

Key Points
Low MS4A3 is a common mechanism among LSPC quiescence, BCR-ABL1-independent primary TKI resistance, and blastic transformation.
MS4A3 controls LSPC sensitivity to differentiating cytokines by regulating βc receptor endocytosis and signaling.

Abstract
The chronic phase of chronic myeloid leukemia (CP-CML) is characterized by the excessive production of maturating myeloid cells. As CML stem/progenitor cells (LSPCs) are poised to cycle and differentiate, LSPCs must balance conservation and differentiation to avoid exhaustion, similar to normal hematopoiesis under stress. Since BCR-ABL1 tyrosine kinase inhibitors (TKIs) eliminate differentiating cells but spare BCR-ABL1-independent LSPCs, understanding the mechanisms that regulate LSPC differentiation may inform strategies to eliminate LSPCs. Upon performing a meta-analysis of published CML transcriptomes, we discovered that low expression of the MS4A3 transmembrane protein is a universal characteristic of LSPC quiescence, BCR-ABL1 independence, and transformation to blast phase (BP). Several mechanisms are involved in suppressing MS4A3, including aberrant methylation and a MECOM-C/EBPε axis. Contrary to previous reports, we find that MS4A3 does not function as a G1/S phase inhibitor but promotes endocytosis of common β-chain (βc) cytokine receptors upon GM-CSF/IL-3 stimulation, enhancing downstream signaling and cellular differentiation. This suggests that LSPCs downregulate MS4A3 to evade βc cytokine-induced differentiation and maintain a more primitive, TKI-insensitive state. Accordingly, knockdown (KD) or deletion of MS4A3/Ms4a3 promotes TKI resistance and survival of CML cells ex vivo and enhances leukemogenesis in vivo, while targeted delivery of exogenous MS4A3 protein promotes differentiation. These data support a model in which MS4A3 governs response to differentiating myeloid cytokines, providing a unifying mechanism for the differentiation block characteristic of CML quiescence and BP-CML. Promoting MS4A3 reexpression or delivery of ectopic MS4A3 may help eliminate LSPCs in vivo.
Introduction
BCR-ABL1 is typically the only driver mutation detectable at chronic myeloid leukemia (CML) diagnosis and is necessary and sufficient to induce the chronic phase of CML (CP-CML) in mouse models.1,2 In most CP-CML patients, hematopoietic progenitor cells are almost exclusively BCR-ABL1-positive, while long-term culture-initiating cells (LTC-ICs) are predominantly BCR-ABL1-negative, indicating that a relatively small population of CML stem cells (LSCs) feeds a large progenitor cell compartment that drives the clinical disease.3 In mouse models, BCR-ABL1 does not confer self-renewal to myeloid progenitor cells and may even enhance differentiation at the expense of self-renewal.4,5 Nonetheless, spontaneous remissions in CML are exceedingly rare,6 raising the question of how human LSCs escape exhaustion in vivo.
While BCR-ABL1 tyrosine kinase inhibitors (TKIs) are highly effective in CP-CML, resistance is common upon progression to blast phase CML (BP-CML), suggesting that differentiation capacity and TKI resistance are inversely correlated.7,8 TKI resistance bifurcates into 2 major categories: (1) BCR-ABL1–dependent resistance due to kinase reactivation, typically through BCR-ABL1 kinase domain mutations that impair TKI binding9,10; (2) BCR-ABL1-independent resistance caused by alternative pathways, accounting for most primary TKI resistance cases (the failure to achieve a meaningful response in the first place).11 We previously reported overlap between the transcriptomes of CD34+ cells from BP-CML and TKI-naïve CP-CML patients who failed subsequent imatinib therapy,12-14 suggesting that BCR-ABL1 independence, primary TKI resistance, and the differentiation blockade of BP-CML share common mechanisms.
The majority of CML patients experience disease recurrence after TKI discontinuation.15-17 Recurrence originates from quiescent, innately TKI-resistant LSCs that persist despite effective BCR-ABL1 inhibition.18,19 Several pathways implicated in persistence also drive BP-CML progression, such as β-catenin activation and PP2A suppression,20-26 suggesting that quiescent LSCs and BP-CML cells use overlapping BCR-ABL1-independent strategies to escape differentiation into TKI-sensitive cells. To identify shared mediators of these phenotypes, we performed a meta-analysis of published CML transcriptomes and found that low expression of MS4A3, a member of the membrane-spanning 4-domains A (MS4A) family of proteins, is characteristic of quiescence, BCR-ABL1-independent TKI resistance, and BP-CML. We demonstrate that MS4A3 promotes myeloid differentiation by enhancing cellular response to GM-CSF/IL-3 through increasing receptor endocytosis and signaling. These data implicate MS4A3 as a functional tumor suppressor in CML, whose downregulation retains LSPCs in a differentiation-blocked, TKI-insensitive state.
Methods
Patient samples
Blood samples (supplemental Table S1, available on the Blood Web site) were separated on Ficoll-Hypaque (GE Healthcare), and CD34+ cells using an autoMACS system (Miltenyi Biotec). All patients gave informed consent in accordance with the Declaration of Helsinki, and all human specimen studies were approved by The University of Utah (UU) Institutional Review Board (IRB).
Mice
Scl-tTA+;TRE-BCR-ABL1+ mice were provided by Emmanuelle Passegué.27Ms4a3-null (Ms4a3tm1.1(KOMP)Vlcg) mouse sperm was obtained from NIH KOMP program. All animal experiments were approved by UU Institutional Animal Care and Use Committee (IACUC).
qRT-PCR and immunoblotting
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) and immunoblotting were performed as previously described.28 Supplemental Table S2 lists primers used; supplemental Table S3 lists antibodies used.
Colony formation and LTC-IC
Clonogenic assays were previously described.29 For LTC-IC assays, CP-CML CD34+ cells were lentivirally transduced with shMS4A3-GFP. GFP+ cells were fluorescence-activated cell sorting-sorted and plated as described.3,30,31BCR-ABL1+ colonies were identified by fluorescence in situ hybridization (FISH) as previously described.32
Digital PCR for BCR-ABL1
Nanofluidic digital polymerase chain reaction was used to quantify BCR-ABL1 transcripts in the bone marrow (BM), peripheral blood (PB), and spleen of xenografted mice.33
DNA bisulfite conversion and patch PCR sequencing
These were performed as described.34MS4A3 promoter CpG islands were analyzed between the transcription start site and 2000 bp upstream. Supplemental Table S4 lists oligonucleotides used.
Chromatin immunoprecipitation (ChIP)-seq
Fluorescence-activated cell sorting and flow cytometry
These were performed as described.28 Data were analyzed using FlowJo V10.
Receptor endocytosis
Cells were stimulated with cytokine and incubated for 5 to 15 minutes at 37°C. Cells were chilled on ice water to stop cellular activities, washed with ice-cold phosphate-buffered saline (PBS) (+0.5% bovine serum albumin [BSA]), and stained with CD116-APC antibody (clone REA211, Miltenyi Biotec) or CD123-BV711 antibody (clone 9F5, BD), and analyzed by flow cytometry.
Nanoparticles
Nanoparticles were collected from the culture supernatant of LAMA-84 cells overexpressing MS4A3-EGFP or LAMA-84 cells with CRISPR-mediated MS4A3 knockout. Nanoparticles were pelleted by adding 10% PEG-8000 and 0.3 M NaCl, resuspended in RPMI1640 to achieve 100 times concentrated, and stored at -80°C. Nanoparticles were added to CD34+ cell cultures at 50 μL/mL. To facilitate nanoparticle uptake, 10 μg/mL polybrene was added for 12 hours, then diluted or washed off. Mock treatment is polybrene only.
