

Key Points
A menin-MLL1 inhibitor halts leukemogenesis in models of NUP98-rearranged leukemias.
Inhibition of menin-MLL1 impairs leukemogenic gene expression and disrupts chromatin binding of menin, MLL1 and NUP98 fusion proteins.

Abstract
Translocations involving the NUP98 gene produce NUP98-fusion proteins and are associated with a poor prognosis in acute myeloid leukemia (AML). MLL1 is a molecular dependency in NUP98-fusion leukemia, and therefore we investigated the efficacy of therapeutic blockade of the menin-MLL1 interaction in NUP98-fusion leukemia models. Using mouse leukemia cell lines driven by NUP98-HOXA9 and NUP98-JARID1A fusion oncoproteins, we demonstrate that NUP98-fusion-driven leukemia is sensitive to the menin-MLL1 inhibitor VTP50469, with an IC50 similar to what we have previously reported for MLL-rearranged and NPM1c leukemia cells. Menin-MLL1 inhibition upregulates markers of differentiation such as CD11b and downregulates expression of proleukemogenic transcription factors such as Meis1 in NUP98-fusion-transformed leukemia cells. We demonstrate that MLL1 and the NUP98 fusion protein itself are evicted from chromatin at a critical set of genes that are essential for the maintenance of the malignant phenotype. In addition to these in vitro studies, we established patient-derived xenograft (PDX) models of NUP98-fusion-driven AML to test the in vivo efficacy of menin-MLL1 inhibition. Treatment with VTP50469 significantly prolongs survival of mice engrafted with NUP98-NSD1 and NUP98-JARID1A leukemias. Gene expression analysis revealed that menin-MLL1 inhibition simultaneously suppresses a proleukemogenic gene expression program, including downregulation of the HOXa cluster, and upregulates tissue-specific markers of differentiation. These preclinical results suggest that menin-MLL1 inhibition may represent a rational, targeted therapy for patients with NUP98-rearranged leukemias.
Introduction
Acute myeloid leukemia (AML) driven by chromosomal translocations involving the nucleoporin 98 (NUP98) gene on chromosome 11p15 is the most common genotype among children with relapsed and refractory disease, representing a high-risk group of patients with extremely poor outcomes.1-3,NUP98 translocations generate NUP98 fusion proteins that join the N-terminal domain of NUP98 with various C-terminal partners that include epigenetic modifiers and transcription factors with homeobox domains such as the HOX genes.4,5 Previous efforts to model NUP98-rearranged hematologic malignancies have revealed that NUP98-JARID1A, NUP98-NSD1, NUP98-HOXA9, and NUP98-HOXD13 fusion proteins are potent drivers of leukemia development.6-9 While these fusions are potent oncoproteins, accompanying mutations in genes such as WT1 or FLT3 internal tandem duplications confer an even more dismal prognosis.10 There is an urgent clinical need to understand the mechanisms by which NUP98 fusion proteins drive leukemogenesis.
NUP98 fusion proteins promote leukemogenesis through their interaction with histone-modifying chromatin complexes.11-13 Previous studies have implicated histone methylation and acetylation at the Hoxa/b clusters, as well as the Meis1 locus, as a mechanism that supports alterations in gene expression that promote leukemia development. Through its interaction with Wdr8, the NUP98-NSD1 fusion recruits the Wdr82–Set1A/COMPASS (complex of proteins associated with Set1) complex, which deposits an activating mark on histone H3 lysine 4 (H3K4me3) across the HOXa cluster and at the Meis1 promoter.11 NUP98 fusions also interact with the non-specific lethal (NSL) and mixed-lineage leukemia (MLL1; also known as KMT2A) chromatin complexes. NUP98-HOXA9 colocalizes with MLL1 at the Hoxa/b clusters, and loss of MLL1 delays onset of disease in a NUP98-HOXA9 mouse leukemia model, demonstrating that MLL1 is a molecular dependency in NUP98-rearranged leukemia.13
The multiple endocrine neoplasia 1 (MEN1) gene encodes the protein menin, which is a member of the MLL1 and MLL2 complexes. Menin is critical for the proliferation and survival of MLL-rearranged (MLL-r) and NPM1-mutant (NPM1c) leukemia.14,15 MLL1 and menin directly interact via a 5 amino acid stretch of MLL1 that binds to a well-defined pocket on menin. This interaction is critical for menin function in leukemia, and small molecules that compete for the menin binding pocket are being developed as therapeutics in MLL-r and NPM1c AML.16-18 Recently, a selective and orally bioavailable small molecule (VTP50469) targeting the menin-MLL1 interaction has been effective in the treatment of MLL-r16,17 and NPM1c mutant leukemia.18 In those leukemia subtypes, treatment with VTP50469 led to the loss of MLL1 and menin binding at specific loci on chromatin and consequent downregulation of leukemogenic gene expression.17,18 Three different menin-MLL1 inhibitors (SDNX-5613, JNJ-75276617, and KO-539) are currently in early-phase clinical trials. Since MLL1 is a molecular dependency in NUP98-rearranged leukemia,13 we hypothesized that disruption of the menin-MLL1 chromatin complex may be a rational, targeted therapy for this group of patients whose AML is typically resistant or refractory to conventional chemotherapy.
Methods
Animals
For mouse leukemia models: Lin-, cKit+, Sca-1+ (LSK) cells were isolated from C57BL/6NCrl mice (C57BL/6, Charles River Laboratories) or Mx1-Cre Wt1 flox/flox mice that have been previously described.19,20 Cells were transplanted into CD45.1 recipients (B6.SJL, Taconic). For patient-derived xenograft (PDX) experiments: NOG-EF NOG-sp/sp;ko/ko (NOG) mice (Taconic) were used as recipients. Animal experiments were performed with the approval of the Institutional Animal Care and Use Committee at Dana-Farber Cancer Institute, Memorial Sloan-Kettering Cancer Center, and Nemours Children’s Health Center.
