

Through genome-wide association studies (GWAS) and fine-mapping, Karnes et al,1 in this issue of Blood, have shown that having platelet factor 4 (PF4)/heparin antibodies in the presence of type O blood predisposes one to heparin-induced thrombocytopenia (HIT).
Unfractionated heparin (UFH) was first isolated in 1916 and entered clinical practice after its structure was determined in the 1930s. It remains one of the oldest drugs still in widespread clinical use.2 Of all the anticoagulants, heparin’s unique pharmacological properties will ensure its continued therapeutic use. However, HIT, first described in 1958, remains a significant iatrogenic complication of heparin use.3
HIT is a pathological prothrombotic syndrome caused by an immunoglobulin G (IgG)-mediated immune response to complexes of PF4 bound to heparin. HIT antibodies can activate platelets, resulting in procoagulant platelet membrane changes that enhance thrombin generation.4 HIT occurs in ∼2.4% of patients receiving UFH and low-molecular-weight heparin.1 The mortality rate of HIT is high, and the prevalence of thromboembolic complications in these patients has been put at 60%.5 Currently, it is impossible to know before administration of heparin which patients will develop HIT.6 Furthermore, HIT has similarities to adenoviral vector SARS-CoV-2 vaccine-induced thrombotic thrombocytopenia (VITT); thus, discoveries related to HIT could aid in management of VITT (see figure).
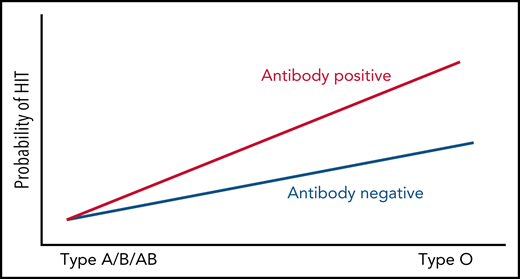
Despite extensive research in the field, molecularly distinguishing pathogenic vs nonpathogenic PF4/heparin antibodies is uncertain. This paper addressed this problem using a genome-wide association study (GWAS) and fine-mapping approach where the phenotype in cases is both a positive heparin-induced platelet activation (HIPA) assay and a PF4/heparin-positive functional assay, and controls are divided into the following 2 groups: (1) antibody-positive (functional assay-negative) with negative HIPA and positive PF4/heparin antibodies; and (2) PF4/heparin antibody-negative controls with both negative functional assay (HIPA) and PF4/heparin antibody-negative results. Because of the experimental design of the control groups, they were importantly able to discover that in the presence of PF4/heparin antibodies, the rs8176719 C deletion encoding the O blood group is a novel risk factor for both platelet reactivity and HIT.
The strength of GWAS and other hypothesis-agnostic genetic approaches lies in their unbiased nature, especially in cases such as HIT where few candidate genes exist. Prior GWAS of patients with HIT were extremely underpowered, lacked appropriate control groups, and had no associations that passed genome-wide significance thresholds.7 Karnes et al performed a large case-control GWAS with the above 2 stated control groups. Polymerase chain reaction–based sequencing was then performed on the ABO gene, and fine-mapping algorithms were used for determining causal single nucleotide polymorphisms. Proper quality control steps and statistical adjustments for genetic ancestry were taken, and results were replicated in an independent cohort.
That the rs8176719 C deletion allele, which encodes for the O blood group, was a significant risk factor for functional assay-positive status when compared with antibody-positive controls but not when compared with antibody-negative controls implies there is an interaction between O blood type and having these antibodies that cause the functional assay to become positive. Thus, having PF4/heparin antibodies in the presence of type O blood predisposes one to thrombocytopenia. Furthermore, the outcome of thrombocytopenia from PF4/heparin antibodies may not be due to type O blood group directly, but rather to one or more of the many relevant phenotypes/biomarkers that have been previously associated with the ABO locus, including p-selectin, a marker of platelet activation, von Willebrand factor antigen levels, factor VIII levels, and the IgG receptor, FcγRIIa. In fact, there are established pathogenic mechanisms directly linking IgG-mediated activation of platelet and monocyte FcγRIIa receptors to thrombin generation,3 and polymorphisms in protein tyrosine phosphatase receptor type J (PTPRJ), also known as CD148, have been linked to risk of HIT.8 Furthermore, loss-of-function variants in this gene have been linked to a new form of inherited thrombocytopenia.9
It is clear that their choice of experimental design and genetic methods paid off. The clinical implications are significant in that it can now be deemed advisable to be more vigilant for signs of HIT in patients receiving heparin with type O blood. In addition, these results could be translatable to VITT. The authors state in the introduction that they wish to discover novel biomarkers for HIT, which could potentially be clinically actionable. They have done this and have provided several promising avenues to pursue next. Future studies could, as suggested by the authors, involve Mendelian randomization (MR) to parse potential relationships between these biomarkers that are all associated with the ABO locus. MR is a well-accepted method using measured variation in genes to interrogate the causal effect of an exposure on an outcome. In addition, in the era of big data, other future avenues of exploration could involve analysis of noncoding DNA, methylation, proteomics, and other high-throughput approaches.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal