

Key Points
Low flow and leukocyte-induced ICAM1 clustering synergize to mechanically activate endothelial PIEZO1.
Activation of PIEZO1 initiates signaling processes that result in opening of the endothelial barrier and leukocyte extravasation.

Abstract
The extravasation of leukocytes is a critical step during inflammation that requires the localized opening of the endothelial barrier. This process is initiated by the close interaction of leukocytes with various adhesion molecules such as ICAM-1 on the surface of endothelial cells. Here we reveal that mechanical forces generated by leukocyte-induced clustering of ICAM-1 synergize with fluid shear stress exerted by the flowing blood to increase endothelial plasma membrane tension and to activate the mechanosensitive cation channel PIEZO1. This leads to increases in [Ca2+]i and activation of downstream signaling events including phosphorylation of tyrosine kinases sarcoma (SRC) and protein tyrosine kinase 2 (PYK2), as well as of myosin light chain, resulting in opening of the endothelial barrier. Mice with endothelium-specific Piezo1 deficiency show decreased leukocyte extravasation in different inflammation models. Thus, leukocytes and the hemodynamic microenvironment synergize to mechanically activate endothelial PIEZO1 and subsequent downstream signaling to initiate leukocyte diapedesis.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 291.
Disclosures
Laurie Barclay, MD, freelance writer and reviewer, Medscape, LLC, has disclosed the following relevant financial relationships: stock, stock options, or bonds: AbbVie Inc. (former).
Learning objectives
Upon completion of this activity, participants will:
- 1.
Describe how low flow and leukocyte-induced intercellular adhesion molecule 1 clustering interact to mechanically activate endothelial PIEZO1, based on a mouse model
- 2.
Identify the role of PIEZO1 activation in signaling processes leading to opening of the endothelial barrier and leukocyte extravasation, based on a mouse model
- 3.
Determine pathophysiologic and clinical implications of how molecular mechanisms underlying the initial interactions between leukocytes and endothelial cells are linked to opening of the endothelial barrier, based on a mouse model
Release date: July 21, 2022; Expiration date: July 21, 2023
Introduction
The endothelial cell layer is a tight barrier for cells in the circulation. However, during inflammation, leukocytes can transmigrate the endothelium and extravasate in the perivascular space, a process that involves a well-coordinated cascade of events. This includes initial leukocyte capture and rolling, firm adhesion, and crawling, which are then followed by breaching of the endothelial barrier and the extravasation.1-3 The molecular mechanisms that control and mediate the initial interactions between leukocytes and endothelial cells are well characterized and involve interactions between endothelial selectins and glycoproteins of leukocytes during capture and rolling steps, whereas arrest, firm adhesion, and crawling are mediated mainly by integrins on leukocytes that bind to endothelial ICAM-1 and VCAM-1 and induce their clustering.2,4-6 How these initial processes are linked to the opening of the endothelial barrier, which requires the remodeling of endothelial adherens junctions and endothelial cell contraction,7-12 is, however, poorly understood.
Opening of endothelial junctions and endothelial cell contraction during leukocyte transmigration require activation of endothelial signaling pathways, and several studies have shown that leukocytes induce an increase in the cytosolic Ca2+ concentration in endothelial cells.13-18 This calcium signal is not necessary for leukocyte adhesion but is required to induce transendothelial migration.13-15 ICAM-1 has been shown to be involved in lymphocyte-induced Ca2+ transients in endothelial cells,15 and, more recently, the transient receptor potential channel C6 (TRPC6) has been shown to be required for endothelial calcium transients induced by neutrophils and for transendothelial migration,19 but how leukocytes induce endothelial Ca2+ transients is still unclear.
The Piezo proteins PIEZO1 and PIEZO2 are mechanically activated cation channels that form homotrimeric complexes,20-22 which are sufficient to mediate mechanically induced currents.20 PIEZO1 has been shown to be gated directly by changes in membrane tension23-25 and to mediate multiple cellular processes including endothelial flow sensing.26-28 In this study, we found that PIEZO1 is required for leukocyte extravasation by coincidentally sensing increased membrane tension induced by flow and ICAM-1 clustering. The subsequent activation of signaling pathways then results in the localized opening of the endothelial barrier.
Methods
Immunoblot analysis
Cells were lysed in cell lysis buffer (9803; Cell Signaling) containing 1% triton X-100 or in radioimmunoprecipitation assay (RIPA) buffer (9806; Cell Signaling) supplemented with protease and phosphatase inhibitors (5872; Cell Signaling). Lysates were centrifuged at 10 000g at 4°C for 10 minutes. Supernatants were then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were probed with primary and horseradish peroxidase–conjugated secondary antibodies (8884 and 7076, respectively; Cell Signaling) and were developed using the enhanced chemiluminescence (ECL) detection system (Pierce).
Determination of [Ca2+]i
For the determination of the intracellular Ca2+ concentration, endothelial cells were loaded with 5 μM Ca2+-sensitive dye Fluo-4 AM (F14201; Molecular Probes) or 5 μM Fura-2 AM (F1221; Molecular Probes) in Hanks balanced salt solution supplemented with 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) for 30 minutes at 37 °C and were then washed with Hanks balanced salt solution 3 times at room temperature. Live-cell images were acquired with an IX81 microscope (Olympus) at a frequency of 1 Hz. Fluo-4 fluorescence was measured by using excitation at 488 nm and emission collected at 500 to 550 nm. Fura-2 was monitored by digital fluorescence 340/380-nm ratio imaging. In experiments in which polymorphonuclear leukocytes (PMNs) were added to endothelial cells, only endothelial cells to which PMNs were attached were used for the analysis.
Fluorescein isothiocyanate-dextran permeability assay
A total of 1.5 × 104 human umbilical venous endothelial cells (HUVECs) were seeded per well of a collagen-coated transwell plate (3-μm pore size; Corning) and were cultured with daily medium changes until reaching confluency. For knockdown experiments, 8000 cells were transfected using Lipofectamine RNAiMAX with the indicated small interfering RNAs (siRNAs). For permeability assay, the medium of the upper insert was removed and replaced with medium containing 250 µg/mL fluorescein isothiocyanate (FITC)-conjugated dextran (relative molecular mass, 40 kDa; Molecular Probes). The permeability was determined by passage of FITC-dextran through the endothelial monolayer into the lower chamber using FlexStation-3 (Molecular Devices).
Electrophysiology
Whole cell patch-clamp recordings were performed at room temperature using an EPC10/2 amplifier with Pulse software (HEKA Electronik GmbH, Lambrecht, Germany). Pipette resistance was between 3 and 4 MΩ, and membrane potential was clamped at −80 mV. Normal external solution contained 140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1mM MgCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4. The intracellular pipette solution contained 95 mM l-aspartate, 40 mM CsCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 0.1 mM guanosine triphosphate, pH 7.2. Local low flow at indicated shear rate was generated with a multichannel microperfusion system (LEAD-2, LONGER). Mechanical stimulation was applied to cells using a fire-polished glass pipette with a 3- to 5-μm tip diameter. Pipette movement was controlled by PCS-5000 Patch-Clamp micromanipulator (Burlegh). The micromanipulator uses the solid-state stability of piezoelectric technology to maintain smooth and predictable pipette motion without drift. Whole cell current was analyzed with IGOR Pro software (WaveMetrics). Experiments were performed with dissociated single cells. In experiments in which PMNs were added to endothelial cells, only endothelial cells to which PMNs were attached were used for the analysis.
PMN application and ICAM-1 clustering
We routinely applied 2 × 105 PMNs per milliliter to cells. For antibody-mediated clustering of ICAM-1, sheep anti-mouse IgG-coupled dynabeads (M280; Invitrogen) were coated with mouse anti-human ICAM-1 antibody (BBIG-I1; R&D Systems) or immunoglobulin IgG1 (IgG1) control (MAB002; R&D Systems) overnight at 4°C according to the manufacturer’s protocol. To induce clustering, 1.5 × 106 antibody-coated beads/mL was added to a tumor necrosis factor α (TNFα)-stimulated HUVEC monolayer cultured in a 6-well dish and incubated for 15 minutes. For some experiments, antibody-coated beads (5 × 105/mL) were injected into the ibidi perfusion system containing HUVECs to induce ICAM-1 clustering on HUVECs under physiologic flow conditions. Alternatively, ICAM-1 was ligated with 15 μg/mL mouse monoclonal antibodies (R&D Systems, catalog no. BBIG-I1) for 30 minutes, followed by washing and ICAM-1 cross-linking with 50 μg/mL mouse secondary antibody (R&D Systems, catalog no. AF007) for 20 minutes at 37°C. Live cell imaging of membrane tension and intracellular Ca2+ during ICAM-1 clustering were performed using a Leica SP8 or an Olympus IX81 microscope (see above). For immunoblot analysis, beads were isolated using a magnetic holder (Miltenyi Biotec), and cells were lysed with RIPA buffer as described above.
Lipopolysaccharide (LPS)-induced extravasation
EC-Piezo1-knockout (KO) and control mice were injected intraperitoneally with 200 µL phosphate-buffered saline (PBS) alone or containing 1 mg/kg body weight lipopolysaccharide (LPS) (L4391; Sigma). After 4 hours, animals were euthanized, and lungs were collected for immunostaining with anti-MPR14 and anti–platelet endothelial cell adhesion molecule 1 (PECAM-1) antibodies. For quantification of extravasating PMNs, cryosections were analyzed in XYZ views on a Leica SP8 confocal microscope by the following criteria: PMNs directly surrounded by PECAM-1 staining (ie, blood vessel) were scored as intravascular, whereas cells outside blood vessels were scored as extravascular.
TNFα-induced peritonitis and flow cytometry
Wild-type or EC-Piezo1-KO mice were injected intraperitoneally with 100 µL PBS containing no or 500 ng TNFα prewarmed to 37°C. After 60 minutes, animals were euthanized, and cells in the peritoneal cavity were collected by flushing with 5 mL ice-cold PBS. Peritoneal cells were filtered using a 70-μm strainer and analyzed by flow cytometry (BD FACS Canto II). The following antibodies were used: FITC conjugated anti-mouse CD11b (101205; BioLegend) and allophycocyanin (APC)-conjugated anti-mouse Ly6G (127614; Biolegend).
Other reagents and antibodies
Yoda1 (5586) was from Tocris Bioscience. Cytochalasin D (C8273), Blebbistatin (B0560), and PF431396 (PZ0185) were from Sigma. PP2 (529576) was from Merck Chemicals. Anti–PIEZO1-antibody was from Proteintech (15939-1-AP). Anti-GAPDH (catalog no. 5174), anti-protein tyrosine kinase 2 (PYK2) (catalog no. 3292), anti–p-PYK2 (Tyr402; 3291), antisarcoma (SRC) (2109), anti–p-SRC (Tyr416; 6943), anti-MLC (3672), and anti–p-MLC (Ser19; 3675) were from Cell Signaling Technology. Anti–endomucin antibody was from Santa Cruz (sc-65495). Gd3+ (439770) was from Sigma, and GsMTx4 (4912) was from Tocris.
Statistical analysis
Trial experiments or experiments done previously were used to determine sample size with adequate statistical power. Samples were excluded in cases where RNA/cDNA quality or tissue quality after processing was poor (below commonly accepted standards). Data are presented as means ± standard error of the mean (SEM). Comparisons between 2 groups were performed with the unpaired, 2-tailed Student t test, and multiple group comparisons at different time points were performed by 1- or 2-way analysis of variance (ANOVA). A value of P ≤ .05 was considered statistically significant.
Results
PIEZO1 is required for leukocyte transendothelial migration in vitro
In a screen to identify endothelial transmembrane proteins involved in the transendothelial migration of leukocytes, we identified the mechanosensitive cation channel PIEZO1 (Figure 1A; supplemental Table 1, available on the Blood Web site). The siRNA-mediated knock down of PIEZO1 in HUVECs or in the mouse brain endothelial cell line bEnd.3 strongly reduced endothelial transmigration of PMNs and peripheral blood mononuclear cells (Figure 1B-D). Similarly, PMN transmigration through mouse lung endothelial cells (MLECs) from mice with endothelium-specific loss of Piezo1 (EC-Piezo1-KO)28 was strongly reduced compared with wild-type MLECs (Figure 1E; supplemental Figure 1A-B). Basal and TNFα-induced expression of endothelial adhesion molecules was not affected by loss of PIEZO1 (supplemental Figure 1C). Both PMN transmigration of human and murine endothelial cells could be stimulated by Yoda1, an activator of PIEZO1, and this effect was not seen after knockdown of PIEZO1 in endothelial cells (Figure 1D-G). PMN rolling on and adhesion to endothelial cells was not affected by loss of endothelial Piezo1 expression (Figure 1B-C), and endothelial barrier function analyzed by measuring the electrical impedance of the endothelial cell layer in vitro or by determining the permeability of the endothelial layer for FITC-labeled dextran in vivo was normal after loss of PIEZO1 (Figure 1H-I).
![PIEZO1 mediates leukocyte transendothelial migration in vitro. (A) HUVECs pretreated with 10 ng/mL TNFα were transfected with 360 siRNAs pools against RNAs encoding transmembrane proteins expressed in endothelial cells and were then exposed to THP-1 monocytic cells for 3 hours. Shown is the ratio of THP-1 cells that transmigrated the HUVEC monolayer transfected with a particular siRNA pool and with control siRNA. The plot shows the ranked average ratios of 3 independent experiments. (B) HUVECs were transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1), and rolling, adhesion, and transmigration of human PMNs applied together with flow (1.2 dynes/cm2) were analyzed (n = 8 independent experiments per group). Cells treated with control siRNA were set as 100%. (C-I) The indicated endothelial cells were transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) or were left untransfected (E). (C) Rolling, adhesion, and transmigration of mouse PMNs (n = 8 per group) applied together with flow (1.2 dynes/cm2) to a bEnd.3 cell monolayer. Cells treated with control siRNA were set as 100%. (D,F,G) Transmigration of human peripheral blood mononuclear cells (D) (n = 4 independent experiments per group), human PMNs (F) (n = 6 independent experiments per group), or mouse PMNs (G) (n = 6 independent experiments per group) across HUVECs (D,F) or bEnd.3 cells (G) pretreated without or with 1 μM Yoda1 for 15 minutes. (E) MLECs were isolated from EC-Piezo1-KO and control mice, and transmigration of mouse PMNs was determined after pretreament without or with 1 μM Yoda1 for 15 minutes (n = 5 independent experiments). (H) HUVEC barrier integrity was assessed using an electric cell-substrate impedance sensing (ECIS) system in the absence or presence of 1 μM Yoda1 (n = 8 independent experiments per group). (I) Paracellular permeability of the endothelial monolayer cultured in transwell plates was determined using 40 kDa FITC-dextran (n = 5 independent experiments per group; a.u., arbitrary units). Shown are mean values ± SEM; *P ≤ .05; **P ≤ .01; ***P ≤ .001 (unpaired t test [B-H], 2-way ANOVA [I]).