Chromatin immunoprecipitation and sequencing (ChIPseq)
ChIPseq for MLL1, menin, and H3K4me3 was performed in NUP98-rearranged mouse leukemia cells as previously described.17,18,21,22 Cells were crosslinked, and nuclei were isolated and sonicated using the E100S (Covaris) to shear chromatin into DNA fragments ranging from 200 to 400 base pairs. Drosophila DNA (Active Motif 53083) was added to sonicated chromatin prior to pulldown and removal of the input fraction according to the manufacturer’s instructions. Between 1 and 5 mg of chromatin was subjected to immunoprecipitation using the anti-MLL antibody (Bethyl A300-086A), anti-menin antibody (Bethyl A300-105A), anti-H3K4me3 (Abcam 8580), or Drosophila DNA internal control antibody (Active Motif 61686). Immunoprecipitated DNA fragments were eluted, decrosslinked, and purified using Agencourt AMPure XP beads for the generation of Illumina-compatible sequencing libraries.
ChIPseq of biotinylated NUP98 fusion proteins (BioChIPseq)
BioChIPseq for NUP98-HOXD13bio was performed as previously described.13 Chromatin was subjected to immunoprecipitation for biotinylated NUP98-HOXD13bio using Dynabeads M-280 Streptavidin (Thermo Fisher 11205D).
PDX experiments
PDX experiments were conducted in NOG mice. Cells were injected from NUP98-rearranged AML patient samples, and the presence of each fusion and cooccurring mutation was confirmed by next-generation sequencing (supplemental Table 3 available on the Blood Web site). For survival experiments, engraftment was confirmed on day 21 following transplantation when human CD45+ cells in the blood reached 1% to 5%. Mice were subsequently randomized into groups to receive control or VTP50469 chow. Mice were bled weekly to assess leukemia burden. Some mice were killed at day 14 or 36 to assess leukemia burden in marrow and spleens.
Results
MLL1 and menin are molecular dependencies in NUP98-rearranged leukemia
We generated murine models of NUP98-rearranged leukemia using retroviral transduction of mouse LSK cells with a vector system containing a fluorescent reporter (pMSCV-IRES-GFP or pMSCV-IRES-TdTomato). We focused on NUP98-HOXA9 and NUP98-JARID1A (also known as KDM5A) since the ability of these fusion proteins to transform mouse LSK cells without cooperating mutations had been previously described.9,23 To confirm that the retrovirally transduced mouse LSK cells harboring these fusion oncogenes are fully transformed and give rise to de novo leukemia in vivo, we transplanted the cells into sublethally irradiated syngeneic recipient mice. Engraftment of LSK cells transduced with either NUP98-HOXA9 or NUP98-JARID1A fusion proteins resulted in relatively rapid onset of disease at 112 days and 85 days, respectively (supplemental Figure 1A,B). As expected, disease latency was further reduced after secondary transplantation (NUP98-HOXA9: 42 days; NUP98-JARID1A: 56 days), indicating disease acceleration (supplemental Figure 1A-D, black lines). Disease progression was accompanied by a rise in transplanted cells in the peripheral blood (supplemental Figure 1C,D). At the time point of euthanasia, bone marrow (BM) and spleens of the primary recipients were highly infiltrated with leukemia cells (supplemental Figure 1E-H).
Our previous work has shown that MLL-r and NPM1c leukemia are dependent on the menin-MLL1 interaction and respond to VTP50469, a small molecule that inhibits the interaction between menin and MLL1.17,18 Since NUP98 fusion proteins interact with MLL1,13 we reasoned that these leukemias might also depend on the menin-MLL1 interaction and may be responsive to treatment with VTP50469. To test this hypothesis, we performed genetic inactivation of menin and MLL1 using CRISPR-Cas9 technology. We engineered NUP98-HOXA9 and NUP98-JARID1A mouse leukemia cells to constitutively express the Cas9 enzyme and introduced small guide RNA (sgRNA) targeting MLL1 or menin using vectors containing an RFP or GFP reporter (supplemental Table 1). We confirmed CRISPR editing at the menin and MLL1 locus by CRISPRseq and immunoblotting for menin protein (supplemental Figure 2A,B). Next, we assessed the ability of cells expressing these sgRNAs to compete with nontransduced cells in an in vitro cell competition assay. We found that sgRNA targeting the menin or MLL1 gene results in rapid depletion of cells compared to the common essential gene Rpa3 (Figure 1A), indicating a strong dependency of NUP98-rearranged leukemia cells on the MLL1-complex, and more specifically on both MLL1 and menin.

MLL and menin are molecular dependencies in NUP98-rearranged leukemia. (A) NUP98-HOXA9 (top panel) and NUP98-JARID1A (bottom panel) mouse leukemia cells engineered to constitutively express Cas9 were transduced with sgRNA targeting MLL1, menin, RPA3 (positive control) and luciferase (negative control). Cells expressing sgRNA also express an RFP or GFP reporter, and the y-axis quantifies the percent RFP+ or GFP+ cells at each time point. Bars represent mean ± SEM. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. Data are representative of 3 independent experiments. (B,C) Mouse leukemia cells expressing NUP98-HOXA9 (B) or NUP98-JARID1A (C) were treated with an escalating dose curve of VTP50469. Data are representative of 5 independent experiments. (D,E) Expression of CD11b on NUP98-HOXA9 (D) or NUP98-JARID1A (E) mouse leukemia cells treated with an escalating dose curve of VTP50469. Data are representative of 5 independent experiments. Bars represent mean ± SEM. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ***P = .002, ****P < .0001.
MLL and menin are molecular dependencies in NUP98-rearranged leukemia. (A) NUP98-HOXA9 (top panel) and NUP98-JARID1A (bottom panel) mouse leukemia cells engineered to constitutively express Cas9 were transduced with sgRNA targeting MLL1, menin, RPA3 (positive control) and luciferase (negative control). Cells expressing sgRNA also express an RFP or GFP reporter, and the y-axis quantifies the percent RFP+ or GFP+ cells at each time point. Bars represent mean ± SEM. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. Data are representative of 3 independent experiments. (B,C) Mouse leukemia cells expressing NUP98-HOXA9 (B) or NUP98-JARID1A (C) were treated with an escalating dose curve of VTP50469. Data are representative of 5 independent experiments. (D,E) Expression of CD11b on NUP98-HOXA9 (D) or NUP98-JARID1A (E) mouse leukemia cells treated with an escalating dose curve of VTP50469. Data are representative of 5 independent experiments. Bars represent mean ± SEM. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ***P = .002, ****P < .0001.
To determine whether we could exploit this dependency on MLL1 and menin therapeutically, we treated NUP98-JARID1A and NUP98-HOXA9 mouse leukemia cells with the menin-MLL1 inhibitor VTP50469 in vitro and quantified live cell numbers after 9 days of treatment. Both NUP98-JARID1A and NUP98-HOXA9 leukemia cells demonstrated sensitivity to menin-MLL1 inhibition, with an IC50 of 84 nM and 517 nM, respectively (Figure 1B,C). Mouse LSK cells transduced with a control empty vector (EV) showed no response to VTP50469. We observed a dose-dependent upregulation of the differentiation marker CD11b (Figure 1D,E), while acute induction of apoptosis was not detected (supplemental Figure 2C,D). Cytologically, treatment of NUP98-JARID1A and NUP98-HOXA9 leukemia cells with VTP50469 resulted in phenotypic changes consistent with myeloid differentiation (supplemental Figure 2E,F). These experiments show that NUP98-rearranged leukemia requires the menin-MLL1 interaction for the maintenance of cell division and maintenance of an undifferentiated leukemia phenotype.
VTP50469 treatment impairs leukemogenic gene expression and disrupts chromatin binding of MLL1 and NUP98 fusion proteins
Next, we sought to perform a comprehensive assessment of the molecular effects of menin-MLL1 inhibition in NUP98-fusion leukemia. To understand how menin-MLL1 inhibition influences gene expression, we performed RNAseq in NUP98-fusion mouse leukemia cells treated with VTP50469. After 3 days of treatment, we observed remarkably specific changes in gene expression in NUP98-HOXA9 and NUP98-JARID1A mouse leukemia cells (Figure 2A,B, top panels). At this early time point, expression of proleukemogenic transcriptional regulators, such Meis1, Eya1, and Pbx3, was significantly repressed. After 6 days of treatment with VTP50469, we observed >Log2 fold downregulation of more than 250 to 300 genes, which is consistent with our observation of phenotypic differentiation at later time points (Figure 2A,B, bottom panels). GSEA-based comparison changes in gene expression demonstrated that this pattern of transcriptional reprogramming is similar to that seen in MLL-r and NPM1c leukemia cells treated with VTP5046917,18 (Figure 2C,D), suggesting that inhibition of menin-MLL1 impacts a program that is central to maintaining an undifferentiated stem cell state across multiple types of leukemia.


MLL1 is displaced from chromatin upon treatment with VTP50469 at a subset of critical genes. (A,B) Gene expression changes at day 3 (top panels) and day 6 (bottom panels) of treatment with 2 μM VTP50469 in mouse leukemia cells expressing NUP98-HOXA9 (A) or NUP98-JARID1A (B). n = 3 replicates per time point. (C,D) Gene-set enrichment analysis of gene expression changes in NUP98-HOXA9 (C) or NUP98-JARID1A (D) mouse leukemia cells treated with VTP50469 compared with that seen in MLL-r (MA9) and NPM1c mouse leukemia cells. (E) Tornado plots and average signal plots depicting genome-wide MLL1, menin, and H3K4me3 chromatin changes upon treatment with VTP50469 as determined by ChIPseq in mouse leukemia cells expressing NUP98-HOXA9. (F) Gene tracks of MLL1 chromatin occupancy in NUP98-HOXA9 mouse leukemia cells treated with VTP50469 as determined by ChIPseq in reads per million (rpm) at selected genes. (G) Violin plots depicting change in MLL1 occupancy for genes that display changes in gene expression vs all genes in NUP98-HOXA9 mouse leukemia cells after 3 days (left panel) or 6 days (right panel) of treatment with VTP50469. Data are representative of 2 independent experiments. One-way ANOVA was performed.
MLL1 is displaced from chromatin upon treatment with VTP50469 at a subset of critical genes. (A,B) Gene expression changes at day 3 (top panels) and day 6 (bottom panels) of treatment with 2 μM VTP50469 in mouse leukemia cells expressing NUP98-HOXA9 (A) or NUP98-JARID1A (B). n = 3 replicates per time point. (C,D) Gene-set enrichment analysis of gene expression changes in NUP98-HOXA9 (C) or NUP98-JARID1A (D) mouse leukemia cells treated with VTP50469 compared with that seen in MLL-r (MA9) and NPM1c mouse leukemia cells. (E) Tornado plots and average signal plots depicting genome-wide MLL1, menin, and H3K4me3 chromatin changes upon treatment with VTP50469 as determined by ChIPseq in mouse leukemia cells expressing NUP98-HOXA9. (F) Gene tracks of MLL1 chromatin occupancy in NUP98-HOXA9 mouse leukemia cells treated with VTP50469 as determined by ChIPseq in reads per million (rpm) at selected genes. (G) Violin plots depicting change in MLL1 occupancy for genes that display changes in gene expression vs all genes in NUP98-HOXA9 mouse leukemia cells after 3 days (left panel) or 6 days (right panel) of treatment with VTP50469. Data are representative of 2 independent experiments. One-way ANOVA was performed.
To determine whether treatment with VTP50469 disrupted the menin-MLL1 chromatin complex in NUP98-HOXA9 and NUP98-JARID1A mouse leukemia cells, we performed ChIPseq for menin and MLL1. Menin chromatin occupancy was decreased genome-wide, consistent with our previous results showing global loss of menin upon treatment with VTP5046917,18 (Figure 2E). By contrast, we observed no changes in genome-wide MLL1 chromatin binding or H3K4me3 marks in cells treated with VTP50469 (Figure 2E, supplemental Figure 3A), consistent with our previous studies.17,18 We observed loss of MLL1 chromatin association at specific genes which were also among those with the most significantly reduced gene expression, such as Meis1, Eya1, and Pbx3 (Figure 2F,G, supplemental Figure 3B,C).
Based on previous observations in other leukemias, we hypothesized that changes in MLL1 chromatin occupancy would correlate with gene expression changes induced by VTP50469. At both early and late time points of drug treatment, we observed that significantly repressed gene expression (defined as those genes with >Log1.5 fold change in expression) is correlated with loci that demonstrate significant changes in MLL1 occupancy (Figure 2F,G, supplemental Figure 3B,C, and supplemental Tables 2 and 3). For example, genes such as Meis1 demonstrate almost complete loss of MLL1 chromatin binding at the promoter and also demonstrate significantly reduced gene expression upon menin-MLL1 inhibition.