Immunofluorescent staining and confocal microscopy
Please refer to the supplemental Methods for details.
Statistical analysis
A 2-tailed t test (or with Welch’s correction when standard deviation [SD] is unequal) was used for comparing 2 groups of observations, and Mann-Whitney nonparametric test was used if values did not conform to a normal distribution. One-way ANOVA was used for comparing multiple groups of observations, and Fisher’s exact test was used for analysis of contingency. Three independent experiments were performed unless otherwise noted. Correlation of MS4A3 and survival in CML was analyzed with 35 patients from McWeeney et al.14
Results
Low MS4A3 in CML CD34+ LSPCs is associated with primary TKI resistance, blastic transformation, and lack of differentiation
We previously reported a gene expression classifier in TKI-naïve CP-CML CD34+ cells that predicted primary resistance to imatinib.14 We also demonstrated overlapping transcriptomic features among primary TKI resistance, rapid transformation to blast phase (BP) (within 3 years after diagnosis),12 and BP-CML.13 We first validated randomly selected classifier genes in CD34+ cells from additional patients with CP-CML, accelerated phase CML (AP-CML), or myeloid BP-CML (supplemental Figure 1). As primary TKI resistance is rarely associated with BCR-ABL1 mutations, reminiscent of the innate TKI resistance of primitive LSCs,18,19,39 we incorporated comparisons of quiescent vs proliferating CML CD34+ cells,40 and LSCs vs hematopoietic stem cells (HSCs)41 for a comprehensive meta-analysis. We identified 6 genes (ALOX5AP, AZU1, CTSG, ELANE, MPO, and MS4A3) that were uniformly downregulated in primary TKI resistance, BP-CML, and leukemic stem/progenitor cells (LSPCs) (Figure 1A). In normal hematopoiesis, all 6 genes are expressed from common myeloid progenitors (CMPs) to myelocytes/neutrophils (supplemental Figure 2).42 The gene products of AZU1, CTSG, ELANE, and MPO are neutrophil peroxisome proteases or pseudoproteases,43 and their synchronized downregulation is consistent with blocked differentiation. ALOX5AP encodes ALOX5 activating protein. Genetic ablation of ALOX5 decreased leukemic stem cell (LSC) survival in a CML mouse model.44 However, low ALOX5 in human CML CD34+CD38− cells and their insensitivity to ALOX5 inhibitors argue against a critical role of ALOX5AP in human CML.45 MS4A family proteins function as cell surface signaling and/or intracellular adaptor proteins in immune and epithelial cells that can be upregulated with differentiation.46-48 For instance, MS4A1 (CD20), the target of rituximab, is upregulated during B-cell activation and proliferation.49,50 MS4A3 was recently reported as an early marker of myeloid lineage commitment.51,52 Using published data, we confirmed that MS4A3 expression increases with myeloid differentiation (Figure 1B),42 suggesting that aberrantly low MS4A3 expression in CML may contribute to leukemogenesis and TKI resistance.

MS4A3low is a common feature of CML blastic transformation, primitive leukemic stem cell, and primary TKI resistance. (A) Meta-analysis with transcriptomic datasets comparing CP-CML vs BP-CML (CD34+),13 long CP (>3 years) vs short CP (≤3 years) (CD34+),12 LSCs vs HSCs (CD34+CD38−),41 imatinib responders vs nonresponders (pretherapy CD34+),14 proliferating (CD34+Hoechst+/Pyronine+) vs quiescent (CD34+Hoechstlo/Pyroninelo)40 CML cells. Venn diagram shows the number of genes with a >1.5-fold expression difference. (B) MS4A3 expression on a hierarchical differentiation tree, based on BloodSpot42 illustration of the dataset GSE24759.82 (C) MS4A3 mRNA was quantified by qRT-PCR in CD34+ cells from normal CB (n = 4), adult BM (from femoral head, n = 2), CP-CML (n = 13), BP-CML (n = 4), or TKI-resistant (TKI-R, n = 3) CP-CML patients with unmutated BCR-ABL1. Data were normalized to FH controls. (D) MS4A3 expression was quantified by Affymetrix HG-U133A arrays in 2 independent datasets comparing CD34+ cells from CP-CML (n = 7) and BP-CML (n = 7) patients.53 (E) Flow cytometry analysis of MS4S3 in CB, adult BM, and CP-CML samples (n = 3). (F) OS of CP-CML (n = 35) patients with MS4A3 mRNA expression in CD34+ cells (prior to imatinib therapy) above (high, n = 21) or below (low, n = 14) the value at a bimodal separation. *P < .05, **P < .01, ***P < .001.
MS4A3low is a common feature of CML blastic transformation, primitive leukemic stem cell, and primary TKI resistance. (A) Meta-analysis with transcriptomic datasets comparing CP-CML vs BP-CML (CD34+),13 long CP (>3 years) vs short CP (≤3 years) (CD34+),12 LSCs vs HSCs (CD34+CD38−),41 imatinib responders vs nonresponders (pretherapy CD34+),14 proliferating (CD34+Hoechst+/Pyronine+) vs quiescent (CD34+Hoechstlo/Pyroninelo)40 CML cells. Venn diagram shows the number of genes with a >1.5-fold expression difference. (B) MS4A3 expression on a hierarchical differentiation tree, based on BloodSpot42 illustration of the dataset GSE24759.82 (C) MS4A3 mRNA was quantified by qRT-PCR in CD34+ cells from normal CB (n = 4), adult BM (from femoral head, n = 2), CP-CML (n = 13), BP-CML (n = 4), or TKI-resistant (TKI-R, n = 3) CP-CML patients with unmutated BCR-ABL1. Data were normalized to FH controls. (D) MS4A3 expression was quantified by Affymetrix HG-U133A arrays in 2 independent datasets comparing CD34+ cells from CP-CML (n = 7) and BP-CML (n = 7) patients.53 (E) Flow cytometry analysis of MS4S3 in CB, adult BM, and CP-CML samples (n = 3). (F) OS of CP-CML (n = 35) patients with MS4A3 mRNA expression in CD34+ cells (prior to imatinib therapy) above (high, n = 21) or below (low, n = 14) the value at a bimodal separation. *P < .05, **P < .01, ***P < .001.
We first validated the MS4A3low feature from the meta-analysis in additional samples. Compared with cord blood (CB) or normal adult BM, MS4A3 mRNA in CP-CML CD34+ cells was greatly reduced, with additional reduction in myeloid BP-CML and TKI-resistant CP-CML (Figure 1C). We confirmed downregulation of MS4A3 in CD34+ cells from BP-CML vs CP-CML patients in an independent microarray dataset53 (Figure 1D). Flow cytometry revealed low MS4A3 in CB and adult BM CD34+CD38− cells, with strong upregulation in CD38+ cells (Figure 1E). By contrast, MS4A3 was consistently lower in CML, with the greatest difference in CD38+ cells (Figure 1E). Lower MS4A3 expression in CD34+ cells from a cohort of TKI-naïve CP-CML patients was associated with shorter overall survival (OS) (Figure 1F).