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/3/10.1182_blood.2021014614/5/m_bloodbld2021014614f1.png?Expires=1769086547&Signature=StEH0DbNlD6OAe2UR2j7lcMagJEFqmsskgeDdev8IoXuijT~rI8xuNVlt0IRgLudkBzAlFXJnNv6bsFEt0Oz05i8j~EW196QK87H9rqhiN5Qm3NXj9DWtjc6XQGirWGyT3tuCDIisrYdqmed5fkUtb4j7zRlCEu45MwWmVqa9~6Md7ArXS8glRuUB5uamS32hgFPMnFiNqFJp8qr8K4XJ0U4Fqs2FXle~kjX13iC9EGWp1049-qyfDNMgPQ77njcmSNlCfSMav2DEqLW52hxnL5l4byqXrKV-m9zhs3LJB33p0xaS94dQnbWfjmx2JeGguJLJxNqdcYu-jZpkHhPvg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PIEZO1 mediates leukocyte transendothelial migration in vitro. (A) HUVECs pretreated with 10 ng/mL TNFα were transfected with 360 siRNAs pools against RNAs encoding transmembrane proteins expressed in endothelial cells and were then exposed to THP-1 monocytic cells for 3 hours. Shown is the ratio of THP-1 cells that transmigrated the HUVEC monolayer transfected with a particular siRNA pool and with control siRNA. The plot shows the ranked average ratios of 3 independent experiments. (B) HUVECs were transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1), and rolling, adhesion, and transmigration of human PMNs applied together with flow (1.2 dynes/cm2) were analyzed (n = 8 independent experiments per group). Cells treated with control siRNA were set as 100%. (C-I) The indicated endothelial cells were transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) or were left untransfected (E). (C) Rolling, adhesion, and transmigration of mouse PMNs (n = 8 per group) applied together with flow (1.2 dynes/cm2) to a bEnd.3 cell monolayer. Cells treated with control siRNA were set as 100%. (D,F,G) Transmigration of human peripheral blood mononuclear cells (D) (n = 4 independent experiments per group), human PMNs (F) (n = 6 independent experiments per group), or mouse PMNs (G) (n = 6 independent experiments per group) across HUVECs (D,F) or bEnd.3 cells (G) pretreated without or with 1 μM Yoda1 for 15 minutes. (E) MLECs were isolated from EC-Piezo1-KO and control mice, and transmigration of mouse PMNs was determined after pretreament without or with 1 μM Yoda1 for 15 minutes (n = 5 independent experiments). (H) HUVEC barrier integrity was assessed using an electric cell-substrate impedance sensing (ECIS) system in the absence or presence of 1 μM Yoda1 (n = 8 independent experiments per group). (I) Paracellular permeability of the endothelial monolayer cultured in transwell plates was determined using 40 kDa FITC-dextran (n = 5 independent experiments per group; a.u., arbitrary units). Shown are mean values ± SEM; *P ≤ .05; **P ≤ .01; ***P ≤ .001 (unpaired t test [B-H], 2-way ANOVA [I]).
PIEZO1 mediates leukocyte transendothelial migration in vitro. (A) HUVECs pretreated with 10 ng/mL TNFα were transfected with 360 siRNAs pools against RNAs encoding transmembrane proteins expressed in endothelial cells and were then exposed to THP-1 monocytic cells for 3 hours. Shown is the ratio of THP-1 cells that transmigrated the HUVEC monolayer transfected with a particular siRNA pool and with control siRNA. The plot shows the ranked average ratios of 3 independent experiments. (B) HUVECs were transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1), and rolling, adhesion, and transmigration of human PMNs applied together with flow (1.2 dynes/cm2) were analyzed (n = 8 independent experiments per group). Cells treated with control siRNA were set as 100%. (C-I) The indicated endothelial cells were transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) or were left untransfected (E). (C) Rolling, adhesion, and transmigration of mouse PMNs (n = 8 per group) applied together with flow (1.2 dynes/cm2) to a bEnd.3 cell monolayer. Cells treated with control siRNA were set as 100%. (D,F,G) Transmigration of human peripheral blood mononuclear cells (D) (n = 4 independent experiments per group), human PMNs (F) (n = 6 independent experiments per group), or mouse PMNs (G) (n = 6 independent experiments per group) across HUVECs (D,F) or bEnd.3 cells (G) pretreated without or with 1 μM Yoda1 for 15 minutes. (E) MLECs were isolated from EC-Piezo1-KO and control mice, and transmigration of mouse PMNs was determined after pretreament without or with 1 μM Yoda1 for 15 minutes (n = 5 independent experiments). (H) HUVEC barrier integrity was assessed using an electric cell-substrate impedance sensing (ECIS) system in the absence or presence of 1 μM Yoda1 (n = 8 independent experiments per group). (I) Paracellular permeability of the endothelial monolayer cultured in transwell plates was determined using 40 kDa FITC-dextran (n = 5 independent experiments per group; a.u., arbitrary units). Shown are mean values ± SEM; *P ≤ .05; **P ≤ .01; ***P ≤ .001 (unpaired t test [B-H], 2-way ANOVA [I]).
Endothelial PIEZO1 is critically involved in leukocyte extravasation in vivo
To study leukocyte extravasation in vivo, we injected TNFα into the peritoneal cavity and determined the number of CD11b+/Ly6G+ myeloid cells in the peritoneal cavity 6 hours later. Whereas TNFα induced a significant influx of cells into the peritoneal cavity of wild-type mice compared with untreated controls, the effect of TNFα was strongly reduced in EC-Piezo1-KO mice (Figure 2A). We then studied the role of endothelial PIEZO1 in a model of acute dermatitis of the ear by applying croton oil to the ear surface and found that the total number of neutrophils seen in sections of ears from wild-type and EC-Piezo1-KO mice was identical (supplemental Figure 1D). However, when analyzing postcapillary venules characterized by a diameter of 20 to 30 µm, the primary site of leukocyte extravasation, we found that most neutrophils had completed extravasation and were found in the perivascular space in wild-type mice, whereas about 25% to 30% of the leukocytes were found in the lumen of vessels (Figure 2B-D). However, in EC-Piezo1-KO mice, a significantly reduced portion of leukocytes had completed extravasation, and the majority, about 70% of cells, showed arrest at the luminal surface of the endothelium (Figure 2B,D), suggesting that they adhered to the endothelium but were not able to initiate the process of endothelial transmigration. Similarly, LPS-induced extravasation of neutrophils into the lung parenchyma was strongly reduced in EC-Piezo1-KO mice (Figure 2E-F). Staining and intravital microscopy of the cremaster of EC-Piezo1-KO mice revealed a reduced extravasation of neutrophils compared with controls after intrascrotal injection of interleukin 1β (IL-1β; Figure 2G-I). Hemodynamic parameters were similar in both mouse types, and there was no significant different in leukocyte rolling and adhesion within venules (supplemental Figure E-I). Also, basal extravasation of Evans blue and extravasation after subcutaneous injection of histamine or vascular endothelial growth factor (VEGF) were indistinguishable between wild-type and EC-Piezo1-KO mice (Figure 2J), indicating that vascular permeability was unchanged. Expression of genes encoding proteins involved in endothelial functions was not changed in endothelial cells from EC-Piezo1-KO mice (supplemental Figure 1J-K).