Our previous studies suggest that NUP98 fusion proteins associate with the MLL chromatin complex,13 and therefore, we questioned whether the NUP98 fusion itself was evicted from chromatin upon treatment with VTP50469. To address this hypothesis, we employed a mouse leukemia model that expresses a C-terminal Flag-Avi tagged NUP98 fusion protein in cells that stably express bacterial BirA biotin ligase. As a result, NUP98 fusion proteins are biotinylated and can be immunoprecipitated from chromatin using streptavidin beads. This strategy was used to generate mouse leukemia cells expressing NUP98-HOXD13 and has been previously described.13 We confirmed that these cells, hereafter referred to as NUP98-HOXD13bio, are sensitive to menin-MLL1 inhibition and upregulated markers of differentiation such as CD11b (supplemental Figure 4A,B). Using this model, we found that treatment with VTP50469 did not induce global changes in NUP98-HOXD13bio chromatin occupancy (Figure 3A). In previous studies, we defined a subset of NUP98-HOXD13bio gene targets,13 and we were interested in whether VTP50469 induced chromatin changes at these loci. At NUP98-HOXD13 targets, treatment with VTP50469 led to the eviction of MLL1, menin, and the NUP98-HOXD13bio fusion protein itself (Figure 3B). For example, genes such as Meis1, Eya1, and Mef2c (all NUP98-HOXD13 targets that function as transcriptional regulators that maintain an undifferentiated stem cell phenotype) were among those that displayed the most significant loss of MLL1, menin, and NUP98 fusion chromatin binding (Figure 3B).

NUP98-fusion proteins dissociate from chromatin upon treatment with VTP50469 at a subset of critical genes. (A) Tornado plot and average signal plot depicting genome-wide NUP98-HOXD13bio chromatin occupancy as determined by ChIPseq in mouse leukemia cells expressing NUP98-HOXD13bio. (B) Gene tracks of MLL1, Menin, and NUP98-HOXD13bio ChIPseq signal (rpm) at selected genes. (C) Violin plot depicting changes in NUP98-HOXD13bio occupancy for previously defined NUP98-HOXD13 target genes13 that display >log2 fold change in gene expression (dark blue, n = 19 genes) vs all NUP98-HOXD13 targets (light blue, n = 295 genes) vs all genes (gray, n = 616 genes). (D) Scatterplot depicting all genes that display changes in both NUP98-HOXD13bio (x-axis) and MLL1 (y-axis) chromatin occupancy as measured by ChIPseq in NUP98-HOXD13bio mouse leukemia cells. Data are representative of 2 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed.
NUP98-fusion proteins dissociate from chromatin upon treatment with VTP50469 at a subset of critical genes. (A) Tornado plot and average signal plot depicting genome-wide NUP98-HOXD13bio chromatin occupancy as determined by ChIPseq in mouse leukemia cells expressing NUP98-HOXD13bio. (B) Gene tracks of MLL1, Menin, and NUP98-HOXD13bio ChIPseq signal (rpm) at selected genes. (C) Violin plot depicting changes in NUP98-HOXD13bio occupancy for previously defined NUP98-HOXD13 target genes13 that display >log2 fold change in gene expression (dark blue, n = 19 genes) vs all NUP98-HOXD13 targets (light blue, n = 295 genes) vs all genes (gray, n = 616 genes). (D) Scatterplot depicting all genes that display changes in both NUP98-HOXD13bio (x-axis) and MLL1 (y-axis) chromatin occupancy as measured by ChIPseq in NUP98-HOXD13bio mouse leukemia cells. Data are representative of 2 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed.
Overall, loss of NUP98 fusion protein chromatin occupancy was most significant at those NUP98 fusion target loci that also displayed >Log2 fold change in gene expression (Figure 3C, supplemental Table 4). To understand whether loss of NUP98-HOXD13bio was also associated with loss of MLL1, we performed MLL1 ChIPseq in NUP98-HOXD13bio cells. We observed that genes which demonstrate the most significant loss of MLL1 also demonstrate the most significant loss of NUP98 fusion protein (Figure 3B,D). We did not observe significant loss of MLL1, menin, and NUP98-HOXD13bio chromatin occupancy at the HOXa cluster, nor did we observe changes in HOXa gene expression (supplemental Figure 4C). Changes at the HOXa cluster are sluggish compared with loci such as Meis1, and these results are consistent with our previous findings in MLL-r and NPM1c leukemias.17,18 These results suggest that menin-MLL1 inhibition destabilizes chromatin complexes containing NUP98 fusion proteins, leading to the downregulation of a proleukemogenic gene expression program.
Rescue of Meis1 gene expression abrogates sensitivity to VTP50469
Since Meis1 was one of the critical loci consistently impacted by treatment with VTP50469, we questioned whether restoration of Meis1 expression was sufficient to rescue proliferation of NUP98 fusion cells in the presence of VTP50469. First, we used retroviral overexpression of Meis1 in NUP98-JARID1A and NUP98-HOXA9 mouse leukemia cells. We confirmed that Meis1 mRNA and protein expression increased above baseline, compared with EV controls (Figure 4A,B). Meis1 expression remained high in cells overexpressing exogenous Meis1 despite treatment with VTP50469 (Figure 4A,B). Cell proliferation assays demonstrated a lack of response to menin-MLL1 inhibition and failure to upregulate markers of differentiation such as CD11b in cells overexpressing Meis1 (Figure 4C,D).
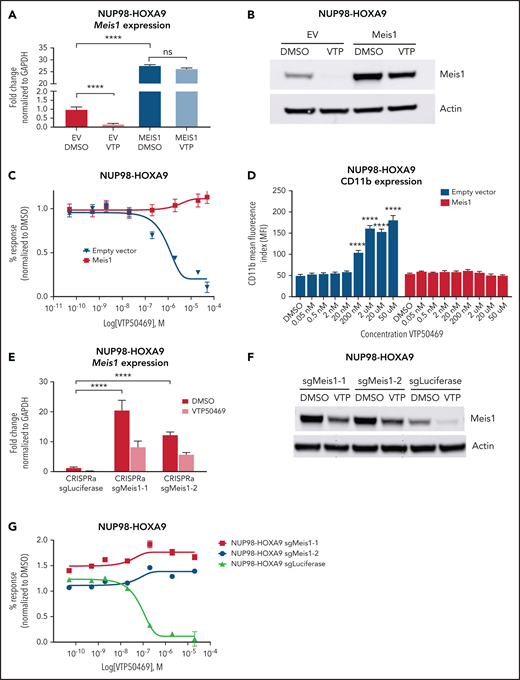
Rescue of Meis1 gene expression abrogates sensitivity to menin-MLL1 inhibition in NUP98-fusion leukemia. (A) Quantitative PCR for Meis1 mRNA in NUP98-HOXA9 mouse leukemia cells engineered to overexpress Meis1 and treated with DMSO or 2 μM VTP50469. Data are representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. (B) Immunoblot for Meis1 protein in NUP98-HOXA9 mouse leukemia cells engineered and treated as in (A). (C) NUP98-HOXA9 mouse leukemia cells engineered as in (A) and treated with an escalating dose curve of VTP50469. Data are representative of 3 independent experiments. (D) Expression of CD11b on NUP98-HOXA9 mouse leukemia cells overexpressing Meis1 and treated with an escalating dose curve of VTP50469. Data are representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. (E) Quantitative PCR for Meis1 mRNA in NUP98-HOXA9 mouse leukemia cells engineered to constitutively express dead Cas9 (dCas9) and transcriptional activators MS2, p65, and HSF1 with small guide RNAs targeting Meis1 or luciferase and treated with 2 μM VTP50469. Data are representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. (F) Immunoblot for Meis1 protein levels in cells engineered and treated as in (E). (G) NUP98-HOXA9 mouse leukemia cells engineered as in (E) and treated with an escalating dose curve of VTP50469. Data are representative of 3 independent experiments.
Rescue of Meis1 gene expression abrogates sensitivity to menin-MLL1 inhibition in NUP98-fusion leukemia. (A) Quantitative PCR for Meis1 mRNA in NUP98-HOXA9 mouse leukemia cells engineered to overexpress Meis1 and treated with DMSO or 2 μM VTP50469. Data are representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. (B) Immunoblot for Meis1 protein in NUP98-HOXA9 mouse leukemia cells engineered and treated as in (A). (C) NUP98-HOXA9 mouse leukemia cells engineered as in (A) and treated with an escalating dose curve of VTP50469. Data are representative of 3 independent experiments. (D) Expression of CD11b on NUP98-HOXA9 mouse leukemia cells overexpressing Meis1 and treated with an escalating dose curve of VTP50469. Data are representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. (E) Quantitative PCR for Meis1 mRNA in NUP98-HOXA9 mouse leukemia cells engineered to constitutively express dead Cas9 (dCas9) and transcriptional activators MS2, p65, and HSF1 with small guide RNAs targeting Meis1 or luciferase and treated with 2 μM VTP50469. Data are representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test was performed, ****P < .0001. (F) Immunoblot for Meis1 protein levels in cells engineered and treated as in (E). (G) NUP98-HOXA9 mouse leukemia cells engineered as in (E) and treated with an escalating dose curve of VTP50469. Data are representative of 3 independent experiments.
Next, in order to achieve more physiologic restoration of Meis1 gene expression, we used the CRISPRa system24 with sgRNA targeting the endogenous Meis1 locus to enhance transcriptional activity at the Meis1 promoter (supplemental Table 5). CRISPRa-induced upregulation of Meis1 resulted in increased expression at the transcript and protein level compared with the luciferase control (Figure 4E,F). CRISPRa-induced expression of Meis1 blunted the suppressive effects of VTP50469 on Meis1 transcription and translation, as Meis1 mRNA and protein levels remained higher than the EV DMSO control despite VTP50469 treatment (Figure 4E,F). Similarly, we found that CRISPRa mediated upregulation of Meis1 rescued the phenotype and rendered NUP98-HOXA9 mouse leukemia cells unresponsive to VTP50469 (Figure 4G). Rescue of Meis1 expression in NUP98-JARID1A mouse leukemia cells had significant, albeit more modest effects, on responsiveness to menin-MLL1 inhibition (supplemental Figure 5A-G). These results using 2 independent methods show that the menin-MLL1 chromatin complex maintains the malignant phenotype in part through activation of proleukemogenic transcription factors such as Meis1. Loss of NUP98 fusion and MLL1 binding results in decreased Meis1 expression, and failure to suppress Meis1 renders NUP98-fusion leukemia cells unresponsive to menin-MLL1 inhibition.
Menin-MLL1 inhibition eradicates disease in vivo in NUP98-rearranged models of AML
Our in vitro studies suggest that menin-MLL1 inhibition may be a rational, targeted therapeutic strategy for patients with NUP98-rearranged leukemia. Therefore, we investigated whether VTP50469 would be effective in treating NUP98-rearranged leukemia in vivo in mice. First, we treated our mouse model of NUP98-JARID1A leukemia with chow containing 0.1% VTP50469 and monitored disease (TdTomato+CD45.2+ cells) in peripheral blood and survival. Remarkably, treatment with VTP50469 led to a survival advantage of 56 days (P = .0016) (Figure 5A). Whereas the percent of transplanted leukemia cells rose rapidly in control mice between 30 and 50 days after transplant, VTP50469-treated mice demonstrated significantly longer disease latency and onset of clinical illness (Figure 5B).
