

Key Points
AML with NPM1 mutation can relapse with NPM1wt, which could prevent MRD detection.
The majority of comutations persist in molecular remission and at NPM1wt relapse, suggestive of clonal hematopoiesis driving AML relapse.

Abstract
Acute myeloid leukemia (AML) with NPM1 mutation (NPM1mut) defines a World Health Organization entity. Absence of minimal residual disease (MRD) following induction chemotherapy is associated with an excellent prognosis. Data are conflicting on NPM1mut AML relapsing with wild-type NPM1 (NPM1wt). We analyzed 104 paired samples of NPM1mut AML patients with relapse and identified 14/104 that relapsed with NPM1wt AML. Blood counts at diagnosis differed significantly between patients with NPM1mut and NPM1wt relapse (median white blood cell count, 30 vs 3 × 109/L, P = .008; platelet count, 66 vs 128 × 109/l, P = .018). NPM1mut relapse occurred significantly earlier than NPM1wt relapse (14 vs 43 months, P = .004). At diagnosis, FLT3-ITD were more frequent in patients with NPM1mut relapse (P = .029), whereas DNMT3A mutations were more frequent in patients with NPM1wt relapse (P = .035). Sequencing analysis of paired samples at diagnosis, molecular remission, and NPM1wt relapse identified cooccurring mutations that persist from diagnosis throughout remission and at relapse, suggestive of a preexisting clonal hematopoiesis. We provide evidence that AML relapsing with NPM1wt is a distinct disease and that initial leukemia and relapse potentially arise from a premalignant clonal hematopoiesis.
Introduction
Acute myeloid leukemia (AML) with NPM1 mutation (NPM1mut) is the most common AML with normal karyotype and NPM1mut defines a World Health Organization category with good prognosis in the absence of concomitant FLT3-ITD mutations.1,2 There is conflicting data if NPM1mut AML can relapse with NPM1 wild-type (NPM1wt) disease following intensive chemotherapy.3,4 Here we report on 14/104 patients with initial NPM1mut AML that relapsed with NPM1wt AML. Analyzing those patients at diagnosis and at relapse provides evidence of molecular features suggestive of premalignant clonal hematopoiesis driving initial AML and relapse.
Patients and methods
Patients
All patients gave their written informed consent for scientific evaluations. The study was approved by the internal review board and adhered to the tenets of the Declaration of Helsinki. Between 2006 and 2017, we investigated a total of 104 intensively treated patients with NPM1mut AML who attained a complete molecular remission (CMR, absence of NPM1 transcripts; sensitivity, 0.001%) and finally had a hematologic relapse (Table 1).
Patient characteristics
| NPM1mut relapse | NPM1wt relapse | P | |
|---|---|---|---|
| Total, n | 90 | 14 | |
| Sex, n (%) | |||
| Female | 52 (58) | 7 (50) | NS |
| Male | 38 (42) | 7 (50) | NS |
| Age, median (range), y | 59 (22-82) | 62 (41-72) | NS |
| AML subtype, n (%) | |||
| M0 | 2 (2) | — | NS |
| M1 | 36 (40) | 8 (57) | NS |
| M2 | 21 (23) | 3 (21) | NS |
| M4 | 22 (23) | 2 (14) | NS |
| M5a | 1 (6) | 1 (7) | NS |
| M5b | 4 (4) | — | NS |
| M6 | 1 (1) | — | NS |
| Not specified | 4 (4) | — | NS |
| Initial blast count in BM, median (range), % | 76 (21-99) | 84 (26-90) | NS |
| Leukocyte count, median (range), ×109/L | 30 (1-213) | 3 (0.9-51) | .008 |
| Hemoglobin, median (range), g/dL | 9.4 (4-16) | 9.8 (8-13) | NS |
| Platelet count, median (range), ×109/L | 66 (9-1100) | 128 (32-267) | .018 |
| NPM1mut relapse | NPM1wt relapse | P | |
|---|---|---|---|
| Total, n | 90 | 14 | |
| Sex, n (%) | |||
| Female | 52 (58) | 7 (50) | NS |
| Male | 38 (42) | 7 (50) | NS |
| Age, median (range), y | 59 (22-82) | 62 (41-72) | NS |
| AML subtype, n (%) | |||
| M0 | 2 (2) | — | NS |
| M1 | 36 (40) | 8 (57) | NS |
| M2 | 21 (23) | 3 (21) | NS |
| M4 | 22 (23) | 2 (14) | NS |
| M5a | 1 (6) | 1 (7) | NS |
| M5b | 4 (4) | — | NS |
| M6 | 1 (1) | — | NS |
| Not specified | 4 (4) | — | NS |
| Initial blast count in BM, median (range), % | 76 (21-99) | 84 (26-90) | NS |
| Leukocyte count, median (range), ×109/L | 30 (1-213) | 3 (0.9-51) | .008 |
| Hemoglobin, median (range), g/dL | 9.4 (4-16) | 9.8 (8-13) | NS |
| Platelet count, median (range), ×109/L | 66 (9-1100) | 128 (32-267) | .018 |
BM, bone marrow; NS, not significant.
Next-generation sequencing
Unique molecular identifier based sequencing of 63 genes associated with hematological malignancies was performed at diagnosis and for patients with NPM1wt relapse in paired samples at diagnosis, CMR, and at clinical relapse. Digital error suppression was applied to follow mutations in up to 1/500 molecules as follows. In short, all samples were analyzed by a gene panel containing ASXL1, ASXL2, ATM, BCL2, BCOR, BCORL1, BIRC3, BRAF, BTK, CALR, CBL, CSF3R, CSNK1A1, CXCR4, DNMT3A, EGR2, ETNK1, ETV6, EZH2, FBXW7, FLT3, FOXO1, GATA1, GATA2, ID3, IDH1, IDH2, JAK2, KIT, KLF2, KRAS, MAP2K1, MPL, MYC, MYD88, NF1, NFKBIE, NOTCH1, NOTCH2, NPM1, NRAS, PHF6, PIGA, PLCG2, POT1, PTPN11, RAD21, RUNX1, SAMHD1, SETBP1, SF3B1, SRSF2, STAG2, STAT3, STAT5B, TCF3, TET2, TP53, U2AF1, UBR5, WT1, XPO1, and ZRSR2. The library of 63 genes was generated with a TruSeq Custom Amplicon Low Input Kit (Illumina, San Diego, CA) following the manufacturer’s protocol. The library contained molecular tags (unique molecular identifiers) that allow the detection and quantification of the individual molecule of each template DNA fragment. This tag was incorporated and sequenced, enabling the accurate detection of true variants with high resolution up to 0.2% variant allele frequency (VAF) as polymerase chain reaction duplicates can be identified and discarded. The library was sequenced and demultiplexed on a Nextseq instrument (Illumina) as described previously.5 The FASTQ files were further processed using Sequence Pilot software, version 4.3.1, Build 502 (JSI Medical Systems, Ettenheim, Germany), for alignment and variant calling. Analysis parameters were set according to the manufacturer’s default recommendation. Validity of the somatic mutations was checked against the publicly accessible COSMIC v78 (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) and ClinVar databases. Functional interpretation was performed using SIFT 1.03 (http://sift.jcvi.org) and PolyPhen 2.0 (http://genetics.bwh.harvard.edu/pph2).6 Additionally, TP53 variants were verified using the International Agency for Research on Cancer repository.7 Single-nucleotide polymorphisms were annotated according to the National Center for Biotechnology Information dbSNP (http://www.ncbi.nlm.nih.gov/snp; Build 147) and ExAC population frequency database. Variants of uncertain significance were excluded from statistical analyses. FLT3-ITD was analyzed by gene scan.
Statistical analysis
Statistical analysis was performed using Prism 7.03 software by GraphPad (La Jolla, CA). Survival differences were calculated by log-rank test and for group comparisons the χ2 test was used.
Results
We identified 14/104 patients with a hematologic relapse that was NPM1wt. At first diagnosis. there was no significant distribution of age, sex, or AML subtype in the group with NPM1mut or NPM1wt relapse. However, in the subgroup with NPM1mut relapse, we observed a significant higher white blood cell count (WBC; median, 30 vs 3 × 109/L, P = .008) and significant lower platelet count (median platelet count 66 vs 128 × 109/L, P = .018; Table 1). Bone marrow blast count and hemoglobin were comparable (Table 1).
For 90/104 patients with an NPM1mut relapse, the median time to relapse was 14 months (range, 3-77). In contrast, for the 14 patients with an NPM1wt relapse, the median time to relapse was significantly longer, at 43 months (range, 4-66, P = .004; Figure 1A). There was no significant difference in overall survival: at 2 years following relapse, overall survival was 48% (95% confidence interval, 12-77) vs 54% (14-83) for patients with NPM1mut relapse and NPM1wt relapse, respectively (NS; Figure 1B).
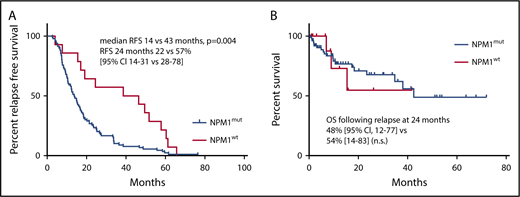
AML relapse with NPM1mutoccurs earlier than relapse with NPM1wt. (A) RFS of patients with NPM1mut relapse and NPM1wt relapse. (B) OS following relapse of patients with NPM1mut relapse and NPM1wt relapse. 95% CI, 95% confidence interval; OS, overall survival; RFS, relapse-free survival.
AML relapse with NPM1mutoccurs earlier than relapse with NPM1wt. (A) RFS of patients with NPM1mut relapse and NPM1wt relapse. (B) OS following relapse of patients with NPM1mut relapse and NPM1wt relapse. 95% CI, 95% confidence interval; OS, overall survival; RFS, relapse-free survival.
Diagnostic samples were available for sequencing in 61/90 patients with NPM1mut relapse and 11/14 patients with NPM1wt relapse. The mutational landscape of these patients revealed recurrent comutations in 18 different genes in 62/72 patients. DNMT3A, TET2, FLT3-ITD, NRAS, IDH1, IDH2, FLT3-TKD, and WT1 were mutated in >5% of cases (Figure 2). FLT3-ITD mutations were significantly more frequent in patients with NPM1mut relapse (analysis available for 104 patients, 33/90 vs 1/14, P = .029), whereas DNMT3A mutations were significantly more frequent in the cohort of patients with NPM1wt relapse (analysis available for 72 patients, 22/59 vs 9/13, P = .035). At diagnosis, a median of 2 genes per patient were comutated with NPM1 (range, 0-5); only 7/72 patients had no cooccurring mutation detected, all of which relapsed with NPM1mut AML (11% vs 0%, NS).
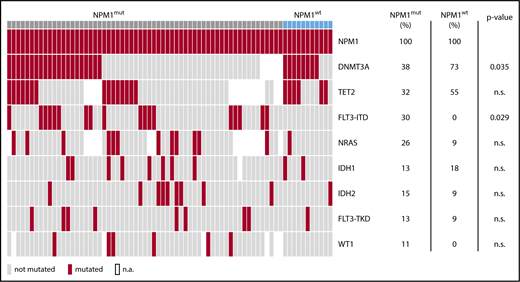
DNMT3A mutations at diagnosis are associated with NPM1wtrelapse. Cooccurring mutations with NPM1 at diagnosis of all patients. For 61/90 patients with NPM1mut relapse and for 11/14 patients with NPM1wt relapse, the diagnostic sample was available for 63 gene panel sequencing. Shown are mutations in genes affected in >10% of cases. DNMT3A mutations are significantly more frequent in patients that relapse with NPM1wt disease. FLT3-ITD mutations are significantly more frequent in patients that relapse with NPM1mut disease. n.a., not available.
DNMT3A mutations at diagnosis are associated with NPM1wtrelapse. Cooccurring mutations with NPM1 at diagnosis of all patients. For 61/90 patients with NPM1mut relapse and for 11/14 patients with NPM1wt relapse, the diagnostic sample was available for 63 gene panel sequencing. Shown are mutations in genes affected in >10% of cases. DNMT3A mutations are significantly more frequent in patients that relapse with NPM1wt disease. FLT3-ITD mutations are significantly more frequent in patients that relapse with NPM1mut disease. n.a., not available.
For the same 11/14 cases with NPM1wt relapse, the relapse samples were also available for 63 gene panel sequencing. At diagnosis, we identified a total of 30 comutations in addition to NPM1 in a total of 11 different genes in 11/11 patients (DNMT3A [n = 8], TET2 [8], SRSF2 [3], IDH1 [2]), KRAS [2], PTPN11 [2], ASXL1 [1], FLT3-TKD [1], IDH2 [1], NRAS [1], and STAG2 [1]). At relapse, we identified a total of 32 comutations in 12 different genes in 10/11 patients with a median of 3 mutations per patient (range, 0-5). An overview of mutated genes detected in each patient at diagnosis vs relapse is given in Figure 3A. The color code depicts lost and gained mutations at relapse. Twelve mutations in 8 genes (TET2, PTPN11, DNMT3A, FLT3-TKD, KRAS, NRAS, SRSF2, and STAG2) were lost at relapse and 14 mutations in 9 genes (TP53, RUNX1, IDH1, IDH2, NF1, NRAS, SRSF2, TET2, and WT1) were gained at relapse. We used digital error correction to reassess these mutations in the diagnostic sample. With a detection limit of up to 0.2% VAF (1/500 molecules), none of the newly gained mutations was detectable at diagnosis. Interestingly, 18 mutations in 7 genes persisted over time, with a median of 2 persistent mutations per patient (range, 0-4; Figure 3B): at relapse, we observed persistent mutations in DNMT3A, TET2, IDH1, SRSF2, ASXL1, IDH2, and KRAS (Figure 3C). DNMT3A, the most prevalent mutation, was lost in only 1 patient (8/11 at diagnosis, 7/11 at relapse), whereas TET2 mutations persisted in 3 patients, IDH1 and SRSF2 mutations in 2 patients (Figure 3C, right) and ASXL1, IDH2, and KRAS mutations in 1 patient. When persistent, the VAF of mutated genes was generally higher at relapse than at diagnosis; however, because of low numbers, no statistical significance was observed for single genes (Figure 3D and Figure 4C). Interestingly, the VAF at diagnosis was lowest for ASXL1, IDH2, and KRAS.
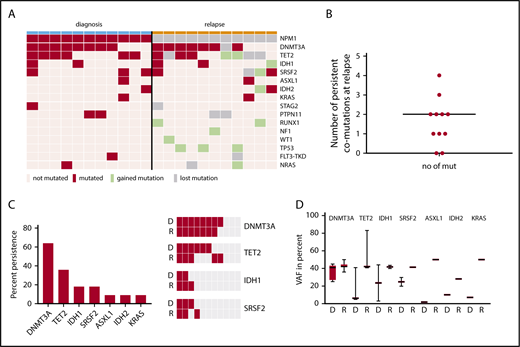
NPM1wtrelapse is associated with persistent mutations. (A) For 11/14 patients with NPM1wt relapse, paired samples at diagnosis and relapse were available for sequencing. Shown are genes mutated at least once in the analyzed samples. Genes with >1 mutation/patient are shown once. (B) Number of persistent mutations at relapse; median, 2 (range, 0-4). (C) Left: mutations in 7 genes detected at diagnosis were found to persist at relapse. Fraction of persistent mutations in relation to all 11 patients is shown. Right: The 4 most frequently mutated genes at D and R are shown as heat maps to visualize persistence of mutations. DNMT3A is the most prevalent mutation and also the most stable mutation over time. (D) The variant allele frequency of the indicated genes is given at D and R. D, diagnosis; R, relapse.
NPM1wtrelapse is associated with persistent mutations. (A) For 11/14 patients with NPM1wt relapse, paired samples at diagnosis and relapse were available for sequencing. Shown are genes mutated at least once in the analyzed samples. Genes with >1 mutation/patient are shown once. (B) Number of persistent mutations at relapse; median, 2 (range, 0-4). (C) Left: mutations in 7 genes detected at diagnosis were found to persist at relapse. Fraction of persistent mutations in relation to all 11 patients is shown. Right: The 4 most frequently mutated genes at D and R are shown as heat maps to visualize persistence of mutations. DNMT3A is the most prevalent mutation and also the most stable mutation over time. (D) The variant allele frequency of the indicated genes is given at D and R. D, diagnosis; R, relapse.
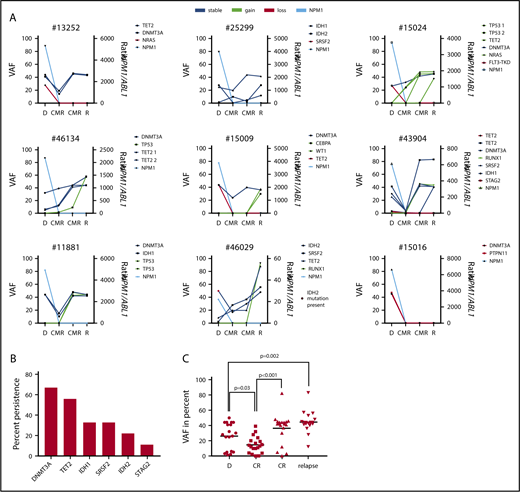
Premalignant clonal hematopoiesis underlies NPM1wtrelapse. (A) For 9/11 patients, 2 remission samples/patient were available for sequencing analysis. Eights of 9 patients showed persistent mutations in CMR and 4 patients acquired additional mutations before the onset of relapse. (B) Mutations in 6 genes detected at diagnosis were found to persist in remission. Shown is the fraction of persistent mutations in relation to all 9 patients. (C) VAF of persisting mutations at diagnosis, CMR, and relapse. Black line, median; CR, complete remission.
Premalignant clonal hematopoiesis underlies NPM1wtrelapse. (A) For 9/11 patients, 2 remission samples/patient were available for sequencing analysis. Eights of 9 patients showed persistent mutations in CMR and 4 patients acquired additional mutations before the onset of relapse. (B) Mutations in 6 genes detected at diagnosis were found to persist in remission. Shown is the fraction of persistent mutations in relation to all 9 patients. (C) VAF of persisting mutations at diagnosis, CMR, and relapse. Black line, median; CR, complete remission.
The high number of persistent mutations raised the possibility of the presence of clonal hematopoiesis as a basis for both initial AML and subsequent relapse. We addressed this point by analyzing remission samples of patients with NPM1wt relapse. For 9/11 patients analyzed at diagnosis and relapse, 2 remission samples/patient were available for sequencing. The progression of individual mutations is shown in Figure 4A. In molecular remission, we detected persistence of comutations (DNMT3A [n = 6], TET2 [5], IDH1 [3], SRSF2 [3], IDH2 [2], and STAG2 [1]) in 8/9 patients (Figure 4B). Only 1 patient showed a loss of all comutations (DNMT3A and PTPN11) at CMR and at relapse. Four patients acquired additional comutations in remission (TP53 [n = 5], TET2 [1], and RUNX1 [1]; Figure 4A). The median VAF of persistent mutations was significantly reduced in the first CMR sample and increased significantly in the second CMR sample and at relapse (Figure 4C).
A recent report showed that a high allelic NPM1 burden was associated with inferior outcome in de novo AML8 ; therefore, we analyzed the allelic burden of NPM1 mutations. NPM1 was sequenced by next-generation sequencing in 13 patients in the cohort with NPM1mut relapse and in 4 patients with NPM1wt relapse. We did not observe a significant difference of NPM1 VAF in the 2 subgroups (Figure 5A). Moreover, we analyzed the VAF of DNMT3A, TET2, and FLT3-TKD at diagnosis and relapse and did not find a significant difference in the 2 subgroups (Figure 5A).
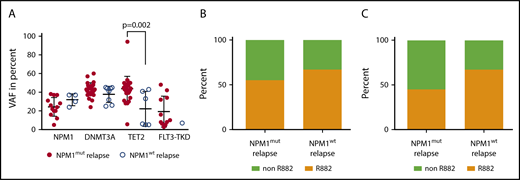
NPM1mutallelic burden and DNMT3A mutation type is not associated with NPM1wtrelapse. (A) The variant allele fraction of NPM1, DNMT3A, TET2, and FLT3-TKD mutations at diagnosis of the indicated cohort. (B) The fraction of DNMT3A R882 mutations at diagnosis for the indicated cohort (n = 22 NPM1mut relapse, n = 9 NPM1wt relapse, NS). (C) At CMR, 11/15 patients in the cohort with NPM1mut relapse showed a persistent DNMT3A mutation and 6/9 in the cohort with NPM1wt relapse. Shown is the fraction of persistent DNMT3A R882 mutations in the respective cohort (NS).
NPM1mutallelic burden and DNMT3A mutation type is not associated with NPM1wtrelapse. (A) The variant allele fraction of NPM1, DNMT3A, TET2, and FLT3-TKD mutations at diagnosis of the indicated cohort. (B) The fraction of DNMT3A R882 mutations at diagnosis for the indicated cohort (n = 22 NPM1mut relapse, n = 9 NPM1wt relapse, NS). (C) At CMR, 11/15 patients in the cohort with NPM1mut relapse showed a persistent DNMT3A mutation and 6/9 in the cohort with NPM1wt relapse. Shown is the fraction of persistent DNMT3A R882 mutations in the respective cohort (NS).
As a control, we analyzed the persistence of the most common comutations in the cohort with NPM1mut relapse. Fifteen of 22 patients that had a DNMT3A mutation at diagnosis were evaluable for DNMT3A mutations at CMR: 11 of those 15 patients had a persistent DNMT3A mutation in CMR (73%). At diagnosis, the frequency of R882 mutations were comparable in the cohorts with NPM1mut and NPM1wt relapse: 55% vs 67%, respectively (NS; Figure 5B). We also analyzed the frequency of persistent R882 mutations in CMR: 45% in the NPM1mut and 67% in the NPM1wt cohort had a persistent R882 mutation (NS; Figure 5C). Moreover, at CMR, 56% of patients had a persistent TET2 mutation (n = 9 patients with initial positivity and analyzed CMR sample) and 60% had a persistent IDH1 (n = 5) mutation and 14% a persistent IDH2 (n = 7) mutation. No patient had a detectable persistent NRAS (n = 7), FLT3-ITD (n = 28), or FLT3-TKD (n = 9) mutation at CMR.
Discussion
We identified 14% of NPM1mut AML patients who achieved CMR following intensive therapy and relapsed with an NPM1wt AML. To our knowledge, this is the largest cohort of NPM1wt AML relapse that has been studied.
In our analysis, NPM1mut relapse occurred significantly earlier than NPM1wt relapse. These findings are in line with a previous study reporting on 10% of NPM1wt relapse with a longer latency.3 Interestingly, in our cohort, patients that relapsed with NPM1wt disease had distinct clinical and molecular features at diagnosis, with significantly lower WBC counts and higher platelet counts, a higher frequency of DNMT3A mutations, and a lower frequency of FLT3-ITD mutations. All patients were treated with induction and consolidation therapy and there was no difference in treatment intensity in the 2 subgroups.
At diagnosis, DNMT3A mutations were the most frequently observed comutations across all patients and significantly more frequent in patients who later relapsed with NPM1wt. DNMT3A was assessed in only 70% of all patients at diagnosis (54% in the NPM1mut relapse; 100% in the NMP1wt relapse cohort), making this observation less reliable. However, DNMT3A mutations most frequently persisted in CMR, and the DNMT3A mutation was lost at relapse in only 1/8 patients. Interestingly, DNMT3A was also the most frequent mutation in the NPM1mut relapse cohort at diagnosis and persisted frequently during remission in this group. Mutated DNMT3A at diagnosis has been described in AML and is only moderately associated with outcome.9 The persistence of DNMT3A mutations in remission has also been addressed by several groups, and was not associated with clinical outcome.10-12 However, the landmark analysis that defined clonal hematopoiesis reported DNMT3A as the most frequently mutated gene.13,14 These analyses finally laid the basis for the definition of clonal hematopoiesis of indeterminate potential (CHIP).15 A recent report investigated persistent mutations at remission of AML patients and showed that DTA mutations (mutations in DNMT3A, TET2, and ASXL1) in remission were not associated with an increased risk of relapse. Interestingly, those mutations are all associated with CHIP or age-related clonal hematopoiesis.16 The biologic role of DTA mutations in leukemogenesis is still under investigation. Preclinical work shows that early purified stem cells in NPM1 mutated AML wild-type for NPM1 carry DNMT3A mutations and display an increased repopulating activity.17 What is more, Single-nucleotide polymorphism array analysis identified that DNMT3A was part of the most commonly focally deleted region in AML, underscoring its role in leukemogenesis.18 Because persistence of somatic mutations per se is associated with relapse,19,20 persistent clonal hematopoiesis in remission might increase repopulating activity sufficient to trigger a treatment related disease or relapse with a preexisting leukemic clone.
Although CHIP does not define a disease entity, there is growing evidence that certain mutations confer a high risk to develop hematologic malignancies.21 The biologic role of mutant DNMT3A or other mutations in NPM1mut AML might therefore cause a clonal hematopoiesis that renders progenitor cells susceptible to transformation. In line with this notion is our observation of persistent mutations in remission and relapse that identify clonal hematopoiesis in the majority of patients. The rising VAF and acquisition of mutations in 4 patients (ie, TP53; Figure 2A) in CMR prove clonal evolution and raise the possibility that chemotherapy could have induced these novel mutations. Moreover, the loss of NPM1 mutation at relapse shows that NPM1 is not the sole driver in NPM1-mutated AML. This is supported by various preclinical models in which transgenic animals and knock-in animals develop a myeloproliferative phenotype or full AML only with a long latency.22-24 In this context, other typical driver mutations such as FLT3 or NRAS were not observed in remission in either cohort. Taken together, this could indicate that those driver mutations are second hits required for NPM1-driven leukemogenesis. Supportive preclinical evidence shows that only compound mice carrying NPM1 and FLT3 or NPM1 and NRAS mutations develop AML with high penetrance and short latency.25 This is especially interesting because we observed a strong enrichment for FLT3-ITD mutations in the cohort of patients that had NPM1mut relapse, indicating that 2 driver mutations cause relapse with the initial aggressive clone. In the same line is our observation that high WBC and low platelet counts, both indicative of highly active disease, are associated with NPM1mut AML relapse. Two reports describe patients with NPM1mut AML that develop NPM1wt myelodysplastic or myelodysplastic/myeloproliferative neoplasms following CMR.26,27 Together with our data, there is growing evidence that clonal hematopoiesis is an underlying phenomenon in this AML entity. Our data expand these findings and suggest that relapse or therapy-related leukemia can arise from preleukemic clones with wild-type NPM1. Recent work established that relapse is driven by drug-resistant AML stem cells.28 If this is the case, mutations present at diagnosis, remission, and relapse should also be present in these stem cells. Leukemia would then arise from a stem cell that drives a premalignant clonal hematopoiesis and give rise to initial leukemia and relapse. Although the NPM1wt relapses occurred significantly later than NPM1mut relapses, we observed some early relapses in the NPM1wt cohort. One of the patients with the earliest relapse (7.7 months, no material available for the patient with a relapse 4.1 months after diagnosis) lost 3 comutations at relapse (KRAS, SRSF2, TET2) and the NPM1 mutation, which could either be clonal evolution of a preexisting clone or argue for a treatment-related disease. DNMT3A mutations are the most common comutations in NPM1mut AML and have also been shown to persist in remission.4,10 Therefore, we analyzed DNMT3A mutations in the cohort of patients with NPM1mut relapse as control. Persistent DNMT3A mutations were detected in 73% of patients in remission. Moreover, TET2 and IDH1 mutations persisted frequently in remission; therefore, clonal hematopoiesis might also contribute to NPM1mut relapse. Interestingly, FLT3-ITD, FLT3-TKD, and NRAS mutations were not observed in CMR in either the cohort with NPM1mut or NPM1wt relapse. Although regularly analyzed in remission, the benefit of minimal residual disease based on those markers is therefore questionable in NPM1-mutated AML. To analyze the cellular compartment from which the NPM1 clone arises and is eventually lost at relapse warrants cell sorting and single-cell sequencing analysis in a prospective manner.
Previous work showed that high allelic NPM1 burden at diagnosis is a negative prognostic factor.8 In our analysis, NPM1 allelic burden was not associated with NPM1wt or NPM1mut relapse. However NPM1 is not routinely analyzed by next-generation sequencing; therefore, the number of evaluable patients for NPM1 VAF is relatively low in our analysis. The DNMT3A mutation subtype has frequently been queried for an association with prognosis, and a recent meta-analysis showed that DNMT3A R882 mutations are significantly associated with reduced relapse-free and overall survival.29 In our analysis, the R882 mutations were slightly more prevalent in the subgroup with NPM1wt relapse; however, this trend was not significant. This could also be due to the low patient number.
Taken together, our data suggest that, in NPM1mut AML, the persistence of mutations defines premalignant clonal hematopoiesis and drives a treatment-related leukemia or relapse (Figure 6). Although we show that a proportion of patients with NPM1mut AML lose the NPM1 mutation at relapse, for the majority of patients, minimal residual disease monitoring by NPM1 quantitative polymerase chain reaction is absolutely mandatory. Although it is not feasible to monitor all NPM1+ AML patients longitudinally with large sequencing panels, there is increasing evidence that supports the screening for persistent mutations in at least 1 remission sample.
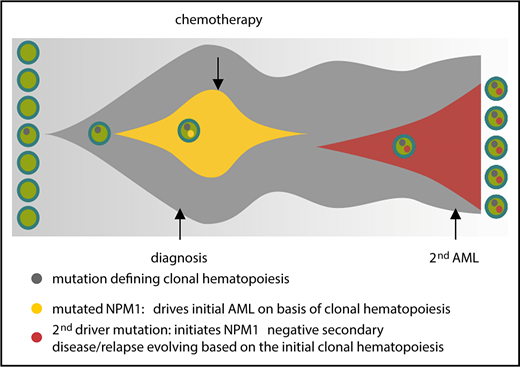
Model for clonal hematopoiesis driving relapse or secondary leukemia. The first leukemia is cured by chemotherapy based on the underlying clonal hematopoiesis; a second driver mutation than leads to overt relapse.
Model for clonal hematopoiesis driving relapse or secondary leukemia. The first leukemia is cured by chemotherapy based on the underlying clonal hematopoiesis; a second driver mutation than leads to overt relapse.
Presented in part in abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9 December 2017.
Acknowledgments
The authors thank all patients and clinicians for their participation in this study and all coworkers in our laboratory for their excellent technical assistance.
Authorship
Contribution: A.H. and T.H. designed the study; A.H. interpreted the data and wrote the manuscript; S.J., M.M., F.D., and N.N. did molecular analyses; T.H. was responsible for cytomorphologic analyses; C.H. was responsible for cytogenetic and fluorescence in situ hybridization analyses; W.K. was responsible for immunophenotyping; and all authors read and contributed to the final version of the manuscript.
Conflict-of-interest disclosure: A.H., M.M., F.D., S.J., and N.N. are employed by MLL Munich Leukemia Laboratory. C.H., W.K., and T.H. have equity ownership of MLL Munich Leukemia Laboratory.
Correspondence: Torsten Haferlach, MLL Munich Leukemia Laboratory, Max-Lebsche-Platz 31, 81377 Munich, Germany; e-mail: torsten.haferlach@mll.com.