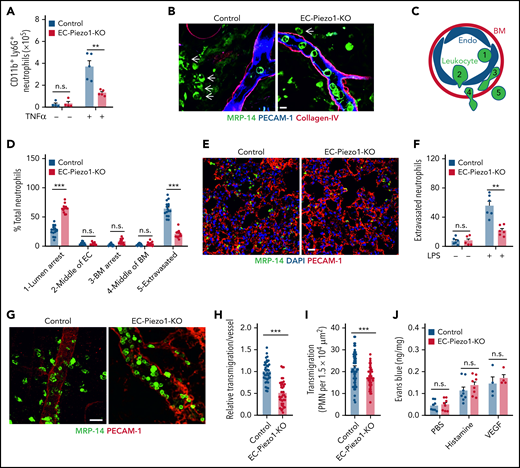
PIEZO1 mediates leukocyte transendothelial migration in vivo. (A) Endothelium-specific PIEZO1-deficient mice (EC-Piezo1-KO) or control animals were injected intraperitoneally with PBS or 500 ng of TNFα, and the number of peritoneal CD11b+;Ly6G+ neutrophils was determined by flow cytometry (n = 4 mice, −TNFα; n = 5 mice, +TNFα). (B-D) EC-Piezo1-KO and control mice were treated with croton oil on 1 ear. Six hours later, animals were euthanized, and ears were immunostained as whole mounts with antibodies against PECAM-1 (blue, endothelium), collagen-IV (red, basement membrane), and MRP14 (green, neutrophil). Arrows indicate neutrophils. Scale bar, 10 μm. (B) Representative images of stained ears. (C) Schematic drawing illustrating the criteria to delineate the 5 positions in which leukocyte are found during extravasation. (D) Distribution pattern of neutrophil positions relative to the endothelium and basement membrane (n = 16 mice, control; n = 14 mice, EC-Piezo1-KO; 3-5 vessels were analyzed per animal). (E-F) Confocal imaging of PMNs and lung microvessels 4 hours after intraperitoneal injection of 1 mg/kg LPS in EC-Piezo-KO and control mice (E). Quantitative analysis of extravasated neutrophils per field (F; n = 6 mice, wild type; n = 6 mice, EC-Piezo1-KO). (G-H) EC-Piezo1-KO and control animals were injected with 50 ng (in 100 µL PBS) IL-1β intrascrotally. After 3 hours, the cremaster muscle was isolated and stained for PECAM-1 and MRP14 (G). The quantitative analysis of extravasated neutrophils per vessel area is shown in panel H (n = 8 mice, control; n = 10 mice, EC-Piezo1-KO; 2-3 vessels were analyzed per animal). (I) EC-Piezo1-KO and control mice were analyzed by intravital microscopy of cremaster venules 4 hours after injection of 50 ng IL-1β for extravasated leukocytes (n = 9 mice per group; 4-10 measurements per animal). (J) Evans blue extravasation was assessed after subcutaneous injection of 20 µL PBS without or with 100 μM of histamine or 100 ng/mL VEGF (n = 8 mice, PBS and histamine; n = 4 mice, VEGF). Shown are mean values ± SEM. n.s., nonsignificant; **P ≤ .01; ***P ≤ .001 (unpaired t test).
PIEZO1 mediates leukocyte transendothelial migration in vivo. (A) Endothelium-specific PIEZO1-deficient mice (EC-Piezo1-KO) or control animals were injected intraperitoneally with PBS or 500 ng of TNFα, and the number of peritoneal CD11b+;Ly6G+ neutrophils was determined by flow cytometry (n = 4 mice, −TNFα; n = 5 mice, +TNFα). (B-D) EC-Piezo1-KO and control mice were treated with croton oil on 1 ear. Six hours later, animals were euthanized, and ears were immunostained as whole mounts with antibodies against PECAM-1 (blue, endothelium), collagen-IV (red, basement membrane), and MRP14 (green, neutrophil). Arrows indicate neutrophils. Scale bar, 10 μm. (B) Representative images of stained ears. (C) Schematic drawing illustrating the criteria to delineate the 5 positions in which leukocyte are found during extravasation. (D) Distribution pattern of neutrophil positions relative to the endothelium and basement membrane (n = 16 mice, control; n = 14 mice, EC-Piezo1-KO; 3-5 vessels were analyzed per animal). (E-F) Confocal imaging of PMNs and lung microvessels 4 hours after intraperitoneal injection of 1 mg/kg LPS in EC-Piezo-KO and control mice (E). Quantitative analysis of extravasated neutrophils per field (F; n = 6 mice, wild type; n = 6 mice, EC-Piezo1-KO). (G-H) EC-Piezo1-KO and control animals were injected with 50 ng (in 100 µL PBS) IL-1β intrascrotally. After 3 hours, the cremaster muscle was isolated and stained for PECAM-1 and MRP14 (G). The quantitative analysis of extravasated neutrophils per vessel area is shown in panel H (n = 8 mice, control; n = 10 mice, EC-Piezo1-KO; 2-3 vessels were analyzed per animal). (I) EC-Piezo1-KO and control mice were analyzed by intravital microscopy of cremaster venules 4 hours after injection of 50 ng IL-1β for extravasated leukocytes (n = 9 mice per group; 4-10 measurements per animal). (J) Evans blue extravasation was assessed after subcutaneous injection of 20 µL PBS without or with 100 μM of histamine or 100 ng/mL VEGF (n = 8 mice, PBS and histamine; n = 4 mice, VEGF). Shown are mean values ± SEM. n.s., nonsignificant; **P ≤ .01; ***P ≤ .001 (unpaired t test).
Leukocytes and low flow induce PIEZO1 activation to stimulate endothelial downstream signaling
Because increases in [Ca2+]i are involved in the initiation of leukocyte transendothelial migration and because leukocyte diapedesis occurs in the presence of low flow in vivo, we studied leukocyte-induced increases in endothelial cytosolic Ca2+ in the absence and presence of flow at a low shear rate (1.2 dynes/cm2). In control HUVECs loaded with the [Ca2+]i indicators Fluo-4 or Fura-2, low flow alone or addition of human neutrophils alone had only a small effect on the cytosolic [Ca2+] (Figure 3A; supplemental Figure 2A). Similar but less pronounced effects were seen with the even lower shear rates of 0.4 and 0.8 dynes/cm2 (supplemental Figure 2B). However, when neutrophils were given together with flow, we observed a strong increase in the endothelial cytosolic Ca2+ concentration (Figure 3A; supplemental Figure 2A). This synergistic effect was rarely seen during rolling or initial arrest of leukocytes but during crawling and during the transmigration phase (supplemental Figure 2C), and it depended on PIEZO1 (Figure 3B; supplemental Figure 2D). Chelation of intra- and extracellular Ca2+ by 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA) and EGTA, respectively, blocked Ca2+ transients (supplemental Figure 2E-F). Whole cell patch-clamp recordings of MLECs showed characteristic inward currents when cells were exposed simultaneously to low flow and PMNs, which were not seen in MLECs from EC-Piezo1-KO animals (Figure 3C-D; supplemental Figure 2G-H). In contrast, exposure of MLECs to flow alone or PMNs alone induced only small currents (Figure 3C-D).
![Leukocytes and flow synergistically induce PIEZO1 activation to stimulate endothelial downstream signaling. (A) HUVECs were preactivated with TNFα, loaded with Fluo-4, and exposed to PMNs alone, low flow (1.2 dynes/cm2) alone, or both. [Ca2+]i was determined as fluorescence intensity (RFU, relative fluorescence units). (B) HUVECs transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) were preactivated with TNFα, loaded with Fluo-4, and exposed to PMNs and low flow (1.2 dynes/cm2) given together. [Ca2+]i was determined as fluorescence intensity. Five traces representative of the traces of 1 experiment are shown in panels A and B, and the time point of addition of PMNs is indicated by an arrow. The bar diagrams in panels A and B show the area under the curve (AUC) of the [Ca2+]i traces from 6 independent experiments (20-40 cells were analyzed per experiment). (C-D) Currents from MLECs of wild-type (control) or EC-Piezo1-KO mice were recorded in the whole cell patch clamp configuration. The holding potential was −80 mV, and the MLECs were exposed to PMNs, low flow (1.2 dynes/cm2), or both (n = 8-9 independent measurements per condition). (E-H) Immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC in lysates of TNFα-activated HUVECs transfected with control siRNA (siCtrl) or siRNA directed against PIEZO1 and incubated without or with human PMNs in the absence or presence of low flow (1.2 dynes/cm2) (E) or without or with 5 μM Yoda1 (G). Immunoblot analysis of PIEZO1 and GAPDH served as controls. Bar diagrams (F,H) show the densitometric analysis of 3 independent experiments. (I) Transmigration of human PMNs across TNFα-activated HUVECs preincubated for 30 minutes with the PYK2 and SRC inhibitors PF431396 (10 μM) and PP2 (10 μM), respectively (n = 5 independent experiments). (J) HUVECs transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) were preactivated with TNFα and exposed to PMNs alone, low flow (1.2 dynes/cm2) alone, or both. After 15 minutes, VE-cadherin internalization was determined as described in Methods (n = 4 independent experiments). Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (1-way ANOVA [A,D]; unpaired t test [B,F,H-J]).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/3/10.1182_blood.2021014614/5/m_bloodbld2021014614f3.png?Expires=1769086547&Signature=FiC8p4I4vBgKcO6kQRRRfS22tFqFFHIfGoxEUH0PYR5BsDQlsyWHXRbMg48uyl9MjyCFlcoWIO2kskaKIioW~mnxrMvNgK3k0m4DD42gMbNd4rh8PR8g4IGtCmRFVK0rNRhDiTZd-18G-Sy8IVtCggdp0WmHOyx3qIIdCxLBgBGoEp7w-KbxugaDHRauicMd2iGbXZA2spitJuwuIHvPbP7nv~XyL-z0xFKqQDcpK-xZ8LJikdsnWMli6DJwKiKF9D2XPHloYYJnZVPs4gHS~5AJbvPou5o-6iFUQuhNzmxzblgJK6cXXkZyoTsslyogqEt~r30rridEvT9z82oDpA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Leukocytes and flow synergistically induce PIEZO1 activation to stimulate endothelial downstream signaling. (A) HUVECs were preactivated with TNFα, loaded with Fluo-4, and exposed to PMNs alone, low flow (1.2 dynes/cm2) alone, or both. [Ca2+]i was determined as fluorescence intensity (RFU, relative fluorescence units). (B) HUVECs transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) were preactivated with TNFα, loaded with Fluo-4, and exposed to PMNs and low flow (1.2 dynes/cm2) given together. [Ca2+]i was determined as fluorescence intensity. Five traces representative of the traces of 1 experiment are shown in panels A and B, and the time point of addition of PMNs is indicated by an arrow. The bar diagrams in panels A and B show the area under the curve (AUC) of the [Ca2+]i traces from 6 independent experiments (20-40 cells were analyzed per experiment). (C-D) Currents from MLECs of wild-type (control) or EC-Piezo1-KO mice were recorded in the whole cell patch clamp configuration. The holding potential was −80 mV, and the MLECs were exposed to PMNs, low flow (1.2 dynes/cm2), or both (n = 8-9 independent measurements per condition). (E-H) Immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC in lysates of TNFα-activated HUVECs transfected with control siRNA (siCtrl) or siRNA directed against PIEZO1 and incubated without or with human PMNs in the absence or presence of low flow (1.2 dynes/cm2) (E) or without or with 5 μM Yoda1 (G). Immunoblot analysis of PIEZO1 and GAPDH served as controls. Bar diagrams (F,H) show the densitometric analysis of 3 independent experiments. (I) Transmigration of human PMNs across TNFα-activated HUVECs preincubated for 30 minutes with the PYK2 and SRC inhibitors PF431396 (10 μM) and PP2 (10 μM), respectively (n = 5 independent experiments). (J) HUVECs transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) were preactivated with TNFα and exposed to PMNs alone, low flow (1.2 dynes/cm2) alone, or both. After 15 minutes, VE-cadherin internalization was determined as described in Methods (n = 4 independent experiments). Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (1-way ANOVA [A,D]; unpaired t test [B,F,H-J]).
Leukocytes and flow synergistically induce PIEZO1 activation to stimulate endothelial downstream signaling. (A) HUVECs were preactivated with TNFα, loaded with Fluo-4, and exposed to PMNs alone, low flow (1.2 dynes/cm2) alone, or both. [Ca2+]i was determined as fluorescence intensity (RFU, relative fluorescence units). (B) HUVECs transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) were preactivated with TNFα, loaded with Fluo-4, and exposed to PMNs and low flow (1.2 dynes/cm2) given together. [Ca2+]i was determined as fluorescence intensity. Five traces representative of the traces of 1 experiment are shown in panels A and B, and the time point of addition of PMNs is indicated by an arrow. The bar diagrams in panels A and B show the area under the curve (AUC) of the [Ca2+]i traces from 6 independent experiments (20-40 cells were analyzed per experiment). (C-D) Currents from MLECs of wild-type (control) or EC-Piezo1-KO mice were recorded in the whole cell patch clamp configuration. The holding potential was −80 mV, and the MLECs were exposed to PMNs, low flow (1.2 dynes/cm2), or both (n = 8-9 independent measurements per condition). (E-H) Immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC in lysates of TNFα-activated HUVECs transfected with control siRNA (siCtrl) or siRNA directed against PIEZO1 and incubated without or with human PMNs in the absence or presence of low flow (1.2 dynes/cm2) (E) or without or with 5 μM Yoda1 (G). Immunoblot analysis of PIEZO1 and GAPDH served as controls. Bar diagrams (F,H) show the densitometric analysis of 3 independent experiments. (I) Transmigration of human PMNs across TNFα-activated HUVECs preincubated for 30 minutes with the PYK2 and SRC inhibitors PF431396 (10 μM) and PP2 (10 μM), respectively (n = 5 independent experiments). (J) HUVECs transfected with control (siCtrl) or PIEZO1-specific siRNA (siPIEZO1) were preactivated with TNFα and exposed to PMNs alone, low flow (1.2 dynes/cm2) alone, or both. After 15 minutes, VE-cadherin internalization was determined as described in Methods (n = 4 independent experiments). Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (1-way ANOVA [A,D]; unpaired t test [B,F,H-J]).
We then tested the potential involvement of PIEZO1 in the induction of downstream signaling events mediating leukocyte-induced opening of endothelial junctions. Whereas low flow alone had hardly any effect on the phosphorylation of PYK2, SRC, and the myosin light chain (MLC) in endothelial cells, addition of PMNs had as small but significant effect. However, application of both flow and PMNs synergistically induced endothelial PYK2, SRC, and MLC phosphorylation to a degree significantly higher than each of the 2 stimuli alone, and this effect was strongly reduced after knockdown of PIEZO1 (Figure 3E-F). The effect of PMNs and flow was mimicked by application of Yoda1, and this effect was again blocked after knockdown of PIEZO1 (Figure 3G-H). Inhibition of endothelial PYK2 or SRC by PF431396 or PP2, respectively, reduced basal transmigration and blocked Yoda1-induced increases in PMN transmigration (Figure 3I). These data strongly indicate that PMNs and low flow synergistically induce downstream signaling events through endothelial PIEZO1, resulting in the opening of endothelial junctions and leukocyte transmigration. Consistent with this, we also observed synergism in the ability of flow and PMNs to induce internalization of vascular endothelial (VE)-cadherin, an effect strongly inhibited after siRNA-mediated suppression of Piezo1 expression (Figure 3J).