![In vivo mouse leukemia models and patient-derived xenograft models of NUP98-fusion leukemia respond to menin-MLL1 inhibition. (A) Survival analysis of CD45.1 mice engrafted with NUP98-JARID1A mouse leukemia cells (CD45.2), treated with control or 0.1% VTP50469 chow (yellow shading) (n = 5 control mice, n = 8 VTP50469-treated mice). (B) Percent TdTomato positive CD45.2 positive NUP98-JARID1A mouse leukemia cells in the peripheral blood of CD45.1 recipient mice treated with control or 0.1% VTP50469 chow (n = 5 control mice, n = 8 VTP50469-treated mice). (C) Survival analysis of NOG mice engrafted with a NUP98-NSD1 PDX, treated with control, 0.05% or 0.1% VTP50469 chow (n = 5 mice per group; P < .001 using Log-rank [Mantel-Cox test]). (D) Percent human CD45 cells in the peripheral blood of NOG mice engrafted with a NUP98-NSD1 PDX, treated with control, 0.05% or 0.1% VTP50469 chow (n = 5 mice per group). Error bars represent S.E.M. (E) Survival analysis of NOG mice engrafted with a NUP98-JARID1A PDX, treated with 0.1% VTP50469 chow for 60 days (yellow shading) (n = 5 mice per group; P < .001 using Log-rank [Mantel-Cox test]). (F) Percent human CD45 cells in the peripheral blood of NOG mice engrafted with a NUP98-JARID1A PDX, treated with control or 0.1% VTP50469 chow for 60 days (yellow shading) (n = 5 mice per group). (G,H) Heatmap of gene expression changes characterized by RNAseq in human CD45 cells isolated from BM of NOG mice bearing a NUP98-NSD1 PDX (G) or NUP98-JARID1A PDX (H). Values in each cell represent the Log2 fold change in gene expression (VTP treated mice/Control mice).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/6/10.1182_blood.2021012806/3/m_bloodbld2021012806f5.png?Expires=1763470684&Signature=lJM0TCsKcXjeO139Pym0QETiAfQ2RNu74XQMhD6uSpgKyBhBw~EJciz0iq9WCX6Pj9MB45RZzUJk72kWMby2wEmeL6ss-q4TNTngNRhOlYH34swVmNrvm58F~okcZLe94Op3WtDPT0A2f1Eyi7Q4411hfiFrVrNZ6R9kOPpQOp4-Lxwgrb241sViNxDTO4i2Md3lfXgtHaZS9uk7dR91MSaB6-ml-0VQ0D6YCFZV48KLJMdtndJfFjmmbxkjXrWOt1JSTv6zLPPO9xG7R~tc6jbJNz8-XiWQMGe6XShXHnXk7VmbyZUCOwGVcxCcHSmcu-BomnLwSAPIFBKk9oeSCw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
In vivo mouse leukemia models and patient-derived xenograft models of NUP98-fusion leukemia respond to menin-MLL1 inhibition. (A) Survival analysis of CD45.1 mice engrafted with NUP98-JARID1A mouse leukemia cells (CD45.2), treated with control or 0.1% VTP50469 chow (yellow shading) (n = 5 control mice, n = 8 VTP50469-treated mice). (B) Percent TdTomato positive CD45.2 positive NUP98-JARID1A mouse leukemia cells in the peripheral blood of CD45.1 recipient mice treated with control or 0.1% VTP50469 chow (n = 5 control mice, n = 8 VTP50469-treated mice). (C) Survival analysis of NOG mice engrafted with a NUP98-NSD1 PDX, treated with control, 0.05% or 0.1% VTP50469 chow (n = 5 mice per group; P < .001 using Log-rank [Mantel-Cox test]). (D) Percent human CD45 cells in the peripheral blood of NOG mice engrafted with a NUP98-NSD1 PDX, treated with control, 0.05% or 0.1% VTP50469 chow (n = 5 mice per group). Error bars represent S.E.M. (E) Survival analysis of NOG mice engrafted with a NUP98-JARID1A PDX, treated with 0.1% VTP50469 chow for 60 days (yellow shading) (n = 5 mice per group; P < .001 using Log-rank [Mantel-Cox test]). (F) Percent human CD45 cells in the peripheral blood of NOG mice engrafted with a NUP98-JARID1A PDX, treated with control or 0.1% VTP50469 chow for 60 days (yellow shading) (n = 5 mice per group). (G,H) Heatmap of gene expression changes characterized by RNAseq in human CD45 cells isolated from BM of NOG mice bearing a NUP98-NSD1 PDX (G) or NUP98-JARID1A PDX (H). Values in each cell represent the Log2 fold change in gene expression (VTP treated mice/Control mice).
In vivo mouse leukemia models and patient-derived xenograft models of NUP98-fusion leukemia respond to menin-MLL1 inhibition. (A) Survival analysis of CD45.1 mice engrafted with NUP98-JARID1A mouse leukemia cells (CD45.2), treated with control or 0.1% VTP50469 chow (yellow shading) (n = 5 control mice, n = 8 VTP50469-treated mice). (B) Percent TdTomato positive CD45.2 positive NUP98-JARID1A mouse leukemia cells in the peripheral blood of CD45.1 recipient mice treated with control or 0.1% VTP50469 chow (n = 5 control mice, n = 8 VTP50469-treated mice). (C) Survival analysis of NOG mice engrafted with a NUP98-NSD1 PDX, treated with control, 0.05% or 0.1% VTP50469 chow (n = 5 mice per group; P < .001 using Log-rank [Mantel-Cox test]). (D) Percent human CD45 cells in the peripheral blood of NOG mice engrafted with a NUP98-NSD1 PDX, treated with control, 0.05% or 0.1% VTP50469 chow (n = 5 mice per group). Error bars represent S.E.M. (E) Survival analysis of NOG mice engrafted with a NUP98-JARID1A PDX, treated with 0.1% VTP50469 chow for 60 days (yellow shading) (n = 5 mice per group; P < .001 using Log-rank [Mantel-Cox test]). (F) Percent human CD45 cells in the peripheral blood of NOG mice engrafted with a NUP98-JARID1A PDX, treated with control or 0.1% VTP50469 chow for 60 days (yellow shading) (n = 5 mice per group). (G,H) Heatmap of gene expression changes characterized by RNAseq in human CD45 cells isolated from BM of NOG mice bearing a NUP98-NSD1 PDX (G) or NUP98-JARID1A PDX (H). Values in each cell represent the Log2 fold change in gene expression (VTP treated mice/Control mice).
Next, we established 3 NUP98-rearranged PDX models by engrafting AML patient samples into immunocompromised NOG mice (supplemental Table 6). We monitored percent human CD45+ cells in the peripheral blood to understand the time course of disease progression. Upon engraftment, we randomized mice into cohorts that received control or VTP50469 chow. Two NUP98-rearranged PDX leukemia models responded to menin-MLL1 inhibition and demonstrated durable remissions. Mice harboring a NUP98-NSD1 PDX (NTPL-511) demonstrated a complete response to menin-MLL1 inhibition and are still alive approximately 250 days after VTP50469 chow was discontinued and with no evidence of peripheral blood disease (Figure 5C,D). Mice harboring a NUP98-JARID1A PDX (CPCT0021) were also successfully treated with VTP50469 and had a median survival advantage of 75 days (P < .0001) (Figure 5E,F). Control mice developed clinical illness and lost approximately 10% of body weight around day 60 following transplantation (supplemental Figure 6A). By contrast, mice treated with VTP50469 survived for an additional 60 days even after treatment was discontinued but ultimately relapsed around day 120 (Figure 5E,F). Similar to the control mice, treated mice demonstrated low burden of disease in the peripheral blood even when they became clinically ill and suffered relapse (Figure 5F, supplemental Figure 6A). In a separate experiment, we harvested control and VTP50469-treated mice after 36 days, and we observed a significantly decreased burden of disease in the BM and spleen of treated mice (supplemental Figure 6B). Altogether, these results in 2 clinically relevant PDX models demonstrate that menin-MLL1 inhibition can slow progression and even eradicate disease in NUP98-rearranged leukemia, although cooccurring mutations might influence response to therapy.
We next tested a second NUP98-NSD1 AML PDX that also harbored cooccurring mutations in ASXL1, IDH1, WT1, FLT3-ITD, and BCORL1 (MSKG5191) (supplemental Table 6). We found that this PDX was unresponsive to VTP50469 (supplemental Figure 7A-C), possibly due to the downstream impact of these cooccurring mutations which have been implicated in clonal hematopoiesis and resistance to both chemotherapy and epigenetic targeted therapies.10,25,26 Since WT1 mutations frequently cooccur with NUP98 translocations in AML, we generated a mouse model of NUP98-NSD1 leukemia on a Wt1 loss of function background (Mx1-Cre Wt1 flox/flox) model that has been previously described.19,20 We found that these cells (NUP98-NSD1, Wt1−/−) were sensitive to VTP50469 with an IC50 of 24 nM, upregulated CD11b, and displayed phenotypic differentiation toward a mature myeloid lineage (supplemental Figure 8A-D). Moreover, we observed significant downregulation of Meis1 gene expression and loss of MLL1 chromatin occupancy at the Meis1 locus (supplemental Figure 8E-G), showing that Wt1 loss does not interfere with the mechanism by which VTP50469 halts a proleukemogenic gene expression program.
Next, we sought to characterize gene expression changes that occur in response to VTP50469 in vivo. We performed RNAseq in human CD45+ leukemia cells isolated from BM of control and treated mice bearing NUP98-NSD1 PDX (NTPL-511) mice after 14 days of VTP50469 treatment. We observed downregulation of the distal HOXA cluster genes, including HOXA9 and HOXA11, in addition to other proleukemogenic genes such as MEIS1 and MEF2C (Figure 5G, supplemental Figure 9A,B). We also observed downregulation of genes with remarkable tissue specificity for B and T lymphocytes, as well as plasmacytoid dendritic cells (Figure 5G, supplemental Figure 9A,C). Previous work suggests that these 3 lineages may share a common hematopoietic precursor.27 Interestingly, we observed upregulation of tissue-specific genes for several mature myeloid lineages, including megakaryocytes, erythrocytes, and granulocytes (Figure 5G, supplemental Figure 9D).
We also characterized gene expression changes at a later time point in our NUP98-JARID1A PDX (CPCT0021). We performed RNAseq from human CD45+ cells isolated from BM after VTP50469 treatment of 36 days. We observed downregulation of the HOXA cluster, including HOXA3, HOXA5, HOXA7, and HOXA9, in addition to other proleukemogenic genes such as CEBPA and ZNF521 (Figure 5H, supplemental Figure 10A,B).28,29 Interestingly, we observed significant upregulation of genes that are exquisitely specific for megakaryocytes and erythroblasts, including von-Willebrand factor (VWF), the platelet glycoprotein IIb/IIIa complex, and hemoglobin E (Figure 5H, supplemental Figure 10C). Furthermore, we observed a striking increase in expression of VWF protein by immunohistochemistry (IHC) in BM of VTP50469-treated mice (supplemental Figure 10D). These results may point to a megakaryocyte-erythroid progenitor as the cell of origin for this patient’s AML. Indeed, this patient’s AML blasts express markers of megakaryocyte and erythrocyte differentiation: CD41, CD61, and CD71 (supplemental Table 6). These results suggest that menin-MLL1 inhibition leads to concomitant downregulation of stem cell-associated genes and upregulation of tissue-specific genes, thus releasing the brakes on differentiation toward a pathway that may have been destined for the leukemia cell of origin.
Discussion
Our studies demonstrate that menin and the menin-MLL1 interaction is a molecular dependency in NUP98-fusion leukemia, and an inhibitor of this interaction halts leukemogenesis and drives differentiation. Treatment of NUP98-JARID1A and NUP98-HOXA9 leukemia with VTP50469 results in changes in gene expression at a critical subset of target genes. In our models of NUP98-rearranged leukemia, treatment with VTP50469 results in a global loss of menin and selective loss of both MLL1 and NUP98 fusion protein chromatin occupancy at a subset of genes that are essential for leukemia maintenance. Among these genes, loss of both MLL1 and the NUP98 fusion is associated with downregulated gene expression, including at the Meis1 locus. Rescue of Meis1 expression via retroviral overexpression or CRISPRa abrogates responsiveness to VTP50469. The central role of Meis1 in leukemogenesis in NUP98-fusion hematologic malignancies suggests that NUP98 and MLL fusion oncoproteins may coopt similar molecular mechanisms of cell transformation. Nonetheless, rescue of Meis1 expression likely activates other proleukemogenic transcriptional networks, and there are probably other equally critical pathways downstream of Meis1 that influence responsiveness to menin-MLL1 inhibition. NUP98-rearranged PDX models are sensitive to VTP50469 in vivo; thus, targeted inhibition of the menin-MLL1 interaction represents a novel, rational therapeutic approach that warrants immediate clinical investigation for patients with NUP98-fusion hematologic malignancies.
These results suggest that NUP98 fusion proteins depend on the MLL1 chromatin complex to maintain a chromatin state that supports NUP98 fusion association with chromatin and, in turn, a proleukemogenic gene expression program. Our data also provides evidence that targeting the menin-MLL1 interaction is sufficient to disrupt MLL1 binding to chromatin. Previous work on NPM1c leukemia demonstrated that targeting the menin-MLL1 interaction leads to eviction of MLL1 from chromatin at a subset of loci that are essential for maintenance of a proleukemogenic gene expression program.18 Our results extend these findings to NUP98-rearranged leukemia. Of particular importance, we show that oncogenic NUP98 fusion proteins are themselves displaced from chromatin. Mechanistically, the downstream impact of inhibiting the menin-MLL1 interaction in NUP98-rearranged AML may be similar to what we have previously reported in NPM1c AML, since both models rely on wildtype MLL1. Furthermore, both NPM1c and NUP98 fusions may be recruited to specific loci by CRM1,30 a finding which is also supportive of the concept that menin-MLL1 may play similar roles in these leukemias.
We have provided evidence that NUP98-fusion proteins with 4 different C-terminal binding partners (HOXA9, HOXD13, JARID1A, and NSD1) are sensitive to VTP50469. This suggests that most NUP98 fusion proteins rely to some extent on the menin-MLL1 interaction, which is in keeping with data that shows the NUP98 portion of the fusion proteins interacts with the MLL1 complex.13 However, previous work from many laboratories suggests that the functions endowed by the COOH-terminal fusion partner in NUP98-rearranged and MLL-rearranged AML drive epigenetic dysregulation and promote leukemogenesis.6,13,23 It will be of interest to see if the function of the COOH-terminal partner influences therapeutic responsiveness. Future studies will help determine whether all or a subset of NUP98-rearranged AML are sensitive to menin-MLL1 inhibition. In addition, cooccurring mutations might also influence the responsiveness of NUP98-rearranged leukemia to menin-MLL1 inhibition. For example, our VTP50469-resistant NUP98-NSD1 PDX model (MSKG5191) harbors several cooccurring mutations (ASXL1, BCORL1, and IDH1) that might influence this PDX model’s responsiveness to menin-MLL1 inhibition. Further understanding of fusion protein biology and the mechanisms by which cooccurring mutations may alter responsiveness to targeted therapies will be essential as we translate menin-MLL1 inhibitors to the clinic.
Phenotypic differentiation toward the megakaryocyte/erythroid lineage in NUP98-rearranged PDX models treated with menin-MLL1 inhibition is unique among other PDX models we have studied (unpublished data). This observation could suggest that the cell of origin for NUP98-rearranged leukemia may be a progenitor that has already committed to the megakaryocyte/erythroid lineage. Indeed, NUP98-JARID1A fusions are found in both acute erythroid and acute megakaryoblastic leukemia (AMKL).31-33 Efforts to model NUP98-JARID1A leukemia in human CD34+ cord blood cells have also produced AMKL.34 In a mouse model, the NUP98-HOXD13 fusion was shown to inhibit megakaryocytic differentiation.7,35 Our RNAseq results from a NUP98-JARID1A PDX model treated with VTP50469 revealed upregulation of a subset of genes expressed almost exclusively by mature megakaryocytes and erythrocytes. We also observed concomitant downregulation of stem cell-associated genes such as ZNF521, a transcription factor that represses erythroid differentiation through its interaction with GATA1.36 These results suggest that menin-MLL1 inhibition drives differentiation toward a specific mature hematopoietic lineage, perhaps one that is defined by the preexisting epigenetic landscape of its cell of origin.
Finally, just as malignant cells develop resistance to conventional chemotherapy, leukemia treated with epigenetic-targeted therapies such as menin-MLL1 inhibitors is also likely to develop resistance if administered as monotherapy. It is likely that alternative mechanisms and pathways might be able to overcome the reliance of NUP98-rearranged leukemia on the menin-MLL1 interaction and downstream gene expression changes. Further studies to understand molecular dependencies and resistance pathways will be of utmost importance in developing combination and salvage therapies. The experiments described here support the inclusion of patients with NUP98-rearranged AML in the early-phase trials of molecules that target the menin-MLL1 interaction.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants CA176745 and CA066996 (S.A.A.), R01 CA204396, and P30 CA008748 (A.K.). S.A.A. and A.K. are both supported by a multi-institutional grant, U54 CA243124. This work was also supported by NIH training grant 5T32HL007574-36 (E.B.H.). This work was supported by grants from the Leukemia and Lymphoma Society (S.A.A). This work was also supported by the Leukemia Research Foundation of Delaware (E.A.K.) and the Lisa Dean Moseley Foundation Award (A.G.). We thank Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core, which provided histology and IHC service. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NIH 5 P30 CA06516. We thank Y. Soto-Feliciano for the ipUSEPR sgRNA expression plasmid. We thank Jennifer Perry (Dana-Farber Cancer Institute) for help with editing the manuscript, and our laboratory manager, Zhaohui Feng (Armstrong Laboratory).
Authorship
E.B.H., A.K., E.A.K., and S.A.A. designed the experiments; E.B.H., J.A.H., E.M.W., S.P.B., A.G., H.X., H.J.U., S.T., Y.K., and A.K. performed the experiments; E.B.H., C.H., Y.W., A.K., G.M.M, E.A.K., and S.A.A. analyzed and interpreted the data; Y.P. provided assistance with obtaining patient samples for PDX experiments; and E.B.H., F.P., H.U., and S.A.A. wrote and edited the manuscript.
Conflict-of-interest disclosure: S.A.A. has been a consultant and/or shareholder for Neomorph Inc, Imago Biosciences, Vitae/Allergan Pharma, Cyteir Therapeutics, C4 Therapeutics, OxStem Oncology, Accent Therapeutics, and Mana Therapeutics. S.A.A. has received research support from Janssen, Novartis, Syndax, and AstraZeneca. S.A.A. is an inventor on patent applications related to Menin inhibition WO/2017/132398A1. G.M.M. is a shareholder of Syndax Pharmaceuticals. A.K. is a consultant and shareholder of Novartis and Rgenta. The remaining authors declare no competing financial interests.
Correspondence: Scott Armstrong, Department of Pediatric Oncology, Dana-Farber Cancer Institute, 360 Longwood Ave, Longwood Center, LC 8214, Boston, MA 02215; e-mail: Scott_Armstrong@dfci.harvard.edu.
BioChIPseq (GSE175594), ChIPseq (GSE175595), and RNAseq (GSE175596) data are available at GEO under superseries accession number GSE175597. Additional methods can be found in the supplemental Materials.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked ”advertisement” in accordance with 18 USC section 1734.