MS4A3low LSPCs exhibit enhanced colony-forming capability and increased persistence ex vivo and in vivo
To test whether MS4A3 is functionally important in CML, we transduced CML CD34+ cells with different doxycycline (dox)-inducible MS4A3 shRNAs (dox-shMS4A3) (supplemental Figure 3A-D) and performed colony assays. MS4A3 knockdown (KD) increased clonogenicity and protected colony formation upon BCR-ABL1 inhibition with imatinib mesylate (IM) in 5 of 6 samples (Figure 2A). Conversely, ectopic MS4A3 (supplemental Figure 3C-D) decreased colony formation and further reduced colonies upon IM exposure (Figure 2B). Manipulating MS4A3 expression had no effect on the colony formation of normal CB or adult BM CD34+ cells (Figure 2C-D; supplemental Figure 3E).
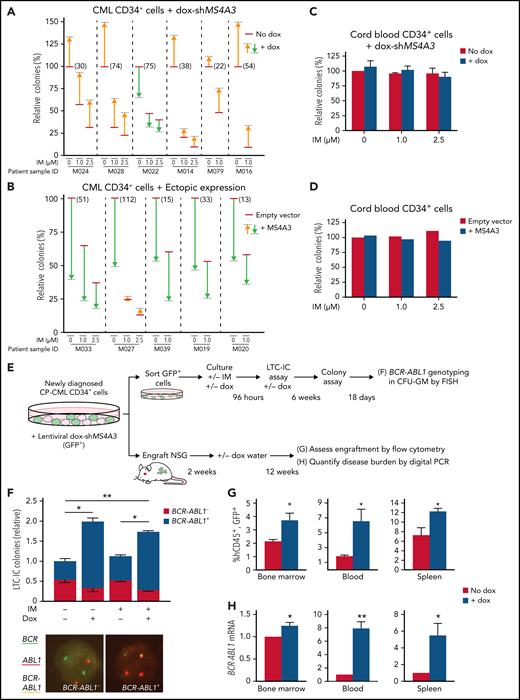
MS4A3 regulates colony formation and TKI resistance of BCR-ABL1+ cells ex vivo, and BCR-ABL1+ cell engraftment in vivo. (A-D) Effects of MS4A3 KD or ectopic MS4A3 expression on colony formation of CD34+ cells of CML samples (A-B) and CB samples (C-D). Colony formation: number of GM-colony formed after culture for 14 days. (E) Design of LTC-IC and xenograft assays. (F) Dox-shMS4A3-GFP+ cells were cultured ±0.1 μg/mL dox in 6-week LTC-IC cultures, then plated in colony assays. Colonies were genotyped by FISH (representative images shown). IM-dox- reference colony count average: 248. (G) Dox-shMS4A3-GFP+ cells were injected into NSG mice that were kept on dox water or normal water. hCD45+GFP+ cells in the BM (left), PB (middle), and spleen (right) were quantified with flow cytometry at 12 weeks postinjection. (H) BCR-ABL1 expression in the BM (left), PB (middle), and spleen (right) of recipient mice were detected by digital PCR at 12 weeks postinjection.
MS4A3 regulates colony formation and TKI resistance of BCR-ABL1+ cells ex vivo, and BCR-ABL1+ cell engraftment in vivo. (A-D) Effects of MS4A3 KD or ectopic MS4A3 expression on colony formation of CD34+ cells of CML samples (A-B) and CB samples (C-D). Colony formation: number of GM-colony formed after culture for 14 days. (E) Design of LTC-IC and xenograft assays. (F) Dox-shMS4A3-GFP+ cells were cultured ±0.1 μg/mL dox in 6-week LTC-IC cultures, then plated in colony assays. Colonies were genotyped by FISH (representative images shown). IM-dox- reference colony count average: 248. (G) Dox-shMS4A3-GFP+ cells were injected into NSG mice that were kept on dox water or normal water. hCD45+GFP+ cells in the BM (left), PB (middle), and spleen (right) were quantified with flow cytometry at 12 weeks postinjection. (H) BCR-ABL1 expression in the BM (left), PB (middle), and spleen (right) of recipient mice were detected by digital PCR at 12 weeks postinjection.
Primitive CML cells are innately TKI insensitive, in accord with the persistence of residual leukemia in most CML patients on TKI therapy.18,19 To assess whether MS4A3 suppression enhances CML cell persistence in vivo, we lentivirally transduced CD34+ cells from newly diagnosed CP-CML patients with dox-shMS4A3-GFP. We cultured GFP+ cells for 96 hours with StemSpan cytokine cocktail ± dox ± 1 μM IM, followed by plating in long-term culture initiating cell (LTC-IC) assays or injection into NSG mice (Figure 2E). MS4A3 KD significantly increased BCR-ABL1+ LTC-ICs without affecting BCR-ABL1− LTC-ICs, while IM had no effect (Figure 2F). In NSG mice, MS4A3 KD enhanced CML engraftment in BM, blood, and spleen (Figure 2G), reflected by an increase of BCR-ABL1 transcripts (Figure 2H).33 These data show that MS4A3 suppression promotes LSPC survival ex vivo and engraftment in vivo, suggesting that low MS4A3 may enhance the persistence of residual CML during TKI therapy.
Ms4a3 depletion enhances persistence of BCR-ABL1+ HSPCs in mice
For additional validation of MS4A3 function, we used a CML mouse model with inducible (Tet-off) BCR-ABL1 expression (Scl-tTA+;TRE-BCR-ABL1+).54Ms4a3 KD increased colony formation by BCR-ABL1+ Lin− BM cells but had no effect on BCR-ABL1− BM cells (Figure 3A-B), confirming the human data. We next generated Ms4a3+/+│+/−│−/−;Scl-tTA+;TRE-BCR-ABL1+ compound transgenic mice. As haploinsufficiency of Ms4a3 was not observed, Ms4a3+/+ and Ms4a3+/− littermates were combined as controls. Upon BCR-ABL1 induction, mice developed leukocytosis with myeloid expansion (Figure 3C-D). At 16 weeks, complete blood counts (CBC) in Ms4a3−/−;Scl-tTA+;TRE-BCR-ABL1+ mice were comparable to controls, but there was a trend toward increased splenomegaly (Figure 3C). The BM of Ms4a3−/−;Scl-tTA+;TRE-BCR-ABL1+ mice showed a trend toward increased short-term HSCs (ST-HSCs) and multipotent progenitor cells (MPPs), and a significant reduction of granulocyte-macrophage progenitors (GMPs) compared with controls (Figure 3E). We next transplanted equal numbers of Lin− BM cells into lethally irradiated nonleukemic (TRE-BCR-ABL1-) Ms4a3+ recipients. By week 6, recipients of Ms4a3−/−;Scl-tTA+;TRE-BCR-ABL1+ cells showed increased granulocytosis (Figure 3F). To test leukemic cell sensitivity to IM treatment, we challenged the compound transgenic mice with daily IM for 1 week, followed by observation for an additional week. Granulocyte counts in Ms4a3−/−;Scl-tTA+;TRE-BCR-ABL1+ mice were preserved compared with Ms4a3+ control mice and rebounded more rapidly after stopping IM (Figure 3G). These data suggest that Ms4a3 depletion impairs the differentiation of BCR-ABL1+ cells, preserving HSC function. In contrast, CBC and BM cell compartments were comparable in nonleukemic Ms4a3+/+ and Ms4a3−/− mice (Figure 3H-J), confirming a CML-specific role of MS4A3 in myelopoiesis.
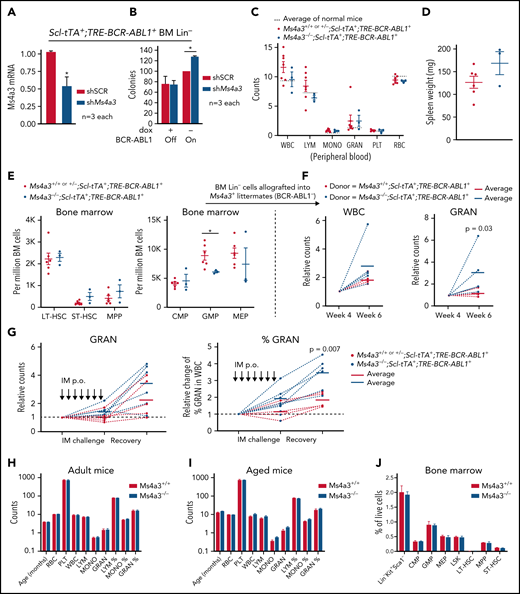
Ms4a3 depletion preserves stemness of BCR-ABL1+ HSPCs, but does not affect normal hematopoiesis in mice. (A-B) BM Lin− cells from Scl-tTA+;TRE-BCR-ABL1+ mice were transduced with shMs4a3 and cultured ex vivo. (A) Ms4a3 expression was quantified by qRT-PCR in cells cultured with dox (BCR-ABL1 off). (B) Colony formation was assessed with or without BCR-ABL1 induction. (C-E) Ms4a3−/−;Scl-tTA+;TRE-BCR-ABL1+ and Ms4a3+/+ or +/−;Scl-tTA+;TRE-BCR-ABL1+ mice were monitored for 16 weeks after BCR-ABL1 induction (dox withdrawal). Blood cell counts (C), spleen weight (D), and BM stem cell and progenitor counts (E) were quantified by veterinary hematology analyzer and flow cytometry. (F) Blood counts of recipient mice after lethal irradiation and allograft of BM Lin− cells from leukemic donors in panel E. Counts are normalized to week 4, the initial successful engraftment time point. (G) Granulocyte counts and percentage of leukemic mice undergoing 7 days of IM challenge (400 mg/kg) and 7 days of IM withdrawal and recovery. IM were administered by oral gavage. (H-I) PB counts in Ms4a3+/+ and Ms4a3−/− mice, of adult (n = 13 and 10) (H) and old (n = 8 and 14) (I) individuals. The cell count units are: 1012/L for red blood cell (RBC); 109/L for white blood cell (WBC), lymphocyte (LYM), monocyte (MONO), granulocyte (GRAN), and platelet (PLT). (J) Flow cytometry analysis of BM stem and progenitor populations in Ms4a3+/+ and Ms4a3−/− mice.
Ms4a3 depletion preserves stemness of BCR-ABL1+ HSPCs, but does not affect normal hematopoiesis in mice. (A-B) BM Lin− cells from Scl-tTA+;TRE-BCR-ABL1+ mice were transduced with shMs4a3 and cultured ex vivo. (A) Ms4a3 expression was quantified by qRT-PCR in cells cultured with dox (BCR-ABL1 off). (B) Colony formation was assessed with or without BCR-ABL1 induction. (C-E) Ms4a3−/−;Scl-tTA+;TRE-BCR-ABL1+ and Ms4a3+/+ or +/−;Scl-tTA+;TRE-BCR-ABL1+ mice were monitored for 16 weeks after BCR-ABL1 induction (dox withdrawal). Blood cell counts (C), spleen weight (D), and BM stem cell and progenitor counts (E) were quantified by veterinary hematology analyzer and flow cytometry. (F) Blood counts of recipient mice after lethal irradiation and allograft of BM Lin− cells from leukemic donors in panel E. Counts are normalized to week 4, the initial successful engraftment time point. (G) Granulocyte counts and percentage of leukemic mice undergoing 7 days of IM challenge (400 mg/kg) and 7 days of IM withdrawal and recovery. IM were administered by oral gavage. (H-I) PB counts in Ms4a3+/+ and Ms4a3−/− mice, of adult (n = 13 and 10) (H) and old (n = 8 and 14) (I) individuals. The cell count units are: 1012/L for red blood cell (RBC); 109/L for white blood cell (WBC), lymphocyte (LYM), monocyte (MONO), granulocyte (GRAN), and platelet (PLT). (J) Flow cytometry analysis of BM stem and progenitor populations in Ms4a3+/+ and Ms4a3−/− mice.
Stepwise downregulation of MS4A3 during CML progression
To understand MS4A3 regulation in CML, we transduced 32D-cl3 mouse myeloid progenitor cells with p210BCR-ABL1 or the kinase-inactive p210BCR-ABL1/K271R mutant, focusing on early passage cells to avoid phenotypic drift. Expression of p210BCR-ABL1 drastically reduced Ms4a3 expression, while kinase-inactive p210BCR-ABL1-K271R had no effect (Figure 4A). Surprisingly, IM inhibition of BCR-ABL1 rescued only <5% of Ms4a3 expression. In Lin− BM cells from Scl-tTA+;TRE-BCR-ABL1+ mice, BCR-ABL1 induction by dox-withdrawal also reduced Ms4a3 expression (Figure 4B). In contrast, MS4A3 expression remained unchanged in CD34+ CP-CML or BP-CML cells treated ex vivo with IM (Figure 4C). These data suggest that reduction of MS4A3/Ms4a3 expression is initially dependent on BCR-ABL1 kinase activity but then becomes uncoupled, decreasing further with CML progression.