Endothelial PIEZO1 is activated by flow-induced ICAM-1 clustering
Because engagement of endothelial ICAM-1 by leukocyte β2 integrins is essential for induction of increases in [Ca2+]i and diapedesis,13,15,29,30 we suppressed expression of endothelial ICAM-1 and found that this strongly inhibited PMN-induced Ca2+ transients and PYK2, SRC, and MLC phosphorylation (Figure 4A-B; supplemental Figure 3A-B). Clustering of ICAM-1 using beads coated with anti–ICAM-1 antibodies mimicked the effect of PMNs and induced Ca2+ transients and phosphorylation of PYK2, SRC, and MLC synergistically with low flow (Figure 4C-D; supplemental Figure 3C), whereas beads coated with a control IgG had no effect (supplemental Figure 3D-E). The effects of ICAM-1 clustering were inhibited after knockdown of PIEZO1 and ICAM-1 (Figure 4C-D). Similar results were obtained when ICAM-1 clustering was induced by cross-linking of bound anti–ICAM-1 antibodies (supplemental Figure 3F-I). When beads coated with anti–ICAM-1 antibodies were given together with low flow, an inward current was induced, which was sensitive to Gd3+ and the PIEZO1 inhibitor GsMTx4, whereas beads and flow alone had hardly any effect (Figure 4E-F). This strongly indicates that clustering and activation of ICAM-1 by leukocytes in the presence of low flow results in PIEZO1-mediated downstream signaling leading to the opening of endothelial junctions.
![Endothelial PIEZO1 activation by leukocytes involves ICAM-1 activation and flow. (A-B) HUVECs were transfected with control siRNA (siCtrl) or siRNA directed against ICAM-1. After treatment with TNFα, cells were exposed to low flow and human PMNs (A) or to flow and PMNs alone or given together (B). Thereafter, the free [Ca2+]i was determined after loading of HUVECs with Fluo4 (A), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC was performed (B). Immunoblot analysis of GAPDH served as control. The bar diagram (A) shows the AUC of the [Ca2+]i-trace from 3 independent experiments. The bar diagram (B) shows the densitometric analysis of 3 independent experiments. The arrow in panel A indicates the time point of addition of PMNs. (C-D) TNFα-activated HUVECs transfected with control siRNA (siCtrl) or siRNA directed against ICAM-1 or PIEZO1 were exposed to low flow and anti–ICAM-1 antibody beads (ICAM-1 beads) given together (C) or to low flow and anti–ICAM-1 beads alone or given together (D). Thereafter, the free [Ca2+]i was determined after loading of HUVECs with Fluo-4 (C), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC (D) was performed. Traces shown in panel C represent signals from 20 to 40 cells, and the time point of addition of beads is indicated by an arrow. In panel D, immunoblot analysis of GAPDH served as controls. Bar diagrams (C) show the AUC of the [Ca2+]i traces from 3 independent experiments. Bar diagrams (D) show the densitometric analysis of 3 independent experiments. (E-F) Currents from HUVECs pretreated without or with 10 µM Gd3+ or 5 µM GsMTx4 and exposed to low flow (1.2 dynes/cm2), anti–ICAM-1 beads, or both were recorded in the whole cell patch clamp configuration at a holding potential of −80 mV. Shown are characteristic traces (E) and statistical analysis of 5 to 12 independent recordings (F). Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (unpaired t test [A-B]; 1-way ANOVA [C-D,F]).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/3/10.1182_blood.2021014614/5/m_bloodbld2021014614f4.png?Expires=1769086547&Signature=Gf6xVHdemtKpLc0qagzZJ2wuEvy1yYO0OfnRbuQCtzhFBBmBpdyGv1jv8XRFi7i1-7-qf-~1xCR2zShVFPa5Ny2e~a0IbccdpFFG~wwgBSHRgL6xxuqtgsBvCOlohoXBfhrtBjd8DszM-OvU~-3OBf2EsOvlG965VNvKA5-KNvHydfLn-qEczXHTKpkx5-rYQ9EPLT7PGrg519tNY-J-CJv5W6kiEUkW0bMBMJLAgy4d9I80EqigvBIrLXm0~wvq2MS9sgDvFzuDjVJX6RA2N12uXxzKGeOzR6YpqP3u7rOHUyBC4NgDV9NJQmYomAtVNeiICci9NW3Dy6fe1Zvzog__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Endothelial PIEZO1 activation by leukocytes involves ICAM-1 activation and flow. (A-B) HUVECs were transfected with control siRNA (siCtrl) or siRNA directed against ICAM-1. After treatment with TNFα, cells were exposed to low flow and human PMNs (A) or to flow and PMNs alone or given together (B). Thereafter, the free [Ca2+]i was determined after loading of HUVECs with Fluo4 (A), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC was performed (B). Immunoblot analysis of GAPDH served as control. The bar diagram (A) shows the AUC of the [Ca2+]i-trace from 3 independent experiments. The bar diagram (B) shows the densitometric analysis of 3 independent experiments. The arrow in panel A indicates the time point of addition of PMNs. (C-D) TNFα-activated HUVECs transfected with control siRNA (siCtrl) or siRNA directed against ICAM-1 or PIEZO1 were exposed to low flow and anti–ICAM-1 antibody beads (ICAM-1 beads) given together (C) or to low flow and anti–ICAM-1 beads alone or given together (D). Thereafter, the free [Ca2+]i was determined after loading of HUVECs with Fluo-4 (C), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC (D) was performed. Traces shown in panel C represent signals from 20 to 40 cells, and the time point of addition of beads is indicated by an arrow. In panel D, immunoblot analysis of GAPDH served as controls. Bar diagrams (C) show the AUC of the [Ca2+]i traces from 3 independent experiments. Bar diagrams (D) show the densitometric analysis of 3 independent experiments. (E-F) Currents from HUVECs pretreated without or with 10 µM Gd3+ or 5 µM GsMTx4 and exposed to low flow (1.2 dynes/cm2), anti–ICAM-1 beads, or both were recorded in the whole cell patch clamp configuration at a holding potential of −80 mV. Shown are characteristic traces (E) and statistical analysis of 5 to 12 independent recordings (F). Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (unpaired t test [A-B]; 1-way ANOVA [C-D,F]).
Endothelial PIEZO1 activation by leukocytes involves ICAM-1 activation and flow. (A-B) HUVECs were transfected with control siRNA (siCtrl) or siRNA directed against ICAM-1. After treatment with TNFα, cells were exposed to low flow and human PMNs (A) or to flow and PMNs alone or given together (B). Thereafter, the free [Ca2+]i was determined after loading of HUVECs with Fluo4 (A), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC was performed (B). Immunoblot analysis of GAPDH served as control. The bar diagram (A) shows the AUC of the [Ca2+]i-trace from 3 independent experiments. The bar diagram (B) shows the densitometric analysis of 3 independent experiments. The arrow in panel A indicates the time point of addition of PMNs. (C-D) TNFα-activated HUVECs transfected with control siRNA (siCtrl) or siRNA directed against ICAM-1 or PIEZO1 were exposed to low flow and anti–ICAM-1 antibody beads (ICAM-1 beads) given together (C) or to low flow and anti–ICAM-1 beads alone or given together (D). Thereafter, the free [Ca2+]i was determined after loading of HUVECs with Fluo-4 (C), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC (D) was performed. Traces shown in panel C represent signals from 20 to 40 cells, and the time point of addition of beads is indicated by an arrow. In panel D, immunoblot analysis of GAPDH served as controls. Bar diagrams (C) show the AUC of the [Ca2+]i traces from 3 independent experiments. Bar diagrams (D) show the densitometric analysis of 3 independent experiments. (E-F) Currents from HUVECs pretreated without or with 10 µM Gd3+ or 5 µM GsMTx4 and exposed to low flow (1.2 dynes/cm2), anti–ICAM-1 beads, or both were recorded in the whole cell patch clamp configuration at a holding potential of −80 mV. Shown are characteristic traces (E) and statistical analysis of 5 to 12 independent recordings (F). Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (unpaired t test [A-B]; 1-way ANOVA [C-D,F]).
