mye, myeloid; Mono, monocytes; NK, natural killer cells.
Single-cell transcriptome analysis of JMJD1C Jumonji domain–mutated MLL-AF9 leukemia cells. (A-B) T-distributed stochastic neighbor embedding plot of single-cell gene-expression data of mouse MLL-AF9-Cas9 leukemia cells 7 days after transduction with sgRNA against Renilla or JMJD1C JmjC domain (Jumonji). (A) Cell phenotype. (B) Cell clusters identified within phenotypes. The numbers of cells are indicated in parenthesis. (C) Cyclone cell cycle status. (D) Expression level of Somerville LSC34 signature. (E) Monocle single-cell trajectory analysis. (F) SCENIC regulon analysis. Black bars indicate that a regulon is activated within a cell, across all clusters identified in panel B. (G-H) Decomposed Z score of MAST analysis on C6 oncogenic signature from the Molecular Signature Database (all Renilla cells vs all Jumonji cells). (G) Pink: enrichment in Jumonji; green: enrichment in Renilla sample, and hematopoietic fingerprint gene sets (Jumonji 2 cells vs Jumonji 3 cells). (H) Pink: enrichment in Jumonji 2; green: enrichment in Jumonji 3. GSVA analysis of top KRAS pathway (I; identified in panel G) and RAS/MAPK and JAK-STAT pathways (J). P < .01 for all pairwise comparisons in panel D, with the exception of Renilla 1 vs Renilla 2 (not significant), unpaired 2-tailed Student t test with Bonferroni correction. Gran, granulocytes; Mye, myeloid; Mono, monocytes; NK, natural killer cells.
Next, we performed MAST46 analysis using the C6 oncogenic collection from the Molecular Signature Database.48 We observed an enrichment of similar sets of genes in leukemia cells harboring JmjC sgRNA as we had previously reported in JMJD1C-knockout MLL-AF9 cells, including a HOXA9-regulated gene transcription program (Figure 4G).29 Surprisingly, we uncovered an enrichment of much more concentrated and higher ranked gene signatures in the KRAS pathways within the Jumonji sample compared with previous analysis of bulk RNA-seq data.29 These include multiple KRAS-, RAF-, and MEK-controlled transcription programs (Figure 4G). Further examination using gene set variation analysis (GSVA)49 showed that the Jumonji 2 and Jumonji 3 clusters have the most enrichment of the KRAS pathway (Figure 4I). The enrichment of the RAS pathways led us to examine whether the IL-3 signaling pathway is affected in JMJD1C-mutated cells. The IL-3 signaling pathway plays an important role in myeloid cell proliferation, survival, and differentiation,50 and IL-3 is required for MLLr mouse leukemia cell survival. Two important downstream signaling pathways that mediate the effect of IL-3 signaling include RAS/RAF/ERK and JAK-STAT. Using GSVA, we revealed an enrichment of the RAS/MAPK and JAK-STAT pathways in Jumonji 2 and Jumonji 3 clusters (Figure 4J).
To further examine whether IL-3 signaling pathway is activated in leukemia cells harboring JmjC sgRNAs, we performed western blotting of downstream molecules, including phosphorylated ERK1/2 and STAT5. Our result showed that basal ERK1/2 and STAT5 phosphorylation levels, as well as those in response to IL-3 stimulation, increased in leukemia cells harboring JmjC or ZFD sgRNAs compared with controls (Figure 5A). Moreover, these cells are more sensitive to proliferation inhibition by RAS pathway inhibitors, including tipifarnib (a farnesyltransferase inhibitor) and selumetinib (a MEK1/2 inhibitor) (Figure 5B), as well as to that by the JAK kinase inhibitors ruxolitinib and tofacitinib (Figure 5C). To understand the mechanism underlying IL-3 signaling pathway activation, we examined and found significantly increased expression of mouse IL-3 receptor genes Csf2rb/Csf2rb2 in single-cell data (Figure 5D; supplemental Figure 5A), JMJD1C sgRNA bulk RNA-seq data (supplemental Table 7), and our published bulk RNA-seq data (supplemental Figure 5B). Csf2rb/Csf2rb2 encodes the common β chain for IL-3, IL-5, and granulocyte macrophage colony-stimulating factor and IL-3–specific β chain, respectively. The Il3ra (encodes the α subunit) gene is only increased when comparing Renilla 2 and Jumonji 3 (supplemental Figure 5A) in the single-cell data. Finally, to examine whether the observed phenotype is indeed mediated by mouse IL-3 receptor, we performed a competitive proliferation experiment using sgRNAs against JMJD1C in the presence or absence of sgRNAs against CD131 (encoded by the Csf2rb gene). We observed faster depletion of MLL-AF9-Cas9 cells transduced with sgRNAs against both compared with that of JMJD1C and lentiGuide BFP controls (Figure 5E). Together, these data suggest that mutating the JmjC domain and ZFD resulted in hyperactivation of the IL-3 pathway through upregulation of the IL-3 receptor genes (model in Figure 5F).
![Figure 5. Increased IL-3 signaling in MLL-AF9 cells with mutated Jumonji domain and ZFD. (A) Western blotting of phospho- and total ERK1/2 and STAT5 in MLL-AF9-Cas9 leukemia cells 6 days after transduction with respective sgRNAs. Dose response curve (4-variable slope model) in the presence of RAS/ERK inhibitors (B) or JAK inhibitors (C). Two or 3 independent experiments, each done with technical triplicates. Shown are representative experiments (mean ± standard deviation [SD] of technical triplicates). Bar graphs on the far right show EC50 (mean ± SD of 2-3 independent experiments) of inhibitors normalized to that of Renilla control. Numbers on top of the bar are average EC50 (nM). *P < .05, **P < .01, unpaired 2-tailed Student t test. (D) Violin plot of relative gene expression data of Il3ra/Csf2rb/b2 derived from single-cell transcriptome. *****Adjusted P < .00001 by MAST. (E) Competitive cell-proliferation assay using sgRNAs against JMJD1C in the presence or absence of sgRNA against Csf2rb (marked by BFP). Data are mean ± SD of percentage of BFP+ and TdTomato+ double-positive cells from independently transduced triplicate samples. Curves were fitted with an asymmetric sigmoidal 5 parameter logistical model. (F) Model of transcription regulation of IL-3 receptor gene by JMJD1C.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/9/10.1182_bloodadvances.2018026054/2/m_advances026054f5.png?Expires=1769109307&Signature=0oTyaWACMSEHymRQKYmIvUFyIv78XA9VmPRc3B5F7MmL~HW~dfitIt-yJyrnffUENMEOU~jVN45CBJzjI2S222Dq8jZBDWr-8pdMGro9syKA77~IJ5NyeZyq3ph3zB6D2~pH3s9XK2kz1ZIMq18NjyVbRWzKsAkepoTP2YJ6UnZV1DI~KHttR9iq~9ZEhRm-~NiRpd9ZRbbg8PVcQfB2i99UtEdqxLUq3uvXfMwkxsqrY~sTQBotLLPUIDbviGZ5~rKYXR3jl82egS3FHI5aCrAi9zwgJ9zunaohZBgEQQ8JIZvWhAfIkWXQNgUPhCBm-PQwVpMxqk69ZhN7CnQEug__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Increased IL-3 signaling in MLL-AF9 cells with mutated Jumonji domain and ZFD. (A) Western blotting of phospho- and total ERK1/2 and STAT5 in MLL-AF9-Cas9 leukemia cells 6 days after transduction with respective sgRNAs. Dose response curve (4-variable slope model) in the presence of RAS/ERK inhibitors (B) or JAK inhibitors (C). Two or 3 independent experiments, each done with technical triplicates. Shown are representative experiments (mean ± standard deviation [SD] of technical triplicates). Bar graphs on the far right show EC50 (mean ± SD of 2-3 independent experiments) of inhibitors normalized to that of Renilla control. Numbers on top of the bar are average EC50 (nM). *P < .05, **P < .01, unpaired 2-tailed Student t test. (D) Violin plot of relative gene expression data of Il3ra/Csf2rb/b2 derived from single-cell transcriptome. *****Adjusted P < .00001 by MAST. (E) Competitive cell-proliferation assay using sgRNAs against JMJD1C in the presence or absence of sgRNA against Csf2rb (marked by BFP). Data are mean ± SD of percentage of BFP+ and TdTomato+ double-positive cells from independently transduced triplicate samples. Curves were fitted with an asymmetric sigmoidal 5 parameter logistical model. (F) Model of transcription regulation of IL-3 receptor gene by JMJD1C.
Increased IL-3 signaling in MLL-AF9 cells with mutated Jumonji domain and ZFD. (A) Western blotting of phospho- and total ERK1/2 and STAT5 in MLL-AF9-Cas9 leukemia cells 6 days after transduction with respective sgRNAs. Dose response curve (4-variable slope model) in the presence of RAS/ERK inhibitors (B) or JAK inhibitors (C). Two or 3 independent experiments, each done with technical triplicates. Shown are representative experiments (mean ± standard deviation [SD] of technical triplicates). Bar graphs on the far right show EC50 (mean ± SD of 2-3 independent experiments) of inhibitors normalized to that of Renilla control. Numbers on top of the bar are average EC50 (nM). *P < .05, **P < .01, unpaired 2-tailed Student t test. (D) Violin plot of relative gene expression data of Il3ra/Csf2rb/b2 derived from single-cell transcriptome. *****Adjusted P < .00001 by MAST. (E) Competitive cell-proliferation assay using sgRNAs against JMJD1C in the presence or absence of sgRNA against Csf2rb (marked by BFP). Data are mean ± SD of percentage of BFP+ and TdTomato+ double-positive cells from independently transduced triplicate samples. Curves were fitted with an asymmetric sigmoidal 5 parameter logistical model. (F) Model of transcription regulation of IL-3 receptor gene by JMJD1C.
The observed upregulation of the RAS pathway in response to JMJD1C mutation led us to examine the effect of the loss or mutation of JMJD1C in AML cells that harbor activating RAS mutations. Although typically secondary events late in leukemogenesis,51 mutations in the RAS pathway are found in AML in general and at a high frequency in MLLr leukemia (∼10% NRAS and KRAS mutations),52 and they play a role in disease pathogenesis.53,54 In a negative-selection screen of epigenetic regulators in mouse MLL-AF9 leukemia cells overexpressing an activating form of Ras, NRasG12D, Shi et al26 did not observe depletion of JmjC sgRNAs e20.1, e21.1, e22.1, e23.1, and e23.2. RAS mutations are also found in common AML cell lines, including NOMO1 (KRASG13D) and THP1 (NRASG12D; Broad Institute Cancer Cell Line Encyclopedia). We examined published data sets of short hairpin RNA55 and sgRNA56 screens in human leukemia cell lines and found that MLLr cell lines with RAS mutations are less sensitive to JMJD1C loss or mutation (Figure 6A-B). We performed an sgRNA negative-selection assay in human MLLr cell lines (MOLM13, MONO-MAC-1, THP1, and NOMO1) and non-MLLr cell lines (U937 and OCI-AML3). Our results showed that only MOLM13 and MONO-MAC-1 cells demonstrated significant depletion of the cells expressing sgRNAs against these domains (Figure 6C; sgRNAs 12.5 and 20.1), comparable to that of positive controls BRD4, DOT1, RPA3, and POLRA. Finally, to demonstrate that the observed resistance is indeed mediated by the RAS mutation, we overexpressed a RAS mutant in the mouse MLL-AF9 or human leukemia cell line MOLM13 and performed a similar negative-selection screen. Our results showed that the degree of depletion is significantly attenuated in mouse MLL-AF9 cells overexpressing NRASG12V or KRASG12D or in MOLM13 NRASG12V cells compared with parental cells (Figure 6D-E). Together, these data suggest that the JmjC domain and ZFD of JMJD1C are most critical for its function in human AML and that MLLr leukemias with an activating RAS mutation have reduced or diminished sensitivity to JMJD1C modulation alone.
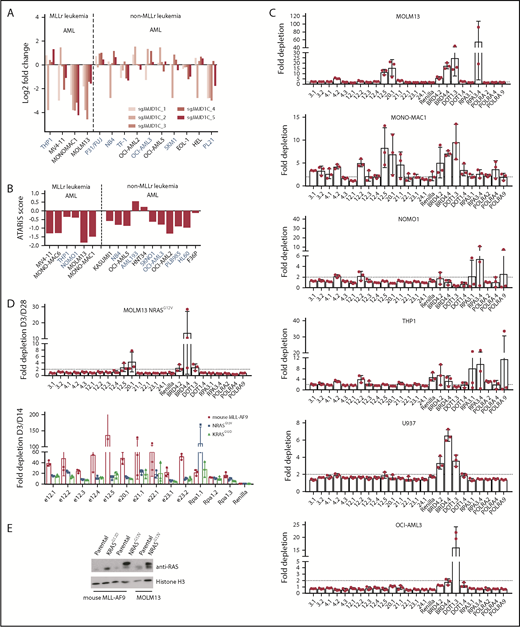
Presence of activating RAS mutations renders cells resistant to JMJD1C loss. Fold change in JMJD1C sgRNA abundance (A)56 and ATARIS score (B)55 in leukemia cell lines. Cell lines in blue bear RAS mutations according to the Broad Institute Cancer Cell Line Encyclopedia. (C) MOLM13, MOMO-MAC-1, NOMO1, THP1, U937, and OCI-AML3 cells were infected with the indicated sgRNA-Cas9-TdTomato viruses and subjected to flow cytometry analysis over a period of 28 days. Fold changes in TdTomato positivity compared with day 3 are plotted. Data are mean ± standard deviation of independently transduced triplicate samples. (D) Fold depletion of sgRNAs in MOLM13 cells overexpressing NRASG12V and in mouse MLL-AF9-Cas9 cells overexpressing NRASG12V and KRASG12D. Data are mean ± standard deviation of independently transduced triplicate samples. (E) Western blotting of NRAS/KRAS expression in cells in panel D.
Presence of activating RAS mutations renders cells resistant to JMJD1C loss. Fold change in JMJD1C sgRNA abundance (A)56 and ATARIS score (B)55 in leukemia cell lines. Cell lines in blue bear RAS mutations according to the Broad Institute Cancer Cell Line Encyclopedia. (C) MOLM13, MOMO-MAC-1, NOMO1, THP1, U937, and OCI-AML3 cells were infected with the indicated sgRNA-Cas9-TdTomato viruses and subjected to flow cytometry analysis over a period of 28 days. Fold changes in TdTomato positivity compared with day 3 are plotted. Data are mean ± standard deviation of independently transduced triplicate samples. (D) Fold depletion of sgRNAs in MOLM13 cells overexpressing NRASG12V and in mouse MLL-AF9-Cas9 cells overexpressing NRASG12V and KRASG12D. Data are mean ± standard deviation of independently transduced triplicate samples. (E) Western blotting of NRAS/KRAS expression in cells in panel D.
Discussion
We have previously showed that JMJD1C is required for AML LSC self-renewal. In this study, we identified the JmjC domain and ZFD within JMJD1C as important for the survival of MLL-AF9 leukemia cells (Figure 1) and demonstrated that they are essential for MLL-AF9 leukemogenesis in vitro and in vivo (Figure 2). The phenotypes observed in mutated cells phenocopied those in knockout cells, including impaired clonogenic activity and proliferation, increased differentiation and apoptosis, and prolonged survival in secondary transplants. Our results established a critical role for the JmjC domain in MLLr AML leukemogenesis and provide rationale for potential therapeutic targeting of this enzymatic domain. However, the mechanism through which ZFD contributes to leukemogenesis is less clear. The ZFD in JMJD1C is a noncanonical C2HC4 type. A similar ZFD in KDM3A has been shown to be required for its enzymatic activity.9 Furthermore, a T667A mutation in the KDM3A ZFD abolished its activity toward H3K9me1, but not H3K9me2, suggesting that the ZFD may aid in differentiating the 2 methylation states.9 Therefore, the ZFD in JMJD1C might be involved in substrate recognition and critical for its enzymatic activity.
The enzymatic activity of JMJD1C remains incompletely characterized. In our current work, we examined changes in H3K27me3 and H3K36me3, as well as H3K9me1 and H3K9me3, in JMJD1C-knockout out cell vs controls. H3K27me3 and H3K36me3 are modifications that are found in other KDMs that also possess activity against H3K9me.30,57 Our results showed that there is no enrichment of JMJD1C-regulated genes in any of these marks, with the exception of H3K36me3 (Figure 3). Given that H3K36me associates with gene activation, this result is consistent with our observation that the loss or mutation of JMJD1C largely results in genes that are upregulated rather than downregulated (211 vs 57 in the knockout data29 and 790 vs 116 in the sgRNA data) (Figure 3). Whether JMJD1C is a demethylase for H3K36me3 needs to be definitively established. Nonetheless, our findings showed that H3K36me3 can be used as a marker for JMJD1C activity.
Our single-cell transcriptome analysis of murine MLL-AF9 leukemia (Figure 4) shows that leukemia cells exist in a simple hierarchy consisting of the more proliferative Renilla 1 cluster and the less proliferative Renilla 2 cluster. Upon mutation of the JmjC domain, leukemia cells can progress toward 2 differentiation states resembling granulocytes or monocytes. During this differentiation process, cells upregulate the IL-3 signaling pathway, as indicated by increased basal activation and sensitivity to IL-3 stimulation of its downstream signaling pathways. This is evident by the increased ERK1/2 and STAT5 phosphorylation, the increased sensitivity to inhibitors against RAS/MEK/ERK and the JAK-STAT pathway, and the increased sensitivity to IL-3 receptor loss in JMJD1C-mutated vs control cells. Moreover, our data showed that the increased IL-3 pathway activation in leukemia cells harboring JmjC sgRNAs is mediated by the upregulation of IL-3 receptor genes (Figure 5). Importantly, our data cannot discern whether Csf2ra/b genes are direct targets of JMJD1C. Nonetheless, given our previous data showing that JMJD1C physically interacts with HOXA929 and that HOXA9 represses the expression of IL-3 receptor genes (Il3ra/Csf2rb/b2),58 it is likely that JMJD1C collaborates with HOXA9 in repressing Csf2rb/b2 gene expression in leukemia cells (model in Figure 5F).
Finally, we showed that human MLLr leukemia cells MOLM13 and MONO-MAC-1 are sensitive to JmjC and ZFD sgRNAs but that the non-MLLr cells (U937 and OCI-AML3) are not. Interestingly, we observed a loss of responsiveness to JMJD1C modulation in leukemia cells bearing activating RAS mutations (Figure 6), suggesting that MLL-AF9 leukemia cells upregulate the IL-3/RAS/JAK pathway as a mechanism to counteract the impairment of survival and proliferation resulting from loss of JMJD1C. To this end, we screened MLL-AF9 cells (day 9 after transduction of JMJD1C sgRNAs), as well as BM cells from transplantation, for the presence of RAS mutations but did not observe any (supplemental Tables 5 and 6). This may be due to the relatively short time frame to allow RAS mutations to evolve, undergo selection, and outgrow.
Overall, our results establish the requirement of the JMJD1C JmjC domain and ZFD in MLL-AF9 leukemogenesis. The requirement of the JmjC domain, in particular, provides rationale for the potential development of small molecule inhibitors against its enzymatic activity. Moreover, our results suggest that the presence of RAS mutations in MLLr will render the cells insensitive to JMJD1C loss. These data add further evidence to the importance of targeted therapies that take into account the mutation spectrum of patients.
Chromatin immunoprecipitation sequencing, bulk RNA, and single-cell RNA sequencing data are deposited in the Gene Expression Omnibus database and Sequence Read Archive (accession number GSE129675).
For original data, please contact nan.zhu@bcw.edu.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Prethish Screenivas and Joshua DeJong for technical assistance with the guide RNA screen; David Schauder, Ryan Zander, and Mark Zogg for technical assistance with 10× Genomics sequencing; and Benedetta Bonacci for assistance with cell sorting.
N.Z. was supported by a grant from the National Institutes of Health, National Cancer Institute (R00CA168996), an ASH Scholar Award from the American Society of Hematology, and a grant from the Blood Center Research Foundation.
Authorship
Contribution: N.Z., J.I.-C., and L.C. designed the study, performed research, and wrote the manuscript; J. Schmitz, R.L.M., T.B., J. Shen, and K.-S.C. performed research and analyzed data; R.B. and C.L. analyzed single-cell transcriptome data; R.B. also performed data analysis of RNA-seq, ChIP-seq, and SITE-seq; Y.Z. and D.W. performed and helped with the IL-3 signaling study and provided conceptual input; S.R. provided technical help with SITE-seq; and T.B. and S.R. edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nan Zhu, Blood Research Institute, Versiti, 8727 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: nan.zhu@bcw.edu.