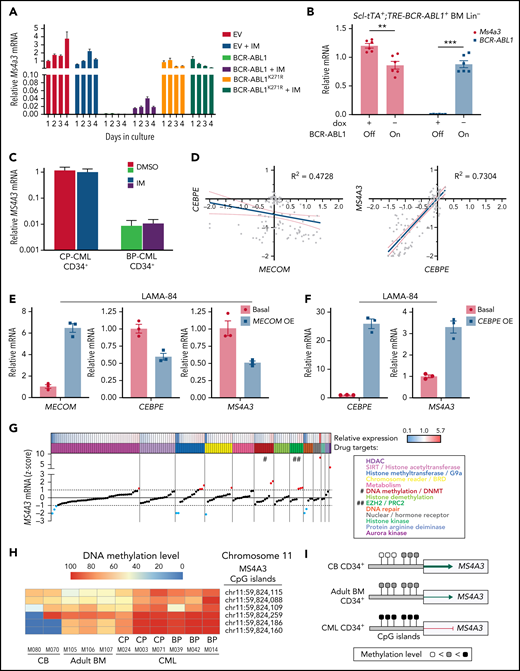
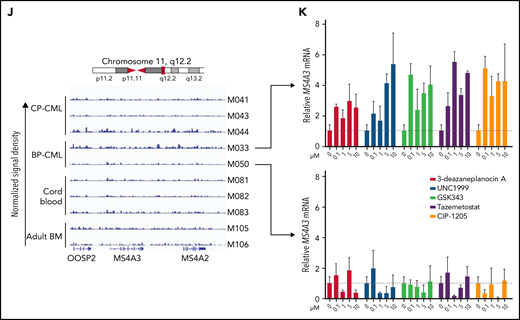
MS4A3/Ms4a3 transcription is regulated by BCR-ABL1, MECOM-C/EBPε axis, and epigenetic factors in CML. (A) Relative Ms4a3 mRNA in 32D-cl3 cells transfected with wild-type p210BCR-ABL1, the kinase inactive K271R mutant of p210BCR-ABL1, or an EV control, after culturing ±1 μM IM for 1 to 4 days. (B) Relative expression of Ms4a3 and BCR-ABL1 mRNA after BCR-ABL1 induction for 72 hours in Lin− BM cells from Scl-tTA+;TRE-BCR-ABL1+ mice. (C) Relative MS4A3 mRNA expression in CD34+ cells from CP-CML or BP-CML patients cultured ±1 μM IM for 24 hours. (D) Correlations between the expression of MECOM, CEBPE, and MS4A3 in CML CD34+ cells from a cohort of CP-, BP-, and AP-CML patients. Analysis was performed using log2 transformed microarray data. (E) Gene expression after overexpression of MECOM (EVI1). (F) Gene expression after overexpression of CEBPE. (G) Relative MS4A3 expression in CP-CML CD34+ cells treated with an epigenetic inhibitor library. Drug targets are color-coded. # and ## denote the classes of interest. (H-I) The heat map and diagram representing CpG methylation levels in the MS4A3 promoter region as detected by DNA bisulfite conversion and patch PCR sequencing on DNA of CD34+ cells from CB, adult BM, CP-CML, and BP-CML samples. (J) H3K27me3 ChIP‐seq signal is shown at the indicated MS4A3 locus in CD34+ cells from CB, adult BM, CP‐CML, and BP‐CML patients. Each ChIP‐seq track is normalized and scaled to the same scale. (K) Relative MS4A3 expression in CD34+ cells from 2 BP-CML patients, after treatment with indicated EZH2 inhibitors. M033 sample had elevated H3K27me3 reads at MS4A3 locus, while M050 had no obvious change compared with CB and adult BM.
MS4A3/Ms4a3 transcription is regulated by BCR-ABL1, MECOM-C/EBPε axis, and epigenetic factors in CML. (A) Relative Ms4a3 mRNA in 32D-cl3 cells transfected with wild-type p210BCR-ABL1, the kinase inactive K271R mutant of p210BCR-ABL1, or an EV control, after culturing ±1 μM IM for 1 to 4 days. (B) Relative expression of Ms4a3 and BCR-ABL1 mRNA after BCR-ABL1 induction for 72 hours in Lin− BM cells from Scl-tTA+;TRE-BCR-ABL1+ mice. (C) Relative MS4A3 mRNA expression in CD34+ cells from CP-CML or BP-CML patients cultured ±1 μM IM for 24 hours. (D) Correlations between the expression of MECOM, CEBPE, and MS4A3 in CML CD34+ cells from a cohort of CP-, BP-, and AP-CML patients. Analysis was performed using log2 transformed microarray data. (E) Gene expression after overexpression of MECOM (EVI1). (F) Gene expression after overexpression of CEBPE. (G) Relative MS4A3 expression in CP-CML CD34+ cells treated with an epigenetic inhibitor library. Drug targets are color-coded. # and ## denote the classes of interest. (H-I) The heat map and diagram representing CpG methylation levels in the MS4A3 promoter region as detected by DNA bisulfite conversion and patch PCR sequencing on DNA of CD34+ cells from CB, adult BM, CP-CML, and BP-CML samples. (J) H3K27me3 ChIP‐seq signal is shown at the indicated MS4A3 locus in CD34+ cells from CB, adult BM, CP‐CML, and BP‐CML patients. Each ChIP‐seq track is normalized and scaled to the same scale. (K) Relative MS4A3 expression in CD34+ cells from 2 BP-CML patients, after treatment with indicated EZH2 inhibitors. M033 sample had elevated H3K27me3 reads at MS4A3 locus, while M050 had no obvious change compared with CB and adult BM.
In acute myeloid leukemia (AML) cell lines, ectopic MECOM (EVI1) was shown to inhibit MS4A3 transcription and C/EBPε (CEBPE) expression.55,56 In support of this, we found that MS4A3 and CEBPE expression are positively correlated in the TCGA AML dataset more significantly than any other transcription factor (supplemental Figure 4A). As high MECOM is associated with progression to BP-CML and TKI resistance,57,58 we hypothesized that a MECOM-C/EBPε axis may regulate MS4A3 transcription in CML. Indeed, we found a negative correlation between MECOM and CEBPE and a strong positive correlation between CEBPE and MS4A3 in primary CML cells53 (Figure 4D). We then used LAMA-84 cells (the only CML cell line with endogenous MS4A3 expression in our panel [supplemental Figure 5B]) to verify this experimentally. Ectopic MECOM expression downregulated both CEBPE and MS4A3 (Figure 4E), and ectopic CEBPE expression upregulated MS4A3 (Figure 4F), consistent with a MECOM-C/EBPε axis regulating MS4A3 expression in CML.
To identify epigenetic regulators of MS4A3, we screened CML CD34+ cells with a library of epigenetic pathway inhibitors.59,60 We prioritized classes of epigenetic targets with at least 2 drugs increasing MS4A3 (z-score >1) and no drug decreasing MS4A3 expression (z-score <−1) (Figure 4G). DNA methyltransferase and EZH2 inhibitors met these criteria. We first analyzed MS4A3 promoter methylation using a patch PCR oligonucleotide library,34 comparing CB, adult BM, CP-CML, and BP-CML CD34+ cells. The 3 proximal CpG islands are methylated to 50% to 90% among most normal and CML samples, with CML showing increased methylation coverage (Figure 4H-I). The more distal 3 CpG islands had low methylation levels in CB, medium levels in adult BM, and high levels in CML samples (Figure 4H-I). There was not a consistent difference between CP-CML and BP-CML. Inhibition of DNA methylation rescued MS4A3 transcription (supplemental Figure 4B-D). Hence, promoter hypermethylation explains MS4A3 downregulation in CML compared with normal CD34+ cells, but not further reduction in BP-CML compared with CP-CML. We next performed ChIP-seq to detect EZH2/PRC2 footprints (H3K27me3) near the MS4A3 locus. Two of 5 CML samples showed increased H3K27me3 compared with CB and adult BM (Figure 4J). When 2 BP-CML samples were treated with EZH2 inhibitors, only the sample with elevated H3K27me3 showed restoration of MS4A3 expression, suggesting PRC2 contributes to MS4A3 suppression in select cases (Figure 4K). Altogether these data show that several mechanisms contribute to an increasingly profound reduction of MS4A3 expression from the initial BCR-ABL1-mediated transformation of HSCs through CP-CML to BP-CML.
MS4A3 shuttles between plasma membrane and endosomes to mediate membrane protein endocytosis
MS4A3 was reported to reduce pCDK2Y160 and inhibit G1/S progression in U937 cells.61 However, high MS4A3 in proliferative progenitors and low MS4A3 in quiescent, undifferentiated cells argue against a cell cycle inhibitory role. Accordingly, modulating MS4A3 had no effect on cell cycle progression in CML cell lines or primary cells (supplemental Figure 5A), and we saw no correlation between MS4A3 and pCDK2Y160 levels (supplemental Figure 5B). When CML CD34+ cells were cultured ex vivo, MS4A3 expression remained stable or increased in postdivision cells (supplemental Figure 5C), arguing against a dominant G1/S inhibitory function and prompting us to search for an alternative MS4A3 function.