Flow and ICAM-1 clustering synergistically increase endothelial membrane tension
ICAM-1 clustering and adhesion of leukocytes to endothelial cells have been shown to induce stiffening of the endothelial surface and to induce traction stress.31-35 We therefore determined membrane tension in response to ICAM-1 clustering and PMNs using the fluorescent lipid tension sensor FliptR36 and the membrane stress sensor (MSS) biosensor35 (supplemental Figure 4A-B). We found that clustering of ICAM-1 or addition of PMNs leads to a small increase in endothelial membrane tension (Figure 5A-D). Low flow, which by itself had no significant effect on endothelial membrane tension, when given together with ICAM-1 clustering agents or PMNs, resulted in a very strong increase in plasma membrane tension (Figure 5A-D). This indicates that low flow and ICAM-1 clustering synergistically increase endothelial membrane tension.

Flow and ICAM-1 clustering synergistically increase endothelial membrane tension. (A-B) Fluorescence lifetime τ1 images of FliptR in TNFα-activated HUVECs kept under static conditions or in the presence of low flow (1.2 dynes/cm2) or exposed to anti–ICAM-1 antibody beads or to anti–ICAM-1 crosslinking antibodies (ICAM-1 XL) without or together with low flow. The color bar corresponds to lifetime in nanoseconds. Scale bar, 15 µm. Corresponding lifetime mean values indicating membrane tension are shown in the bar diagram (B; n = 40 measurements from 5 independent experiments). (C-D) Representative pseudocolored Förster resonance energy transfer (FRET) images of MSS-expressing cells kept under static conditions or in the presence of low flow (1.2 dynes/cm2) or exposed to anti–ICAM-1 crosslinking antibodies (ICAM-1 XL) without or together with low flow (C). The color bar indicates YPet/ECFP emission ratio. Corresponding normalized YPet/ECFP emission ratio of Förster resonance energy transfer (FRET) biosensors indicating membrane tension are shown in the bar diagram (D; n = 15-22 measurements from 3 independent experiments). Shown are mean values ± SEM. n.s., nonsignificant; **P ≤ .01; ***P ≤ .001 (1-way ANOVA).
Flow and ICAM-1 clustering synergistically increase endothelial membrane tension. (A-B) Fluorescence lifetime τ1 images of FliptR in TNFα-activated HUVECs kept under static conditions or in the presence of low flow (1.2 dynes/cm2) or exposed to anti–ICAM-1 antibody beads or to anti–ICAM-1 crosslinking antibodies (ICAM-1 XL) without or together with low flow. The color bar corresponds to lifetime in nanoseconds. Scale bar, 15 µm. Corresponding lifetime mean values indicating membrane tension are shown in the bar diagram (B; n = 40 measurements from 5 independent experiments). (C-D) Representative pseudocolored Förster resonance energy transfer (FRET) images of MSS-expressing cells kept under static conditions or in the presence of low flow (1.2 dynes/cm2) or exposed to anti–ICAM-1 crosslinking antibodies (ICAM-1 XL) without or together with low flow (C). The color bar indicates YPet/ECFP emission ratio. Corresponding normalized YPet/ECFP emission ratio of Förster resonance energy transfer (FRET) biosensors indicating membrane tension are shown in the bar diagram (D; n = 15-22 measurements from 3 independent experiments). Shown are mean values ± SEM. n.s., nonsignificant; **P ≤ .01; ***P ≤ .001 (1-way ANOVA).
Because ICAM-1 clustering has been shown to induce localized actin polymerization, MLC phosphorylation, and actomyosin contractility, which promote junctional opening,18,31 we analyzed the effect of cytochalasin D and blebbistatin on membrane tension and on phosphorylation of PYK2, SRC, and MLC induced by ICAM-1 clustering. Both agents blocked ICAM-1–dependent changes in membrane tension and downstream signaling (Figure 6A-D). We then tested whether increased membrane tension and downstream signaling induced by ICAM-1 clustering involves the actin adapter proteins α-actinin-4 and cortactin, which have been shown to be recruited after clustering of ICAM-1 and be required for ICAM-1–mediated actin filament branching and for ICAM-1–dependent transendothelial migration of neutrophils.37-39 As shown in Figure 6E-G and supplemental Figure 5A-B, siRNA-mediated knockdown of the RNAs encoding α-actinin-4 and cortactin blocked the effect of ICAM-1 clustering on membrane tension and downstream signaling. These data suggest that actin polymerization and actomyosin contractility of the cortical cytoskeleton induced by ICAM-1 clustering and leading to increased cortical tension40,41 directly affect plasma membrane tension41 and thereby induce PIEZO1 activation.
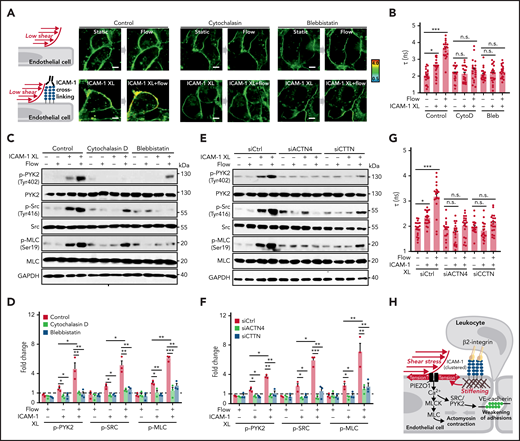
Actin polymerization and actomyosin contractility mediate increase of endothelial membrane tension and downstream signaling induced by ICAM-1 clustering. (A-G) HUVECs were preincubated without or with 10 µM cytochalasin D (CytoD) or 30 µM blebbistatin (Bleb) (A-D) or were transfected with control siRNA (siCtrl) or siRNA directed against the RNA encoding α-actinin-4 (siACTN4) or cortactin (siCTTN) (E-G) and were exposed to low flow alone, anti–ICAM-1 clustering antibodies (ICAM-1 XL) alone or both, and membrane tension was determined using FliptR (A-B,G; n = 20 measurements from 3 independent experiments), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC was performed (C-F). Bar diagrams show lifetime mean values (B,G) or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC (D,F; 3 independently performed immunoblot experiments). (H) Schematic representation showing how fluid shear stress exerted by the flowing blood and leukocyte-induced ICAM-1 clustering synergistically activate PIEZO1 to induce downstream signaling events resulting in opening of the endothelial barrier. Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (1-way ANOVA).
Actin polymerization and actomyosin contractility mediate increase of endothelial membrane tension and downstream signaling induced by ICAM-1 clustering. (A-G) HUVECs were preincubated without or with 10 µM cytochalasin D (CytoD) or 30 µM blebbistatin (Bleb) (A-D) or were transfected with control siRNA (siCtrl) or siRNA directed against the RNA encoding α-actinin-4 (siACTN4) or cortactin (siCTTN) (E-G) and were exposed to low flow alone, anti–ICAM-1 clustering antibodies (ICAM-1 XL) alone or both, and membrane tension was determined using FliptR (A-B,G; n = 20 measurements from 3 independent experiments), or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC was performed (C-F). Bar diagrams show lifetime mean values (B,G) or immunoblot analysis of total and phosphorylated PYK2, SRC, and MLC (D,F; 3 independently performed immunoblot experiments). (H) Schematic representation showing how fluid shear stress exerted by the flowing blood and leukocyte-induced ICAM-1 clustering synergistically activate PIEZO1 to induce downstream signaling events resulting in opening of the endothelial barrier. Shown are mean values ± SEM. *P ≤ .05; **P ≤ .01; ***P ≤ .001 (1-way ANOVA).
