

Key Points
Nonacute NPM1-mutated myeloid neoplasms are biologically distinct from nonacute NPM1 wild-type myeloid neoplasms.
Nonacute NPM1-mutated myeloid neoplasms are associated with poorer survival compared with NPM1-mutated AML and NPM1-WT myeloid neoplasms.

Abstract
NPM1-mutated myeloid neoplasms (NPM1+ MNs) with <20% blood or bone marrow blasts are rare and have been previously shown in limited case series to exhibit an aggressive clinical course. We assembled the largest cohort of NPM1+ MN cases to date (n = 45) and compared it with NPM1− MN (n = 95) and NPM1+ de novo acute myeloid leukemia (AML; n = 119) patients. Compared with NPM1− MN, NPM1+ MN were associated with younger age (P = .007), a normal karyotype (P < .0001), more frequent mutations involving DNMT3A (P = .01) and PTPN11 (P = .03), and fewer involving ASXL1 (P = .003), RUNX1 (P = .0004), and TP53 (P = .02). Mutations involving IDH1 or IDH2 (IDH1/2) (P = .007) and FLT3 (internal tandem duplication, P < .0001; noninternal tandem duplication, P = .01) were less frequent in NPM1+ MN than in NPM1+ AML. In multivariable analyses performed in patients with myelodysplastic syndrome only, total mutation count (hazard ratio [HR], 1.3; P = .05), NPM1 mutation (HR, 3.6; P = .02), TP53 mutation (HR, 5.2; P = .01), and higher International Prognostic Scoring System-R score (HR, 1.7; P = .0003) were independently associated with shorter overall survival, whereas stem cell transplant conferred a favorable effect (HR, 0.1; P < .0001). These data suggest that NPM1+ MN are biologically distinct from NPM1− MN. Similar to NPM1+ AML, patients with NPM1-mutated myelodysplastic syndrome may benefit from more intensive therapeutic regimens.
Introduction
Carboxy-terminal insertion mutations in the nucleophosmin-1 (NPM1) gene represent the most common somatic mutations identified in de novo acute myeloid leukemia (AML), and are typically associated with a favorable prognosis in the absence of unfavorable clinical parameters or FLT3 internal tandem duplication (FLT3-ITD) comutation.1-6 Previous studies have reported NPM1 mutations in 5% to 17% of secondary AML and 1% to 5% of myelodysplastic syndrome (MDS) and/or MDS/myeloproliferative (MDS/MPN) neoplasms.7-16 In these small series, the presence of an NPM1 mutation in the setting of a myeloid neoplasm with fewer than 20% blasts (NPM1+ MN) has been associated with aggressive disease and a relatively rapid progression to overt AML. However, most of these studies have interrogated for NPM1 mutations using limited single-gene assays and have therefore been unable to comprehensively compare the genetic profiles of NPM1+ MN with myeloid neoplasms lacking NPM1 mutation (NPM1− MN).
De novo AML with mutated NPM1 (NPM1+ AML) is classically associated with a normal karyotype and frequent comutations in DNMT3A, IDH1/IDH2, and RAS pathway genes, notably FLT3.1,6,17-20 Interestingly, morphologic dysplasia can be seen in NPM1+ AML, although this finding carries no prognostic significance and should not invoke a diagnosis of AML with myelodysplasia-related changes, as mandated by the 2017 World Health Organization (WHO) classification.1,21 Because it is unknown if NPM1+ MN and NPM1+ AML groups share genetic features, it is conceivable that morphologic dysplasia could be uniformly disregarded in the context of NPM1-mutated disease if they are related; moreover, NPM1+ MN may simply represent NPM1+ AML “caught early.”
MDS is the most common type of non-AML myeloid neoplasm, presents at a median age of 70 years, and is often associated with karyotypic abnormalities and/or 1 or more somatic mutations classically involving the “DTA” genes (DNTM3A, TET2, ASXL1), the splicing factor genes SRSF2 and SF3B1, and/or the transcription factor RUNX1.22 Risk stratification in MDS is performed using the revised International Prognostic Scoring System (IPSS-R) with the primary goal of identifying patients who may benefit from hypomethylating agent (HMA) therapy and/or stem cell transplantation (SCT).23 Whether or not this approach to prognostication and management is appropriate for NPM1+ MN remains unclear; although NPM1-mutated AML has been shown to be sensitive to induction chemotherapy, intensive therapy is not standard treatment of MDS and other non-AML MN.24,25
In this study, we assembled the largest known cohort of NPM1+ MN and compared its clinicopathologic and genetic profile with 2 control groups: NPM1− MN and NPM1+ AML.
Methods
Case selection
The pathology archives of 7 major academic medical centers (Brigham and Women’s Hospital/Dana-Farber Cancer Institute, Massachusetts General Hospital, University of Pennsylvania, Vanderbilt University School of Medicine, Weill Cornell Medical College, Memorial Sloan Kettering Cancer Center, and Yale) were queried for cases of NPM1+ MN, defined by the presence of <20% blood or bone marrow blasts in conjunction with a C-terminal insertion mutation involving NPM1. Separately, the pathology archive at Brigham and Women’s Hospital/Dana-Farber Cancer Institute was queried to generate a control group of NPM1− MN patients diagnosed between 2014 and 2018 that were randomly selected to match the WHO Classification subtypes of NPM1+ MN (Table 1). “Pure” MPNs, defined by the presence of either JAK2, CALR, or MPL mutations in isolation, were excluded. The control cohort was unselected for patient age or other specific mutational profiles. A previously examined cohort of NPM1+ AML was also retrieved for comparison as a second control group.21 Cytogenetic data were available for all cases. Flow cytometric and cytomorphologic data were reexamined in a subset of cases.
Characteristics of NPM1− MN, NPM1+ MN, and NPM1+ AML cohorts
| NPM1− MN (n = 95) | NPM1+ MN (n = 45) | NPM1+ AML (n = 119) | |
|---|---|---|---|
| Patient characteristics | |||
| Median age (range), y | 68 (38-84)* | 63 (36-96) | 61 (15-85) |
| Male:female | 1.9 | 1.0 | 0.75 |
| Clinical parameters | |||
| Hemoglobin, median (range), g/dL | 9.7 (4.8-15.9) | 9.0 (6.1-12.7) | 9.0 (5.7-15) |
| WBC, median (range), ×109/L | 3.5 (0.6-69.4) | 3.3 (1.2-225) | 21 (0.69-340)* |
| Platelet count, median (range), ×109/L | 84 (15-808) | 79 (15-607) | 72 (10-356) |
| Median of BM cellularity (range), % | 70 (10-95) | 80 (10-100) | 90 (30-98)* |
| Median of BM blasts (range), % | 8 (1-18) | 10 (1-19) | 73 (21-96)* |
| Diagnosis, n (%) | |||
| MDS non-EB | 5 (5) | 2 (4) | NA |
| MDS-EB | 55 (58) | 24 (53) | NA |
| CMML | 16 (17) | 9 (20) | NA |
| MDS/MPN (non-CMML) | 8 (8) | 5 (11) | NA |
| t-MN | 11 (12) | 5 (11) | NA |
| AML | NA | NA | 119 (100) |
| IPSS-R scores (MDS cases only), median (range) | 5.0 (1.0-10.0) | 5.0 (1.5-7.0) | NA |
| Outcome | |||
| Median follow-up time (range), mo | 19.4 (0.3-57) | 10 (0.07-70) | 24 (0.13-125) |
| Alive at last follow-up, n (%) | 53 (56) | 29 (64) | 67 (56) |
| Progression to AML, n (%) | 30 (32) | 20 (44) | NA |
| Median time to progression (range), mo | 6.3 (1.7-43) | 5.2 (0.4-17.5) | NA |
| Received upfront HMA therapy, n (%) | 55 (58) | 33 (73) | 5 (4) |
| Received upfront induction chemotherapy, n (%) | 0 (0) | 3 (7) | 113 (95) |
| Received SCT at any time, n (%) | 44 (46) | 19 (42) | 67 (56) |
| NPM1− MN (n = 95) | NPM1+ MN (n = 45) | NPM1+ AML (n = 119) | |
|---|---|---|---|
| Patient characteristics | |||
| Median age (range), y | 68 (38-84)* | 63 (36-96) | 61 (15-85) |
| Male:female | 1.9 | 1.0 | 0.75 |
| Clinical parameters | |||
| Hemoglobin, median (range), g/dL | 9.7 (4.8-15.9) | 9.0 (6.1-12.7) | 9.0 (5.7-15) |
| WBC, median (range), ×109/L | 3.5 (0.6-69.4) | 3.3 (1.2-225) | 21 (0.69-340)* |
| Platelet count, median (range), ×109/L | 84 (15-808) | 79 (15-607) | 72 (10-356) |
| Median of BM cellularity (range), % | 70 (10-95) | 80 (10-100) | 90 (30-98)* |
| Median of BM blasts (range), % | 8 (1-18) | 10 (1-19) | 73 (21-96)* |
| Diagnosis, n (%) | |||
| MDS non-EB | 5 (5) | 2 (4) | NA |
| MDS-EB | 55 (58) | 24 (53) | NA |
| CMML | 16 (17) | 9 (20) | NA |
| MDS/MPN (non-CMML) | 8 (8) | 5 (11) | NA |
| t-MN | 11 (12) | 5 (11) | NA |
| AML | NA | NA | 119 (100) |
| IPSS-R scores (MDS cases only), median (range) | 5.0 (1.0-10.0) | 5.0 (1.5-7.0) | NA |
| Outcome | |||
| Median follow-up time (range), mo | 19.4 (0.3-57) | 10 (0.07-70) | 24 (0.13-125) |
| Alive at last follow-up, n (%) | 53 (56) | 29 (64) | 67 (56) |
| Progression to AML, n (%) | 30 (32) | 20 (44) | NA |
| Median time to progression (range), mo | 6.3 (1.7-43) | 5.2 (0.4-17.5) | NA |
| Received upfront HMA therapy, n (%) | 55 (58) | 33 (73) | 5 (4) |
| Received upfront induction chemotherapy, n (%) | 0 (0) | 3 (7) | 113 (95) |
| Received SCT at any time, n (%) | 44 (46) | 19 (42) | 67 (56) |
BM, bone marrow; EB, excess blasts; NA, not available; t-MN, therapy-related myeloid neoplasm.
P < .05 for difference in comparison with NPM1+ MN group (Mann-Whitney U test).
Next-generation sequencing studies
Next-generation sequencing was performed on either diagnostic blood or bone marrow samples, and data were available for all cases. All sequencing panels (n = 8) included the following common set of genes: DNMT3A, IDH1, IDH2, TET2, ASXL1, KRAS, NRAS, FLT3, PTPN11, RUNX1, SF3B1, and TP53. A large subset of NPM1+ MN (40/45), and all NPM1− MN and NPM1+ AML cases were also evaluated for mutations involving SRSF2.
Statistical analyses
Continuous variables including patient age, white blood cell count (WBC), bone marrow blast percentage, IPSS-R scores, total mutation number at diagnosis, and median time to leukemic transformation were compared between cohorts using the Mann-Whitney U test. Rates for abnormal karyotype and specific somatic mutations were compared using Fisher’s exact test. Statistical significance was set at P < .05.
We evaluated patients’ overall survival (OS) as previously reported.26 Briefly, OS was defined as the time in months from the date of initial diagnosis to last follow-up, or death. Outcome profiles of the study cohorts were compared using Kaplan-Meier curves. Multivariable analyses were performed using a cross-cohort subset of only those patients diagnosed with subtypes of MDS (26 NPM1+, 60 NPM1−; n = 86). Backward elimination was performed on the following variables until only those associated with P < .05 remained in the model: age >60 years, IPSS-R score (continuous), total mutations at diagnosis (continuous), upfront HMA therapy, receipt of SCT at any time, presence of mutations in NPM1, DNMT3A, TET2, ASXL1, SRSF2, RUNX1, and TP53 (each found at >10% of the MDS subgroup).
Statistical analyses were performed using XLSTAT (v2018.7) and Prism 8.0c (GraphPad) software packages.
Results
Patient characteristics
We identified 45 NPM1+ MN, 95 NPM1− MN, and 119 NPM1+ AML. Clinicopathologic features of these cohorts are shown in Table 1. Compared with NPM1− MN, NPM1+ MN were associated with younger patient age (P = .007), and most (73%) received upfront HMA therapy. There were no statistically significant differences between NPM1− MN and NPM1+ MN cohorts with respect to other variables shown, including rates of leukemic progression and median time to progression (P > .05). Compared with NPM1+ AML, NPM1+ MN were associated with lower WBC (P < .0001) and bone marrow cellularity (P = .005) at diagnosis, and only 3/45 patients received upfront induction chemotherapy.
Immunophenotypic findings
Data from flow cytometric immunophenotyping performed on bone marrow aspirate material at the time of diagnosis were reexamined for a subset of the study cohort (n = 13), specifically with respect to CD34 expression on myeloid blasts. Out of this subset, CD34 was positive in 5 cases, negative in 5, and variable in 1. One case had <1% myeloid blasts, but an increased population of immunophenotypically abnormal immature monocytes (CD16+CD64+CD56+). The median blast count was 8% in 5 cases with CD34+ blasts and 10% in 5 cases that were CD34−.
Cytogenetic and comutational variables
NPM1+ MN cases were significantly associated with a normal bone marrow karyotype as compared with NPM1− MN (88% vs 39%, P < .0001), at a rate similar to NPM1+ AML (84%). Compared with the NPM1− MN, NPM1+ MN more frequently harbored mutations involving DNMT3A (15/45 vs 14/95, P = .01) and PTPN11 (5/45 vs 2/95, P = .03), but fewer in ASXL1 (4/45 vs 30/95, P = .003), RUNX1 (0/45 vs 20/95, P = .0004), and TP53 (1/44 vs 15/95, P = .02) (Table 2; Figure 1A). They also exhibited a trend toward fewer TET2 (7/45 vs 29/95, P = .06) and SRSF2 (3/40 vs 20/95, P = .08) mutations. Of note, 2/5 NPM1+ MN diagnosed as therapy-related myeloid neoplasms had a noncomplex abnormal karyotype, and 1/5 harbored a TP53 mutation. Compared with NPM1+ AML, NPM1+ MN were associated with fewer mutations in IDH1 or IDH2 (IDH1/2) (6/45 vs 41/119, P = .007) and FLT3 (ITD: 1/44 vs 36/119, P < .0001; non-ITD: 3/45 vs 29/119, P = .01), and showed a trend toward fewer in KRAS or NRAS (KRAS/NRAS) (5/45 vs 31/119, P = .06).
Genetic features of NPM1− MN, NPM1+ MN, and NPM1+ AML cohorts
| NPM1− MN (n = 95) | NPM1+ MN (n = 45) | NPM1+ AML (n = 119) | |
|---|---|---|---|
| Comutations, by pathway, n (%) | |||
| DNMT3A | 14 (15)* | 15 (33) | 57 (48) |
| IDH1 | 22 (2) | 4 (9)* | 25 (21)* |
| IDH2 | 5 (5) | 2 (4)* | 16 (13)* |
| TET2 | 29 (31) | 7 (16) | 32 (27) |
| ASXL1 | 30 (32)* | 4 (9) | 3 (3) |
| RUNX1 | 20 (21)* | 0 (0) | 1 (0.8) |
| KRAS | 3 (3) | 1 (2)† | 4 (3)† |
| NRAS | 8 (8) | 4 (9)† | 27 (23)† |
| FLT3 (non-ITD) | 1 (1) | 3 (7) | 29 (24)‡ |
| FLT3-ITD | 1 (1) | 1 (2) | 36 (30)‡ |
| PTPN11 | 2 (2)* | 6 (13) | 25 (21) |
| SRSF2 | 20 (21) | 3 (8) | 9 (8) |
| SF3B1 | 9 (9) | 4 (9) | 1 (0.8) |
| TP53 | 15 (16)* | 1 (2) | 0 (0) |
| Mutation count, median (range) | 2 (0-8) | 2 (1-7) | 4 (1-8)§ |
| Abnormal karyotype, n (%) | 57 (61)* | 5 (12) | 18 (16) |
| NPM1− MN (n = 95) | NPM1+ MN (n = 45) | NPM1+ AML (n = 119) | |
|---|---|---|---|
| Comutations, by pathway, n (%) | |||
| DNMT3A | 14 (15)* | 15 (33) | 57 (48) |
| IDH1 | 22 (2) | 4 (9)* | 25 (21)* |
| IDH2 | 5 (5) | 2 (4)* | 16 (13)* |
| TET2 | 29 (31) | 7 (16) | 32 (27) |
| ASXL1 | 30 (32)* | 4 (9) | 3 (3) |
| RUNX1 | 20 (21)* | 0 (0) | 1 (0.8) |
| KRAS | 3 (3) | 1 (2)† | 4 (3)† |
| NRAS | 8 (8) | 4 (9)† | 27 (23)† |
| FLT3 (non-ITD) | 1 (1) | 3 (7) | 29 (24)‡ |
| FLT3-ITD | 1 (1) | 1 (2) | 36 (30)‡ |
| PTPN11 | 2 (2)* | 6 (13) | 25 (21) |
| SRSF2 | 20 (21) | 3 (8) | 9 (8) |
| SF3B1 | 9 (9) | 4 (9) | 1 (0.8) |
| TP53 | 15 (16)* | 1 (2) | 0 (0) |
| Mutation count, median (range) | 2 (0-8) | 2 (1-7) | 4 (1-8)§ |
| Abnormal karyotype, n (%) | 57 (61)* | 5 (12) | 18 (16) |
IDH1 and IDH2 are grouped for the analysis comparing NPM1+ MN with NPM1+ AML (Fisher's exact test).
KRAS and NRAS are grouped for the analysis comparing NPM1+ MN with NPM1+ AML (Fisher's exact test).
Significantly different compared with NPM1+ MN (Fisher's exact test).
P < .05 for difference compared with NPM1+ MN (Mann-Whitney U test).
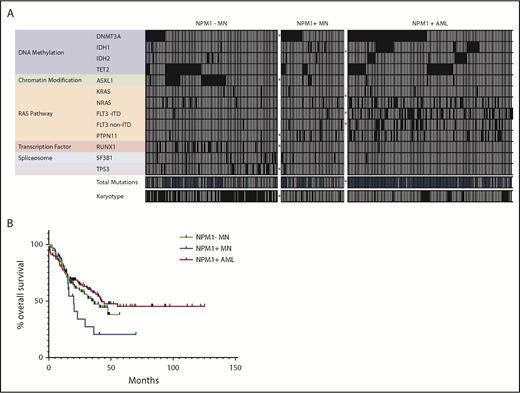
Genetic and clinical outcome characteristics of NPM1−MN, NPM1+MN, and NPM1+AML. (A) Heatmap of commonly evaluated genes, cytogenetic findings, and total mutational counts across all cases of NPM1− MN (n = 95), NPM1+ MN (n = 45), and NPM1+ AML (n = 119). Each column represents an individual patient. All comutations (filled squares = mutation identified) and cytogenetic findings (filled squares = identified deviation from 46,XX or 46,XY) in binary format. Total mutation count per case stratified at greater than (filled square) or less than or equal to 2, by cohort as follows: NPM1− MN (46/95), NPM1+ MN (21/45), and NPM1+ AML (101/119). *Statistically significant differences in proportions between flanking groups (P < .05, Fisher’s exact test). (B) NPM1+ MN (n = 45) exhibit shorter median OS (20 months) than NPM1- MN (36.6 months, n = 95), and NPM1+ AML (42.4 months, n = 119).
Genetic and clinical outcome characteristics of NPM1−MN, NPM1+MN, and NPM1+AML. (A) Heatmap of commonly evaluated genes, cytogenetic findings, and total mutational counts across all cases of NPM1− MN (n = 95), NPM1+ MN (n = 45), and NPM1+ AML (n = 119). Each column represents an individual patient. All comutations (filled squares = mutation identified) and cytogenetic findings (filled squares = identified deviation from 46,XX or 46,XY) in binary format. Total mutation count per case stratified at greater than (filled square) or less than or equal to 2, by cohort as follows: NPM1− MN (46/95), NPM1+ MN (21/45), and NPM1+ AML (101/119). *Statistically significant differences in proportions between flanking groups (P < .05, Fisher’s exact test). (B) NPM1+ MN (n = 45) exhibit shorter median OS (20 months) than NPM1- MN (36.6 months, n = 95), and NPM1+ AML (42.4 months, n = 119).
Clinical outcome
Comparison of all 3 study cohorts revealed shorter OS for NPM1+ MN (20 months) as compared with NPM1− MN (36.6 months) and NPM1+ AML (42.4 months) (Figure 1B). Of 3 NPM1+ MN patients who received upfront induction chemotherapy, none progressed to AML, 2/3 were alive at last follow-up, and 1 died (OS, 20 months). Of 33 NPM1+ MN patients who received upfront HMA therapy, 13 (39%) progressed to AML at a median time of 5.9 months, and 12 ultimately succumbed to their disease. Five of the 13 patients who progressed subsequently received intensive chemotherapy; 3 ultimately died (OS, 20, 23, and 36 months). All 5 NPM1+ MN patients with at least 2 months of follow-up who did not receive either upfront HMA or intensive chemotherapy progressed to AML at a median time of 3 months.
In multivariable analyses performed in patients with a diagnosis of MDS only (26 NPM1+ and 60 NPM1−, n = 86), total mutation count (hazard ratio [HR], 1.3; P = .05), presence of mutations in NPM1 (HR, 3.6; P = .02), or TP53 (HR, 5.2; P = .01), and higher IPSS-R score (HR, 1.7; P = .0003) were factors independently associated with shorter OS, whereas SCT status conferred a favorable effect (HR, 0.1; P < .0001).
Discussion
In this study, we have assembled and performed a comprehensive clinicopathologic and genetic characterization of the largest known cohort of non-AML NPM1-mutated myeloid neoplasms. Most of the cases in our cohort were categorized as either high-grade MDS or chronic myelomonocytic leukemia (CMML) based on current WHO criteria. Our results confirm previously reported findings from limited case series that have correlated the presence of a somatic exon 12 NPM1 insertion mutation with relatively short latency before leukemic transformation. Interestingly, we were unable to establish a statistically significant difference in rate of leukemic transformation, or time to transformation, between our NPM1+ MN and NPM1− MN cohorts. Based on 5 patients who did not receive any upfront therapy and whose disease quickly transformed (median, 3 months), we hypothesize that upfront HMA therapy in the majority of our NPM1+ MN patients may have altered the natural biology and time course of the underlying disease, delaying what might have been more rapid progression to overt AML.
Repeat examination of immunophenotypic findings in a subset of our NPM1+ MN cases revealed heterogeneity in CD34 expression on myeloid blasts, without any clear correlation with the bone marrow blast count or specific WHO diagnostic subtype of disease (data not shown); in contrast, blasts in MDS are classically CD34+.1 The distinct genetic features of our NPM1+ MN group, with poor outcomes despite predominantly normal karyotypes and significantly fewer cooccurring ASXL1 or TP53 comutations, suggest that NPM1+ MN are indeed biologically distinct from NPM1− MN. It is additionally interesting that ASXL1 and RUNX1 mutations, although overrepresented in our NPM1− MN cohort, did not exhibit a statistically significant correlation with adverse outcome (data not shown), as has been reported in the context of AML.
Despite some differences from NPM1+ AML, including fewer IDH1/2 or FLT3 comutations and lower WBC and bone marrow cellularity at diagnosis, NPM1+ MN were associated with similar rates of abnormal karyotype and select gene mutations (eg, DNTM3A, ASXL1, RUNX1, SRSF2). These findings raise the possibility that patients with NPM1+ non-AML MN may benefit from a more intensive therapeutic approach more akin to NPM1+ AML, when appropriate. Certainly, HMA therapy, at least when coupled with venetoclax, has shown efficacy in NPM1-mutated disease27 ; however, we observed overall poor outcomes in our NPM1+ MN cohort, in which the majority of patients received upfront HMA therapy. Of note, 3 patients received upfront induction chemotherapy, and none of them progressed to AML.
Although we have assembled the largest known cohort of NPM1+ non-AML MN, this study is limited by its retrospective nature and the lack of a controlled clinical trial design. Our results might suggest that upfront HMA therapy may have been inadequate in some patients; however, OS can indeed be influenced by many factors, including natural history of the disease, sequential therapy, and SCT. Therefore, confirmation of these findings in the context of a prospective study will be important.
Authorship
Contribution: S.S.P. and O.K.W. designed the study, analyzed the data, and wrote the manuscript; C.H., R.N.P., S.S., A.B., J.T.G., M.L.X., T.P., E.F.M., A.C.S., E.A.M., D.P.S., E.S.W., W.J.W., and R.P.H. contributed patients and diagnostic data; and all authors reviewed and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Olga K. Weinberg, Department of Pathology, Boston Children’s Hospital, 300 Longwood Ave, Bader 126.2, Boston, MA 02115; e-mail: olga.weinberg@childrens.harvard.edu.