We first visualized MS4A3 by expressing MS4A3-EGFP fusion protein in LAMA-84 cells (supplemental Figure 5B). Confocal microscopy revealed that MS4A3-EGFP primarily resides on the plasma membrane and in intracellular vesicles (Figure 5A). To determine which vesicles contain MS4A3, we screened intracellular structure markers in HEK293T cells expressing MS4A3-EGFP (Figure 5B). We found that MS4A3 preferentially colocalizes with Rab7+ late endosomes and LAMP1+ lysosomes (Figure 5B). We then verified that, in CML cells, MS4A3 accumulates in late endosomes but is adjacent to lysosomes (Figure 5C). To assess whether cell surface and endosomal MS4A3 pools communicate, we labeled cell surface MS4A3 with a specific monoclonal antibody (supplemental Figure 6A-B), followed by 37°C incubation. Upon snap fixation and imaging, we observed that a fraction of antibody-labeled surface MS4A3 (red puncta) had joined the pool of MS4A3 in intracellular vesicles (green puncta) (Figure 5D-E). These data suggest that MS4A3 traffics between plasma membrane and endosomes, consistent with an endocytosis-related function. To test this, we labeled surface-bound proteins on LAMA-84 cells ± MS4A3 KD with nonmembrane-permeant sulfo-NHS-SS-biotin. Streptavidin blotting showed that MS4A3 KD increased the presentation of surface proteins (Figure 5F), suggesting that MS4A3 promotes endocytosis of membrane-bound proteins. We next quantified MS4A3-regulated membrane-associated proteins by tandem mass spectrometry. Proteins with the most profound differences between MS4A3 KD and controls included membrane receptors and endocytosis-related proteins (Figure 5G). Notably, cell surface CSF2RB, the common βc of the GM-CSF, IL-3, and IL-5 receptors, was increased upon MS4A3 KD, suggesting MS4A3 may regulate endocytosis of βc receptors. To test this, we coexpressed MS4A3-EGFP and CSF2RB (βc)-RFP fusion proteins in HEK293 and LAMA-84 cells. Confocal microscopy and fluorescence resonance energy transfer (FRET) revealed partial colocalization of MS4A3 and βc in intracellular vesicles (Figure 5H-I). Additionally, MS4A3 and CD116 (GM-CSF receptor α chain) clustered on the cell surface upon GM-CSF stimulation (supplemental Figure 6C). These data indicate that MS4A3 shuttles between plasma membrane and endosomes to escort the endocytosis of membrane proteins, including βc receptors.
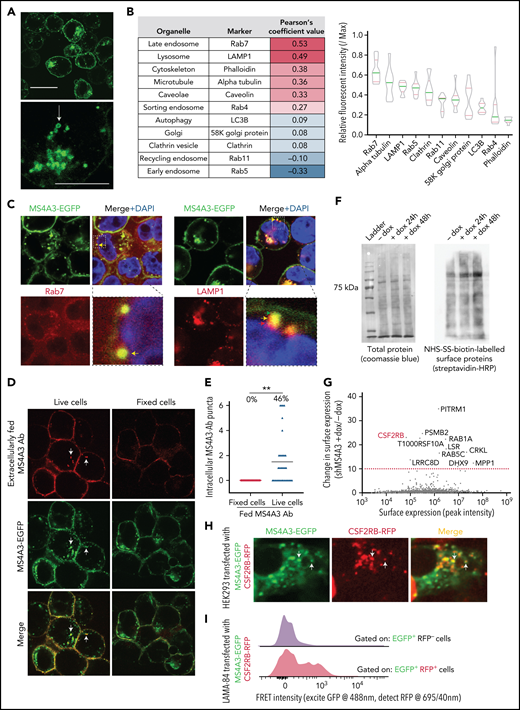
MS4A3 traffics between plasma membrane and endosomes to regulate the endocytosis surface-bound protein. (A) Representative confocal microscopy images of LAMA-84 cells expressing MS4A3-EGFP fusion protein. The arrow depicts a typical intracellular vesicle with MS4A3-EGFP accumulation. Upper scale bar: 10 μm, lower scale bar: 5 μm. (B) Confocal microscopy-based colocalization analysis of MS4A3-EGFP with various intracellular structures in HEK293T cells. Left: extent of colocalization quantified as Pearson’s coefficient value. Right: relative fluorescent intensity of marker proteins in MS4A3-EGFP positive vesicles. (C) Confocal microscopy showing the accumulation of MS4A3-EGFP in late endosomes, and near lysosomes, in LAMA-84 cells. Boxes indicate areas of higher magnification. Color-coded arrows point to representative puncta of overlapping or adjacent signals. (D) Trafficking of cell surface MS4A3 (extracellularly labeled by monoclonal antibody, red) to endosomal MS4A3 pool (shown by MS4A3-EGFP, green puncta) after 5 minutes of incubation at 37°C. Arrows point at representative endosomes with internalized surface MS4A3 (red + green). (E) Quantification of cells containing internalized MS4A3 from experiment in panel D. (F) Streptavidin blotting (left) and Coomassie blue staining (right) of sulfo-NHS-biotin-labeled LAMA-84 cells with or without MS4A3 KD. (G) Tandem MS identified surface-bound proteins whose internalization is mediated by MS4A3. LAMA-84 cells with dox-shMS4A3 were cultured with or without 100 ng/mL dox for 72 hours before sulfo-NHS-SS-biotin labeling, streptavidin-bead pulldown, and MS analysis. (H) HEK293 cells were transfected with MS4A3-EGFP fusion and CSF2RB-RFP fusion protein plasmids, and cells were fixed and imaged by confocal microscopy. Arrows point at intracellular vesicles with overlapping green and red signals. (I) LAMA-84 cells were transfected with MS4A3-EGFP fusion and CSF2RB-RFP fusion protein plasmids by electroporation, and the colocalization between the 2 proteins were analyzed by FRET signals on a flow cytometer with multiple detection filters for each laser.
MS4A3 traffics between plasma membrane and endosomes to regulate the endocytosis surface-bound protein. (A) Representative confocal microscopy images of LAMA-84 cells expressing MS4A3-EGFP fusion protein. The arrow depicts a typical intracellular vesicle with MS4A3-EGFP accumulation. Upper scale bar: 10 μm, lower scale bar: 5 μm. (B) Confocal microscopy-based colocalization analysis of MS4A3-EGFP with various intracellular structures in HEK293T cells. Left: extent of colocalization quantified as Pearson’s coefficient value. Right: relative fluorescent intensity of marker proteins in MS4A3-EGFP positive vesicles. (C) Confocal microscopy showing the accumulation of MS4A3-EGFP in late endosomes, and near lysosomes, in LAMA-84 cells. Boxes indicate areas of higher magnification. Color-coded arrows point to representative puncta of overlapping or adjacent signals. (D) Trafficking of cell surface MS4A3 (extracellularly labeled by monoclonal antibody, red) to endosomal MS4A3 pool (shown by MS4A3-EGFP, green puncta) after 5 minutes of incubation at 37°C. Arrows point at representative endosomes with internalized surface MS4A3 (red + green). (E) Quantification of cells containing internalized MS4A3 from experiment in panel D. (F) Streptavidin blotting (left) and Coomassie blue staining (right) of sulfo-NHS-biotin-labeled LAMA-84 cells with or without MS4A3 KD. (G) Tandem MS identified surface-bound proteins whose internalization is mediated by MS4A3. LAMA-84 cells with dox-shMS4A3 were cultured with or without 100 ng/mL dox for 72 hours before sulfo-NHS-SS-biotin labeling, streptavidin-bead pulldown, and MS analysis. (H) HEK293 cells were transfected with MS4A3-EGFP fusion and CSF2RB-RFP fusion protein plasmids, and cells were fixed and imaged by confocal microscopy. Arrows point at intracellular vesicles with overlapping green and red signals. (I) LAMA-84 cells were transfected with MS4A3-EGFP fusion and CSF2RB-RFP fusion protein plasmids by electroporation, and the colocalization between the 2 proteins were analyzed by FRET signals on a flow cytometer with multiple detection filters for each laser.
MS4A3 mediates GM-CSF/IL-3-induced receptor endocytosis and downstream signaling
Endocytosis of receptor/ligand complexes is required for full activation of signaling from many receptor tyrosine kinases and G-protein-coupled receptors.62,63 To test whether this includes the IL-3 and GM-CSF receptors, we used dynasore to block endocytosis in LAMA-84 CML cells stimulated with IL-3 or GM-CSF, 2 cytokines critically involved in myeloid differentiation.64 Dynasore reduced levels of pERK and pSTAT5Y695, implicating endocytosis in the regulation of signal strength in response to IL-3 and GM-CSF (Figure 6A). We hypothesized that MS4A3 regulates CML differentiation by promoting endocytosis and downstream signaling of GM-CSF receptor (GM-CSFR) and IL-3 receptor (IL-3R). In LAMA-84 CML cell line, MS4A3 KD increased, while overexpression reduced basal cell surface GM-CSFR (shown as CD116, α chain) (Figure 6B). Upon GM-CSF stimulation, CD116 was rapidly internalized through endocytosis (Figure 6B), and this process was inhibited by MS4A3 KD (Figure 6B-C), but not further enhanced by MS4A3 overexpression (Figure 6C). In LAMA-84 cells, GM-CSFR α chain (CD116) and βc (CD131) behave similarly during endocytosis, reflecting their well-established heterodimer conformation (Figure 6D-E). To validate the requirement of MS4A3 for GM-CSFR endocytosis, we generated and expressed a series of MS4A3 mutants, where a potentially critical extracellular or cytoplasmic domain is deleted or replaced by a flexible linker (Figure 6F-G). Replacement of the cytoplasmic domain connecting the second and the third transmembrane domains abrogated MS4A3's control of GM-CSFR endocytosis (Figure 6H-J). These data suggest that proper bundling of the 4 transmembrane domains is critical for MS4A3's control of receptor endocytosis, possibly by facilitating surface receptor clustering. Additionally, deleting the N-terminal cytoplasmic domain mildly reduced MS4A3's function (Figure 6H-J), suggesting that the N-terminal may play a role in the interaction between MS4A3 and GM-CSFR. In analogy to GM-CSFR, MS4A3 KD increased, and overexpression reduced basal cell surface IL-3R (represented by α chain, CD123) (Figure 6K). As expected from low CD123 expression in LAMA-84 cells, IL-3-induced CD123 endocytosis was modest and unaffected by MS4A3 overexpression but still inhibited by MS4A3 KD (Figure 6K). In comparison, modulation of MS4A3 expression did not affect SCF-induced KIT (CD117) endocytosis or G-CSF-induced G-CSFR (CD114) endocytosis, indicating that MS4A3 selectively regulates βc receptors (supplemental Figure 7A-D). We further tested whether MS4A3 controls signal strength downstream of GM-CSF and IL-3. MS4A3 KD abrogated GM-CSF/IL-3-induced full activation of pERK and pSTAT5Y694, while MS4A3 overexpression had opposite effects (Figure 6L). The opposing effects of MS4A3 KD and overexpression strongly argue against artifacts caused by off-target effects of shRNAs and suggest that MS4A3 governs susceptibility of CML cells to GM-CSF and IL-3.
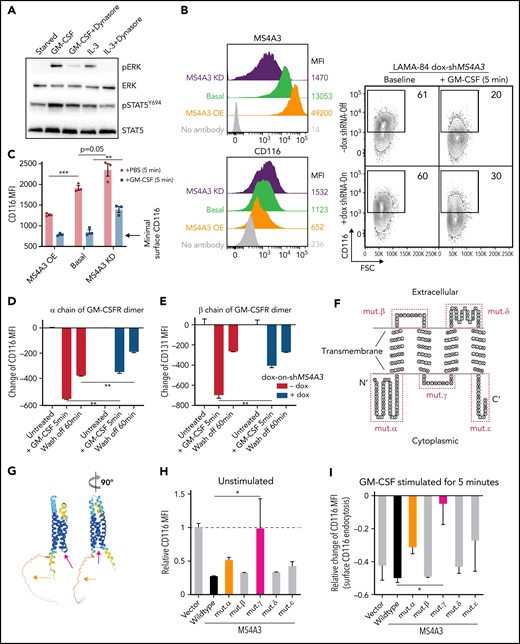
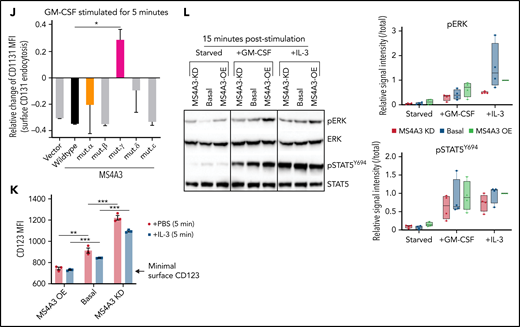
MS4A3 mediates GM-CSF/IL-3-induced receptor endocytosis and downstream signaling in LAMA-84 cells. (A) Western blotting of pERK and pSTAT5Y694 after GM-CSF/IL-3 stimulation, with or without pretreatment of the endocytosis blocker, dynasore (a dynamin inhibitor). (B) Flow cytometry plots of MS4A3 and surface CD116 in LAMA-84 cells with endogenous dox-shMS4A3 (KD), MS4A3 (basal), or MS4A3 overexpression (OE). Right: representative plots of GM-CSF-induced CD116 endocytosis and the effect of MS4A3 KD. (C) GM-CSF-induced CD116 endocytosis analyzed by flow cytometry. MFI: mean fluorescent intensity (surface). (D-E) Endocytosis and recycling of GM-CSFR α chain and β chain after GM-CSF stimulation and wash off. The changes of MFI compared with before stimulation values were plotted. (F-G) Two-dimensional and 3D illustrations of MS4A3 protein structure and features. Two-dimensional structure and transmembrane domain depiction were generated by TOPO2 (Johns S.J., TOPO2, Transmembrane protein display software, http://www.sacs.ucsf.edu/TOPO2/). The 3D structure is predicated by the world-leading AI-based protein folding engine, AlphaFold.83 Color-coding indicates the prediction confidence level, with higher confidence trending blue and lower trending yellow. Arrows point at the color-coded mutant regions in panel H-J. (H) Baseline CD116 surface level in cells transfected with various MS4A3 mutants. (I-J) GM-CSF-induced GM-CSFR endocytosis as shown by relative changes in CD116 and CD131 surface levels in cells transfected with various MS4A3 mutants. (K) IL-3-induced CD123 endocytosis analyzed by flow cytometry. (L) Western blotting of GM-CSF/IL-3-induced signaling activation in cells with endogenous MS4A3, dox-shMS4A3, or MS4A3 overexpression. Quantifications of signal intensities from 4 independent experiments are provided on the right.