Discussion
We report that the mechanosensitive cation channel PIEZO1 plays a critical role in transendothelial migration of leukocytes in vitro and in vivo by integrating low levels of fluid shear stress and mechanical signals induced by clustering of ICAM-1 and thereby mediating increases in [Ca2+]i and a localized opening of the endothelial barrier (Figure 6H). It has been reported that TRPC6 is critically involved in leukocyte-induced increases in endothelial [Ca2+]i during leukocyte transendothelial migration.19 In expression analyses, we were not able to observe expression of TRPC6 in endothelial cells (supplemental Figure 1J). Nevertheless, it could well be that both PIEZO1 and TRPC6 operate in parallel under in vivo conditions or that PIEZO1 is involved in the initiation of leukocyte extravasation by sensing both flow and leukocyte adhesion, whereas TRPC6 mediates increases in [Ca2+]i mainly at later stages of diapedesis.
ICAM-1 is a central endothelial adhesion receptor that functions as a ligand for β2 integrins on leukocytes and promotes leukocyte spreading, migration, and transmigration.29,42 Engagement of ICAM-1 leads to the clustering of ICAM-1 molecules and its redistribution into ring-like structures around adherent leukocytes, which is a requirement for efficient downstream signaling.43,44 ICAM-1 clustering induces cytoskeletal changes such as actin polymerization, MLC phosphorylation, and actomyosin contractility, which promote junctional opening.18,31,42,45 ICAM-1 also promotes increase in [Ca2+]i levels,13,46 which has been shown to lead to activation of SRC via protein kinase C.15 ICAM-1–mediated activation of SRC and PYK2 has been shown to be required for VE-cadherin–dependent leukocyte transendothelial migration.30 This involves direct phosphorylation of VE-cadherin30,47,48 and indirect regulation of VE-cadherin through VE-PTP49 or by phosphorylation of β-catenin.50 How ICAM-1 clustering induces activation of these downstream signaling events resulting in junctional opening and transendothelial migration is poorly understood. Our data indicate that downstream signaling through ICAM-1 requires coactivation of PIEZO1 by fluid shear stress and ICAM-1–induced reorganization of the cortical cytoskeleton.
Various mechanical stimuli acting on cellular membranes have been shown to be able to activate PIEZO1. These include exposure to fluid shear stress, mechanical indentation of the cell surface, cell migration, compression of the cell membrane, or forces generated at the cell–cell or cell–matrix interface.21,51 Our data show that low-level fluid shear stress and interaction of leukocytes with the endothelial surface act in a synergistic manner to activate endothelial PIEZO1 and to initiate leukocyte transendothelial migration. In postcapillary venules, the place where leukocyte extravasation mainly takes place, the shear stress exerted by the flowing blood is relatively low at about 1 to 2 dynes/cm,2,52,53 a shear rate hardly able to induce PIEZO1-mediated signaling.27,28 Consistent with this, we saw only very small increases in [Ca2+]i and no significant increase in the phosphorylation of PYK2, SRC, or MLC in response to fluid shear stress of 1.2 dynes/cm2. Similarly, when ICAM-1 clustering was induced in TNFα-pretreated endothelial cells by PMNs or anti–ICAM-1 antibodies, only small increases in [Ca2+]i and phosphorylation of PYK2, SRC, and MLC could be observed, which were further reduced after suppression of Piezo1 expression. However, when endothelial ICAM-1 clustering was induced while exposing cells to low flow, downstream signaling was strongly activated, and this effect was inhibited by knockdown of PIEZO1. This raised the question as to how ICAM-1 clustering promotes PIEZO1 activation. Both ICAM-1 clustering and adhesion of leukocytes to endothelial cells have been shown to induce stiffening of the endothelial surface and to induce traction stress.31-34,54 These endothelial responses are caused by increased actin polymerization and actomyosin contractility of the cortical cytoskeleton, which lead to increased cortical tension40,41 and require recruitment of the actin adapter proteins α-actinin-4 and cortactin.31,37 Because the plasma membrane and the underlying cortical cytoskeleton are closely interconnected,40,41 changes in the actomyosin cortical tension directly affect plasma membrane tension41 and therefore are likely to regulate PIEZO1 activity. Consistent with this, we found that inhibition of actin polymerization and myosin activity and siRNA-mediated knockdown of α-actinin-4 and cortactin blocked ICAM-1–mediated increases in membrane tension and PIEZO1-dependent downstream signaling required for leukocyte transendothelial migration. We therefore also think that it is the combined effect of low flow and leukocyte-induced ICAM-1 clustering on plasma membrane tension that induces PIEZO1 activation. This is consistent with a series of biophysical studies showing that changes in plasma membrane tension are sufficient to regulate the open probability of PIEZO1 and that no intra- or extracellular interactions are required.23-25 However, we cannot exclude that additional mechanisms are involved. For instance, it has recently been suggested that Piezo1 can be linked to the actin cytoskeleton through members of the cadherin family including VE-cadherin,55 and this mechanism may contribute to Piezo1 activation induced by ICAM-1 clustering and subsequent actin polymerization.
Recent data indicate that changes in plasma membrane tension are restricted to subcellular domains of endothelial cells as local increases in membrane tension lead only to local activation of mechanosensitive ion channels such as PIEZO1.56 The finding that leukocyte-induced endothelial downstream signaling and diapedesis require PIEZO1 and flow is consistent with earlier observations, which showed that fluid shear stress promotes transendothelial leukocyte migration.57-60 Our data identify a novel synergism of local hemodynamic forces and initial endothelial leukocyte adhesion to induce plasma membrane tension and endothelial signaling events that promote leukocyte extravasation. The discovery of a novel mechanosensing and mechanosignaling process required for the initial phase of leukocyte diapedesis may also lead to new anti-inflammatory therapeutic approaches.
Acknowledgments
The authors thank Svea Hümmer for secretarial help; Yin Hao (Instrument Analysis Center of Xi’an Jiaotong University) for assistance with fluorescence-lifetime imaging microscopy; Shuya Liu, Martina Finkbeiner, Ulrike Krüger, and Claudia Ullmann for technical help; and Bo Liu (Dalian University of Technology, Dalian, China) for donating a plasmid encoding the MSS sensor.
This work was supported by the National Key R&D Program of China (2021YFA1301200 [Z.Y.]), the National Natural Science Foundation of China (grants 81870220 and 82122008 [S.W.] and grants 92049203 and 81941005 [Z.Y.]), the Shaanxi Natural Science Fund for Distinguished Young Scholars of China (S2020-JC-JQ-0239 [S.W.]), the Collaborative Research Centre 834 of the German Research Foundation (S.O.), and the Collaborative Research Center 1348 of the German Research Foundation (D.V.).
Authorship
Contribution: S.W. initiated and designed the study, performed experiments, analyzed data, and wrote the manuscript; Y.S., B.W., and C.W. performed in vitro experiments; T.M. and R.I.S. performed in vivo experiments; B.S. helped with in vitro and in vivo experiments; T.L. and Z.Y. helped with in vitro experiments; N.W. supervised part of the study and discussed data; D.V. supervised part of the in vivo experiments and analyzed and discussed data; S.O. designed and supervised the study, discussed data, and wrote the manuscript; and all authors commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: ShengPeng Wang, Department of Cardiology, First Affiliated Hospital, Cardiovascular Research Center, Xi'an Jiaotong University, No. 277 W Yanta Rd, Yanta District 710061, Xi’an, China; e-mail: shengpeng.wang@xjtu.edu.cn; and Stefan Offermanns, Max Planck Institute for Heart and Lung Research, Department of Pharmacology, Ludwigstr 43, 61231 Bad Nauheim, Germany; e-mail: stefan.offermanns@mpi-bn.mpg.de.
Contact the corresponding authors for original data.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal