Key Points
An unbiased transcriptomic analysis revealed a unifying signature across IBMF syndromes.
Specifically, we observed an upregulation of transcripts coding for ribosome, nonsense-mediated decay, and redox metabolism pathways.

Abstract
Gene expression profiling has long been used in understanding the contribution of genes and related pathways in disease pathogenesis and susceptibility. We have performed whole-blood transcriptomic profiling in a subset of patients with inherited bone marrow failure (IBMF) whose diseases are clinically and genetically characterized as Fanconi anemia (FA), Shwachman-Diamond syndrome (SDS), and dyskeratosis congenita (DC). We hypothesized that annotating whole-blood transcripts genome wide will aid in understanding the complexity of gene regulation across these IBMF subtypes. Initial analysis of these blood-derived transcriptomes revealed significant skewing toward upregulated genes in patients with FA when compared with controls. Patients with SDS or DC also showed similar skewing profiles in their transcriptional status revealing a common pattern across these different IBMF subtypes. Gene set enrichment analysis revealed shared pathways involved in protein translation and elongation (ribosome constituents), RNA metabolism (nonsense-mediated decay), and mitochondrial function (electron transport chain). We further identified a discovery set of 26 upregulated genes at stringent cutoff (false discovery rate < 0.05) that appeared as a unified signature across the IBMF subtypes. Subsequent transcriptomic analysis on genetically uncharacterized patients with BMF revealed a striking overlap of genes, including 22 from the discovery set, which indicates a unified transcriptional drive across the classic (FA, SDS, and DC) and uncharacterized BMF subtypes. This study has relevance in disease pathogenesis, for example, in explaining the features (including the BMF) common to all patients with IBMF and suggests harnessing this transcriptional signature for patient benefit.
Introduction
Inherited bone marrow failure (IBMF) is a heterogeneous life-threatening illness characterized by the inability of the patients’ marrow to produce an adequate number of blood cells.1 Recognized IBMF syndromes include Fanconi anemia (FA),2 Shwachman-Diamond syndrome (SDS),3 and dyskeratosis congenita (DC)4 among others. These IBMF syndromes are characterized by genetic lesions that perturb DNA repair in FA, ribosome biogenesis in SDS, and telomere maintenance in DC.5 Defects in hematopoietic stem cell (HSC) renewal that affect blood cell turnover with increased predisposition to blood cancers such as myelodysplastic syndrome (MDS) and acute myeloid leukemia are consistent features of these syndromes.6 Most of the encoded proteins that are mutated in IBMF syndromes seem to be multifunctional, operating in diverse molecular pathways with considerable overlap and interplay between these pathways.7,8
Much has been described in terms of the proximal molecular events in these disorders. Less is understood about the link between these mechanistic events and the clinical presentation. To try to bridge this gap, we have analyzed the transcriptomic profiles of peripheral blood from genetic subtypes of patients with IBMF. Whole-blood gene expression profiling represents an unbiased method for identifying and investigating pathways associated with disease states. Next-generation sequencing platforms adapted for RNA sequencing (RNA-seq) studies have emerged to robustly and efficiently study changes associated with transcriptomic profiles with greater precision and read depth. We hypothesized that this technique would provide unique insight into mechanistic pathways that either overlap or differ between the different genetic subtypes of IBMF syndromes and may therefore direct future study or clinical intervention.
Methods
Patient selection
All patients recruited into this study attended outpatient clinics at The Royal London Hospital (Barts Health National Health Service Trust). Control samples from healthy volunteers were collected contemporaneously. All samples were obtained with written informed consent under the approval of our local research ethics committee (London-City and East). Blood counts and extra hematopoietic features were noted at the time of sampling for all patients involved (Table 1; supplemental Table 1). The study initially focused on a discovery set of patients presenting with FA, SDS, or DC, with mutations in the FANCA and FANCG, SBDS, and DKC1 genes, respectively, with the underlying genetic variant(s) being determined before venesection (Table 1). Six genetically uncharacterized patients were also included. These uncharacterized patients had previously been screened for variants in the known BMF-causing genes using an in-house custom capture panel of 111 disease genes (supplemental Data; supplemental Table 2) and all had a negative result with the chromosome breakage test. They were grouped according to their blood counts as either TRI (trilineage cytopenia with limited extra hematopoietic features) or SNGL (single-lineage cytopeniawith extra hematopoietic features).
PAX Tube blood collection and RNA isolation
Blood was collected in PAXgene Blood RNA Tubes (PreAnalytiX) according to manufacturer’s instructions because this system has been shown to reduce RNA degradation and to inhibit or eliminate RNA induction at the point of sampling.9 Total RNA was extracted from the sample using the PAXgene Blood RNA Kit (QIAGEN). RNA integrity was assessed by using an Agilent 2100 BioAnalyzer with the RNA 6000 Nano Kit (Agilent Technologies, Palo Alto, CA), which provides RNA integrity number (RIN) scores for RNA quality control. A spectrophotometer reading of A260/280 ≥ 1.8 (range, 1.8-2.2) indicates good RNA purity, and an RIN score ≥ 7.0 demonstrates good RNA integrity; only samples that exceeded these thresholds were included in this study. RNA quantity was measured using a Qubit fluorometer.
Library preparation
Libraries were prepared from 500-ng high-quality total RNA using the NEBNext Ultra II directional RNA Prep Kit for Illumina (#E7760; New England Biolabs) with the NEBNext Poly(A) messenger RNA magnetic isolation module (#E7490; New England Biolabs). For sequencing, the libraries were pooled to equimolar amounts and run on the Novoseq sequencing platform. To check the reproducibility of the whole pipeline, a technical replicate was performed (details are provided in the supplemental Data and supplemental Figure 1).
Data analysis
The data were processed through an analysis pipeline using Partek Flow software (build version 6.0.17.0614; Partek, St. Louis, MO) with the following task nodes (non-default parameters are specified in square brackets): Trim bases [both ends with quality encoding Phred+33], align reads using STAR 2.6.1d quantify to transcriptome [Partek E/M using hg38-Ensembl Transcript release 93 as the reference index, with strand specificity auto detect and minimum reads of 20]. The aligned data were further analyzed by using the analysis features of Partek Flow software. Comparisons between the different groups were set up by first filtering the data for each analysis and then performing differential expression analysis using DESeq2 by comparing 3 patients to 3 controls. Significant differential expression was defined as false discovery rate (FDR) adjusted P < .05. Gene sets was analyzed by using gene set enrichment analysis (GSEA; Broad Institute10 ). The whole gene list from each analysis group (FANC, SBDS, DKC1, TRI, and SNGL) was used, and genes were ranked in descending order of fold change (FC) after removing those genes that failed to return an FDR value. GSEA was carried out using the pre-ranked module that had a classic scoring system with KEGG v6.2 and Reactome v6.2 gene sets as references. The Protein Analysis Through Evolutionary Relationships classification system (PANTHER11 ; http://pantherdb.org) was used to perform pathway analysis for lists of specific dysregulated genes.
Validation using quantitative polymerase chain reaction
Complementary DNA was prepared from total RNA using Invitrogen Superscript IV according to the manufacturer’s instructions. A pool of random control complementary DNAs was prepared and serially diluted to form a relative standard curve against which all samples were quantitated. Primers for the genes of interest and the control genes were ordered from the Sigma KiCqStart Sybr green primer range. For each gene, 4 replicates per sample were run on the Roche Lightcycler 480 system. All primers had an amplification efficiency between 90% and 110%. Each gene of interest was normalized against the geometric mean of 3 control genes (UBC, GAPDH, and CSNK2B), and FC between patients and controls was calculated.
Gene selection for signature sets
A stepwise filtering approach was used for all of the group-specific dysregulated genes (both upregulated and downregulated; FDR < 0.05) to obtain a robust list of genes in which the resultant genes had good read depth, were significantly dysregulated, and had a high FC difference that represented each specific disease. First, we removed all genes with a total coverage count <50. Second, we sorted by FDR and selected the top 100 genes. Third, we obtained genes that were group-specific (eg, FANC), and all genes with an absolute FC < 2 were rejected. Fourth, this resultant list was compared with the lists for the other groups, and genes with FC > 2 in the other groups (SBDS and DKC1) were rejected. Fifth, we rejected any genes that were cell type–specific to avoid confounding effects of differences in blood counts. Sixth, the top 30 genes (15 upregulated and 15 downregulated) were selected as being the signature.
Results
Whole-blood transcriptomic signature of FA reveals a skewing toward upregulation
To investigate any potential transcriptome-wide effect of known genetic mutations, RNA-seq analysis was performed on a group of three patients with genetically characterized FA and 3 unrelated healthy controls. Differential expression analysis using DESeq2 showed that there were 875 dysregulated genes with an FDR < 0.05, and of these, 710 were upregulated and 165 were downregulated (Figure 1A; supplemental Table 3). To validate the data, we selected for analysis a small subset of genes relevant to hematopoiesis by using quantitative polymerase chain reaction (qPCR). After normalizing the expression to 3 control genes, we confirmed that the relative levels in the patients were as predicted by the RNA-seq results (Figure 1B). The difference in expression levels as determined by FC detected by qPCR from total RNA correlated very well with the FC recorded in messenger RNA by RNA-seq (supplemental Figure 1D). These data demonstrate a high degree of gene expression dysregulation in the peripheral blood of patients with FA with a highly significant (P < .00001) skewing in favor of upregulated genes.
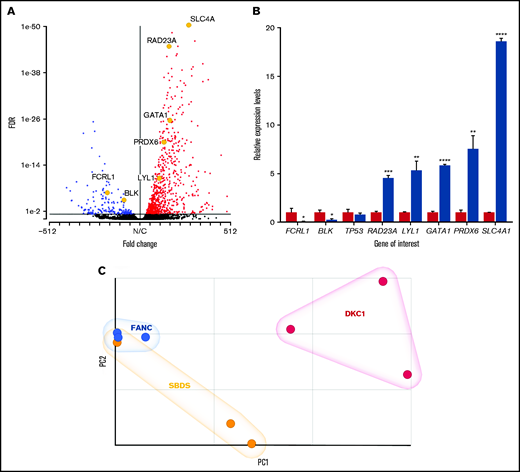
Differential expression in patients with FA. (A) Volcano plot showing a skewed upregulation of genes in patients compared with controls (red dots, genes with a positive FC; blue dots, genes with a negative FC; black dots, genes showing no significant dysregulation at FDR < 0.05. The significantly dysregulated genes analyzed in panel B are highlighted for comparison. (B) Relative expression of a selection of dysregulated genes measured by qPCR in total RNA. Error bars represent standard deviation of the relative expression for the sample groups (either 3 controls or 3 patients). (C) PCA plot for FANC, SBDS, and DKC1 patient groups (blue spheres, FANC; yellow spheres, SBDS; red spheres, DKC1). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Differential expression in patients with FA. (A) Volcano plot showing a skewed upregulation of genes in patients compared with controls (red dots, genes with a positive FC; blue dots, genes with a negative FC; black dots, genes showing no significant dysregulation at FDR < 0.05. The significantly dysregulated genes analyzed in panel B are highlighted for comparison. (B) Relative expression of a selection of dysregulated genes measured by qPCR in total RNA. Error bars represent standard deviation of the relative expression for the sample groups (either 3 controls or 3 patients). (C) PCA plot for FANC, SBDS, and DKC1 patient groups (blue spheres, FANC; yellow spheres, SBDS; red spheres, DKC1). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Similar patterns of dysregulation across different subtypes of IBMF
Having established that a significant number of genes were differentially expressed in patients with FA, we expanded our analysis to include patients who had characterized variants in other genes known to be involved in the IBMF syndromes. The genes selected were SBDS and DKC1. SBDS is the main gene responsible for SDS and is also involved in ribosomal RNA processing and ribosome subunit association.3 DKC1 is one of the key genes responsible for DC and is involved in telomere biology as well as ribosomal RNA processing.12 Samples from 3 patients with SBDS variants and 3 patients with DKC1 variants were assayed (Table 1). Differential expression analysis showed that 452 genes were dysregulated in the SBDS group and 424 genes were dysregulated in the DKC1 group when compared with a set of 3 control samples (supplemental Table 3). The pattern of dysregulation as shown by the volcano plots is also skewed toward upregulation as observed with patients who have FA. However, this is less pronounced because of the reduced number of dysregulated genes observed (Table 2; supplemental Figure 2). Principal component analysis (PCA) showed that all of the patients separate from the controls (supplemental Figure 3) and illustrates the difference between the patient groups (Figure 1C). The patients with FA are more tightly clustered together than those presenting with either SDS or DC. This implies that more variability exists between the patients in the SBDS and DKC1 groups and results in a reduced number of differentially expressed genes. There is also an increased degree of separation between the patients with DC and those presenting with either FA or SDS. This may reflect the blood counts observed between the 3 groups because the white blood cell count and neutrophil levels are both lower than normal in the patients with FA or SDS compared with the patients who had DC (supplemental Table 1). These results show that even when using a small number of well-characterized, genetically defined patients, it is possible to use transcriptomics to demonstrate significant dysregulation compared with that in a set of healthy controls. It was also possible to observe some differences between the patient groups, and further investigation may elucidate pathways or mechanisms that may explain these differences.
GSEA identifies shared pathways
To examine which pathways were dysregulated, we performed GSEA to search for sets of significantly overrepresented genes that function together in known biological pathways. To analyze the pathways obtained by GSEA and determine which might be biologically relevant, we used a more stringent cutoff of a family-wise error rate <0.05, which further limits the probability of false discovery. The resulting pathways were then ordered and ranked by the normalized expression score. Although the downregulated pathways did not suggest a unified picture because of a very limited number of pathways in the SBDS group (n = 4) that reached this cutoff, there was an unexpected preponderance of pathways involved in ribosome structure and function among the upregulated gene sets from the FANC, SBDS, and DKC1 groups (Table 3; supplemental Table 4). Indeed, 6 of 17 of the top shared pathways of the GSEA are involved in protein translation. Additional upregulated gene sets include those involved in nonsense-mediated decay (NMD), RNA metabolism, and redox-related bio-energetic pathways. Together, the upregulation of protein translation, NMD, and redox metabolism pathways at the transcriptional level suggests a stress-related response resulting from genomic instability, which is a feature of the groups under investigation.13-15 These data show that there is an overlap of the upregulated pathways across all 3 subgroups, irrespective of the underlying genetic cause.
With respect to the downregulated pathways, there was no shared overlap among the 3 subtypes. However, there were 3 pathways associated with interferon signaling common to the patients with FANC or DKC1 and 3 pathways associated with ligand and receptor binding common to patients with SBDS or DKC1. Nothing was shared between the FANC and SBDS groups. Together, these results indicate a shared positive drive to the transcriptome in the peripheral blood of patients with IBMF but little commonality among the pathways that are downregulated.
Shared and specifically dysregulated genes among different IBMF syndromes
Having identified shared dysregulated pathways among the 3 classic IBMF subtypes (FA, SDS, and DC), we wanted to identify the specific genes that were responsible for these signals. Of the 4 disease-causing genes in our patient groups, only FANCA showed significant dysregulation in the FANC analysis (FDR, 0.015; FC, −2.4). Neither FANCG, SBDS, nor DKC1 were significantly dysregulated in any of the analyses performed. We then intersected the lists of differentially expressed genes for each subtype, focusing on those that were significantly dysregulated (FDR < 0.05; Figure 2). Of the genes that were significantly dysregulated, we found that 31 genes were shared by all 3 subtypes: 26 were upregulated and 5 were downregulated (Figure 2A-B; supplemental Tables 5 and 6). Among the upregulated genes, 4 are involved in mitochondrial function. However, the standout unifying feature is the presence of a set of 9 genes encoding ribosomal proteins (Figure 2C).
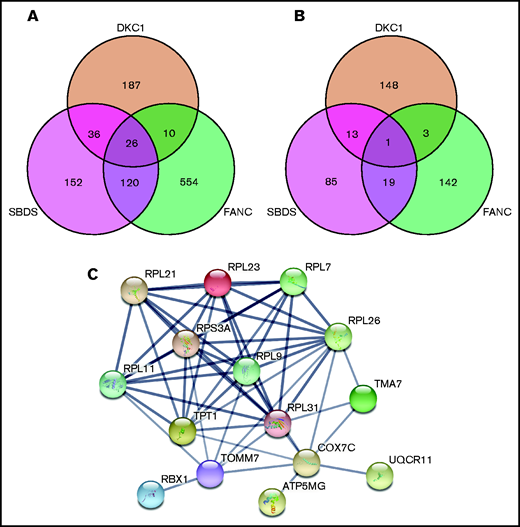
Three-way Venn diagrams detailing the overlap between significantly dysregulated genes in patients with FA, SDS, and DC. Intersections between gene lists from DESeq2 analysis for each patient group in which FDR < 0.05. The Venn diagrams show the intersections between genes that are either all upregulated (A) or all downregulated (B). (C) String analysis of the 26 shared upregulated genes shows strong functional and physical association proteins encoded by the ribosome protein genes as well as those involved with mitochondrial function. Confidence in the interactions was set as high (>0.7) based on curated experimental data and co-expression studies. Only those genes with an interaction are shown.
Three-way Venn diagrams detailing the overlap between significantly dysregulated genes in patients with FA, SDS, and DC. Intersections between gene lists from DESeq2 analysis for each patient group in which FDR < 0.05. The Venn diagrams show the intersections between genes that are either all upregulated (A) or all downregulated (B). (C) String analysis of the 26 shared upregulated genes shows strong functional and physical association proteins encoded by the ribosome protein genes as well as those involved with mitochondrial function. Confidence in the interactions was set as high (>0.7) based on curated experimental data and co-expression studies. Only those genes with an interaction are shown.
PANTHER analysis of the 26 shared upregulated genes showed that there were 7 pathways from the gene ontology (GO) biological process and molecular function annotation sets with an FDR < 0.001 (Table 4; supplemental Table 7), all of which are related to translation. These pathways include translation initiation, cytoplasmic translation, SRP-dependent co-translational protein targeting to the membrane, and structural component of the ribosome. Nine genes, or a subset thereof, are responsible for the signal in each case, all of which encode ribosomal proteins (RPL7, RPL9, RPL11, RPL21, RPL23, RPL26, RPL31, RPL41, and RPS3A). It is intriguing that 8 of 9 of these genes encode components of the 60S subunit. String analysis of proteins encoded by these genes shows the strong association between these ribosomal proteins but also includes additional proteins that are associated with mitochondrial energy production (Figure 2C). Together, these data indicate that the most prominent feature shared across the transcriptome from peripheral blood in patients with FA, SDS, or DC involves the upregulation of ribosome biogenesis.
We also applied PANTHER analysis to the genes in which the dysregulation was specific to each of the disease subtypes (supplemental Table 7) and focused on GO biological processes and molecular functions with a stringent FDR of <0.001. In the FANC set, we did not observe anything at this significance among the upregulated pathways. However, among the FANC-specific downregulated genes, we saw 14 biological processes and 2 molecular functions that have FDR values of <1 × 10−20. These pathways are associated with signaling and second messenger production as part of the immune system (supplemental Table 7). Strikingly, more than a third (52 of 142) of the downregulated genes were immunoglobulins (Ig’s) involving both heavy and light chains and all isotypes (supplemental Table 6). These data agree with previous studies that reported lower Ig levels (IgG, IgA, and IgM) in patients with FA as a result of impaired V-D-J recombination and class switching events in B cells.10 Clinically, this result may not be surprising because all the patients with FANC have exceptionally low B-cell counts as recorded by the CD19+ levels (supplemental Table 1).
In the DKC1-specific upregulated genes, there are 24 biological processes and 4 molecular function groups that are significantly upregulated. We see a considerable strengthening of the upregulation of ribosomal constituents as well as other RNA binding proteins and complement activation (supplemental Table 7). This is a result of the presence of 19 ribosomal protein genes as well as 29 Ig-related genes. By contrast, among the significantly upregulated genes specific to the SBDS data set, PANTHER analysis revealed 15 genes involved in neutrophil degranulation, possibly reflecting a compensatory mechanism for the neutropenia that is commonly observed in these patients. However, PANTHER analysis does not reveal any pathways among the specifically downregulated genes for either the SBDS or DKC1 groups.
One striking result is the overlap between the biological processes observed between the FANC-specific downregulated genes and the DKC1-specific upregulated genes. Fourteen of the GO terms listed are common to both gene groups, and 2 of the molecular functions are also shared (supplemental Table 7). Twenty of the genes involved are the same, but they are dysregulated in the opposite direction (ie, downregulated in FANC but upregulated in DKC1). This could be the result of the exceptionally low levels of B cells reported in the FANC patient group (supplemental Table 1).
Identification of a disease-specific signature
In addition to identifying a shared BMF signature, we were also interested in trying to define a signature of dysregulated genes (both upregulated and downregulated) that was specific to each of the 3 characterized groups. Figure 3 shows heat maps of the 30 genes (15 upregulated and 15 downregulated) selected for each of the patient groups that were significantly dysregulated (FDR < 0.05; absolute FC > 2) that were expressed in multiple tissues and were not specific for blood cell type. Interestingly, for the FANC upregulated signature genes, 13 of 15 showed some degree of downregulation in the group of patients with DKC1. This pattern is reversed in the DKC1 downregulated gene signature in which 14 of 15 patients with FANC showed some level of increased expression. PANTHER analysis of each gene list for the patients with FANC or SBDS did not return any significant pathways, whereas the DKC1 upregulated signature list gave 6 highly significant terms that overlapped with the gene sets listed in supplemental Table 7 for the shared upregulated gene sets because of the presence of 8 ribosome genes.
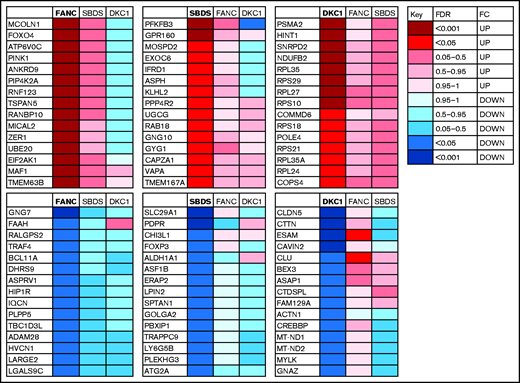
Summary heat maps of the 30 specific signature genes (15 upregulated and 15 downregulated) associated with each characterized disease subtype. For each panel, the specific disease type is highlighted in bold with an indication of the level (FDR) and direction (FC) of dysregulation indicated by the colored box. The corresponding level of dysregulation is also shown for the other study groups.
Summary heat maps of the 30 specific signature genes (15 upregulated and 15 downregulated) associated with each characterized disease subtype. For each panel, the specific disease type is highlighted in bold with an indication of the level (FDR) and direction (FC) of dysregulation indicated by the colored box. The corresponding level of dysregulation is also shown for the other study groups.
Transcriptomics of uncharacterized patients with BMF
Following on from these findings on the transcriptomic profile of IBMF from genetically characterized patients, we performed similar analyses for 6 patients with BMF in whom no disease-causing variants were identified in the known IBMF genes. These patients were divided into 2 groups on the basis of the degree of BMF present. The patients with pancytopenia were grouped together in the TRI category and the remaining samples were classified as SNGL (Table 1; supplemental Table 1). All comparisons were performed with 3 patients against 3 controls; the full differential expression analysis is provided in supplemental Table 3. These analyses revealed a similar significant skewing toward upregulation (Table 2; supplemental Figure 2D-E). PCA of all 5 patient groups again showed that the patients with DKC1 were still the most different from all the other patients (supplemental Figure 2F). Two of the 3 patients with SBDS are also separated from the FANC, TRI, and SNGL groups, which show the greatest degree of overlap (supplemental Figure 2F; PCA of patients vs controls for TRI and SNGL are shown in supplemental Figure 3). Subsequent GSEA analysis also revealed a significant enrichment of gene sets that code for ribosomes, NMD, and redox metabolism (Table 3; supplemental Table 4), indicating a striking overlap of transcriptional dysregulation between these uncharacterized patients and the characterized patients with IBMF.
To identify a list of genes dysregulated in all patients studied, intersections of both significantly upregulated and downregulated genes (FDR < 0.05) were plotted (Figure 4). From the 5-way Venn diagram, we see 22 genes common to all patients with IBMF and to uncharacterized patient groups, nearly half of which are ribosomal subunit genes (supplemental Tables 5 and 6 ). There were no downregulated genes that were common to all 5 groups.
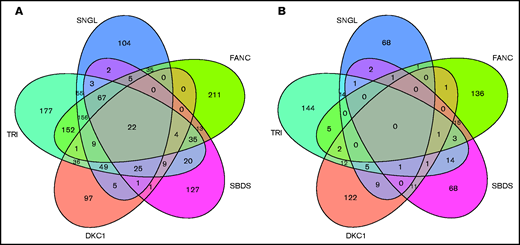
Five-way Venn diagrams detailing the overlap between significantly dysregulated genes in various groups of IBMF. Upregulated genes (A) and downregulated genes (B). All genes had FDR < 0.05. Gene lists for the shared and patient group specific are provided in supplemental Table 5.
Five-way Venn diagrams detailing the overlap between significantly dysregulated genes in various groups of IBMF. Upregulated genes (A) and downregulated genes (B). All genes had FDR < 0.05. Gene lists for the shared and patient group specific are provided in supplemental Table 5.
Finally, we investigated each of the 6 uncharacterized patients independently with respect to the 30 disease-specific signature genes (15 upregulated and 15 downregulated; Figure 3) to determine whether there was any overlap with the identified signatures. The expression data were normalized to 4 genes that were found to be expressed in all samples at similar levels, were ubiquitously expressed, and had an FDR > 0.98 (DDX21, GINM1, ARDM1, MAN2C1). The geometric mean of the read counts for these control genes was used to calculate a normalized read count for all patient and control samples. The FC for each uncharacterized individual was then determined as a patient:control ratio. Table 5 shows the summary of this analysis. In characterized patient groups, we confirmed that the signature genes were informative and discriminatory for their disease group, with the list for the FANC group having the strongest effect. After we looked at the uncharacterized patients individually, none of them showed similarity to the signature of the SBDS group but some did show considerable FCs with respect to both FANC and DKC1 signature genes (Table 5).
Discussion
In this study, we describe the whole-blood transcriptomic profiles of 3 classic types of IBMF arising from mutations in FANCA and FANCG, SBDS, and DKC1, which represent the most common genotypes for the IBMF syndromes FA, SDS, and DC. We hypothesized that exploring these transcriptomic profiles would provide insight into a shared signature that is disease relevant. We have demonstrated that the technique used and the data obtained show a good concordance between RNA-seq and qPCR outputs. Subsequent analysis of whole-blood transcriptomes obtained from genetically uncharacterized patients revealed a significant overlap of upregulated gene subsets and pathways, indicating a common transcriptional drive among these classic and uncharacterized BMF subtypes.
We identified marked skewing in favor of upregulated genes as a common feature across the BMF subtypes we analyzed. Bulk RNA-seq was sensitive enough to identify overlapping dysregulated gene profiles from these different genetic diseases, most strikingly a significant upregulation of genes involved in protein translation. Although this might be expected for the patients with SBDS and DKC1 in whom both genes are known to play a role in ribosome biogenesis, this is not the case for the patients with FA in whom the disease genes are primarily involved in DNA repair. Increased expression in the ribosomal protein genes is a consistent feature in this study because 9 genes are shared among all samples under investigation. Because these data are based on a small number of patients, together with the significant hematologic heterogeneity observed in these syndromes, further studies will be necessary to determine the extent to which these findings can be applied in general to these syndromes.
Differential gene expression analysis has also shown upregulation in NMD activity associated with RNA metabolism and redox-related bioenergetic pathways, which overlap among the FA, SDS, and DC groups and uncharacterized patient subsets. It is widely known that NMD activity controls protein translation by terminating premature transcripts at the stalled ribosomes.16 NMD is programmed to eliminate aberrant transcripts that arise as a result of chromosomal DNA rearrangements and to maintain bona fide hematopoiesis.17 Redox metabolism pathways when upregulated have also been proved to impair hematopoiesis because of an increase in oxidative stress that causes genome instability.18-20 Pluripotent embryonic stem cells are metabolically characterized by high glycolysis activity, whereas primed embryonic stem cells and differentiated cells show increased oxidative phosphorylation activity.21 Manipulation of redox bioenergetics pathways using small molecules has proved to be effective in ameliorating growth defects in cell and mouse models of FA22 and DC.23 Therefore, the combined upregulation of transcripts associated with ribosome structural constituents, NMD activity, and redox metabolism could be a sign of HSC exhaustion in the stressed bone marrow of these patients. These observations may provide additional evidence of HSCs losing their “stemness” and becoming more differentiated in these patients, which is similar to what is seen in early MDS in which patients are characterized by an increased apoptosis in the bone marrow.24 Although MDS is a common feature of all clinical entities under investigation, there were no features of MDS at the time of BMF diagnosis and sampling in our patients.
It is noteworthy that short telomeres are a peripheral blood hallmark of BMF syndromes,25 and patients with DC in particular have very short telomeres when compared with age-matched controls because of defects in telomere maintenance.26,27 Some patients with FA exhibit short telomeres as a result of abnormal DNA damage response mediated by mutant FA proteins in repair and replication of telomeric DNA.28 Likewise, blood cells from some patients with SDS also have short telomeres, and the SBDS protein has recently been shown to function in trafficking of telomerase to telomeres via interaction with the shelterin protein component TPP1.29 However, most of the genes involved in either the telomerase or the shelterin complexes are not significantly dysregulated in any of the 3 classic groups (supplemental Table 3).
In addition to identifying a unified transcriptional drive across the classic (FA, SDS, and DC) and uncharacterized BMF subtypes, this study has identified BMF subtype–specific gene signatures. By focusing on a set of 30 genes (15 of which were the most upregulated and 15 of which were the most downregulated) within each classic BMF subtype, we were able to describe a disease-specific blood gene signature (Figure 3) that is informative and discriminatory for each subgroup. Comparison of the uncharacterized patients showed that even though there was some overlap with the classic IBMF subtypes (Table 5), none of the uncharacterized patients had a profile that matched precisely. This suggests that these uncharacterized patients are likely to represent new genetic categories for FA, SDS, and DC. The disease-specific blood gene signatures will be useful in the research setting but are unlikely to replace the simpler diagnostic tests based on next-generation sequencing gene panels.
It is possible to identify the transcriptomic profiles of subtypes of BMF in the peripheral blood, but this may not completely reflect what is happening in the bone marrow. This is a result of the difference in the cellular composition of these 2 biological sources and the relative maturity of the cell types. Bone marrow has a greater proportion of progenitor cells whereas peripheral blood has more mature blood cells. Therefore, whole blood may give a measure of the overall consequence of disease pathology in the periphery and not be illustrative of how the genetic mutation affects the early hematopoietic and progenitor cells in the bone marrow. This difference between the peripheral blood and bone marrow may also be relevant to data on p53 because we did not observe any significant difference in the expression of p53 transcripts in our patients’ blood (Figure 1B; supplemental Table 3). This lack of significant dysregulation was also observed in many genes that are regulated by p53.30 In future studies, it will be interesting to compare and determine transcriptomic profiles of whole blood and bone marrow from the same individual among the different BMF subtypes. Although it is difficult to predict, it is likely that the bone marrow transcriptomes will show both similarities and differences compared with what has been observed in the peripheral blood.
In conclusion, this study identifies a remarkable unifying signature of gene expression at the transcriptomic level in the classic and uncharacterized patient with BMF subsets. This may help explain some of the features common to the different BMF subtypes, particularly the core BMF. The study also suggests that new approaches capable of normalizing this unified transcriptional drive may be of therapeutic benefit in the clinic.
Acknowledgments
The authors thank all the patients and healthy volunteers for their participation in this study and Dan Pennington for helpful discussions.
This work was supported by grants from the Medical Research Council (MR/P018440/1) and Blood Cancer UK (14032).
Authorship
Contribution: A.J.W. designed the study, performed experiments, analyzed data, and wrote the article; T.V. designed the study, analyzed the data, and wrote the article; F.B.-C. and J.W. analyzed the data; J.A., M.G.B., and B.M. performed the experiments; H.T. analyzed the data and wrote the article; I.D. designed the study, provided clinical samples and details, and wrote the article; and all authors critically reviewed the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Amanda J. Walne, Centre for Genomics and Child Health, Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, 4 Newark St, London E1 2AT, United Kingdom; e-mail: a.walne@qmul.ac.uk.