MS4A3 mediates GM-CSF/IL-3-induced receptor endocytosis and downstream signaling in LAMA-84 cells. (A) Western blotting of pERK and pSTAT5Y694 after GM-CSF/IL-3 stimulation, with or without pretreatment of the endocytosis blocker, dynasore (a dynamin inhibitor). (B) Flow cytometry plots of MS4A3 and surface CD116 in LAMA-84 cells with endogenous dox-shMS4A3 (KD), MS4A3 (basal), or MS4A3 overexpression (OE). Right: representative plots of GM-CSF-induced CD116 endocytosis and the effect of MS4A3 KD. (C) GM-CSF-induced CD116 endocytosis analyzed by flow cytometry. MFI: mean fluorescent intensity (surface). (D-E) Endocytosis and recycling of GM-CSFR α chain and β chain after GM-CSF stimulation and wash off. The changes of MFI compared with before stimulation values were plotted. (F-G) Two-dimensional and 3D illustrations of MS4A3 protein structure and features. Two-dimensional structure and transmembrane domain depiction were generated by TOPO2 (Johns S.J., TOPO2, Transmembrane protein display software, http://www.sacs.ucsf.edu/TOPO2/). The 3D structure is predicated by the world-leading AI-based protein folding engine, AlphaFold.83 Color-coding indicates the prediction confidence level, with higher confidence trending blue and lower trending yellow. Arrows point at the color-coded mutant regions in panel H-J. (H) Baseline CD116 surface level in cells transfected with various MS4A3 mutants. (I-J) GM-CSF-induced GM-CSFR endocytosis as shown by relative changes in CD116 and CD131 surface levels in cells transfected with various MS4A3 mutants. (K) IL-3-induced CD123 endocytosis analyzed by flow cytometry. (L) Western blotting of GM-CSF/IL-3-induced signaling activation in cells with endogenous MS4A3, dox-shMS4A3, or MS4A3 overexpression. Quantifications of signal intensities from 4 independent experiments are provided on the right.
MS4A3 sensitizes primary CML LSPCs to differentiating signals by promoting βc receptor endocytosis
We next assessed βc receptor endocytosis and differentiation upon lentivirally mediated MS4A3 overexpression or KD in primary CP-CML CD34+ cells (Figure 7A). We used GFP label to distinguish between lentiviral transduced (GFP+) cells and nontransduced (GFP-) internal controls, given that GFP expression per se does not significantly alter cell behaviors (supplemental Figure 7E-G). In CD34+CD38- CML cells, MS4A3 KD or overexpression had no significant effect on basal surface CD116/βc (Figure 7B). GM-CSF stimulation reduced surface CD116/βc through endocytosis (Figure 7B). MS4A3 overexpression further enhanced, while MS4A3 KD inhibited, CD116/βc endocytosis (Figure 7B). In unstimulated CD34+CD38+ cells, MS4A3 KD increased surface CD116/βc, while overexpression had no effect, likely reflecting high endogenous MS4A3. GM-CSF-induced endocytosis decreased surface CD116/βc, which was reduced to the level of unstimulated cells by MS4A3 KD, while overexpression had no effect (Figure 7C).
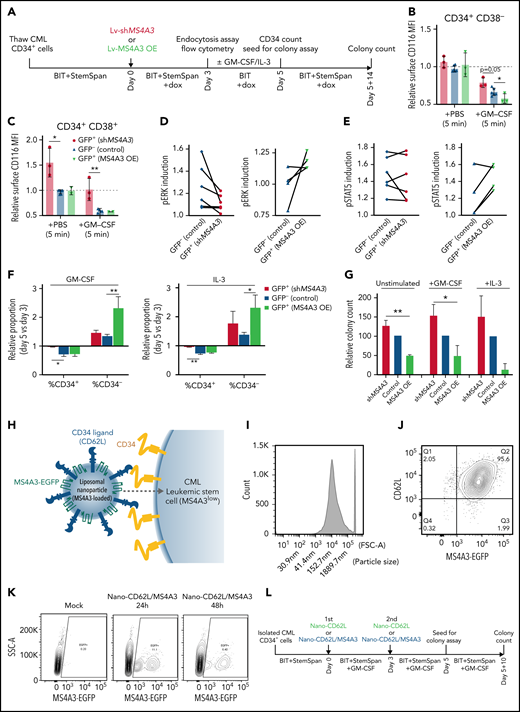
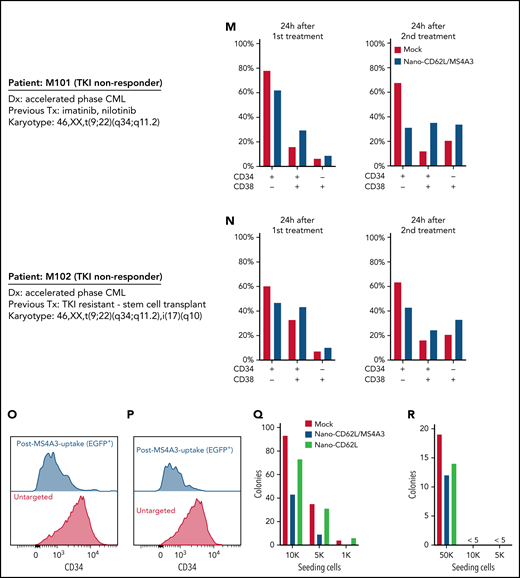
MS4A3 enhances GM-CSF/IL-3-induced receptor endocytosis, signaling, and differentiation in primary CML CD34+ cells. (A) Experimental outline of primary CML sample analyses. Both Lv-MS4A3-OE and Lv-dox-shMS4A3 carry GFP to mark transduced cells. GFP- cells are untransduced internal assay controls in the same samples, receiving the same treatment and analyses. Untransduced cells in both groups are combined for analysis. BIT supplement (BIT9500) contains BSA, human insulin, and human transferrin-Fe. StemSpan (CC100) is a cytokine cocktail for HSC and progenitor expansion. (B,C) Flow cytometry analysis of GM-CSF-induced CD116 endocytosis in CML CD34+ cells (n = 3). (D-E) Intracellular phospho-kinase staining of CML CD34+ cell cultures after GM-CSF and IL-3 stimulation for 30 minutes (n = 6). (F) Relative abundance of CD34+ and CD34- cells in the single cytokine pulsing differentiation assay (n = 3). (G) Colony formation of CML CD34+ cells with or without GM-CSF/IL-3-induced differentiation (n = 3). (H) Illustration of the nanoparticle targeting strategy. (I) Size distribution of the manufactured MS4A3 nanoparticles. Sizes are calculated from a standard curve made by a series of microbeads of known sizes. (J) Contour plot of MS4A3 nanoparticles (EGFP+) carrying CD34 ligand, CD62L. (K) Delivery and retention of MS4A3-EGFP by CD62L-coated nanoparticles, as tested in CD34+ Kasumi-1 cells. (L) Experimental design for testing the effect of nanoparticle-based MS4A3 delivery. (M,N) Proportions of various cell types in stem cell cultures with indicated treatments. (O-P) Flow cytometry detection of CD34 level on groups of cells after indicated treatments. (Q-R) Colony-forming capability of stem cells after indicated treatments.
MS4A3 enhances GM-CSF/IL-3-induced receptor endocytosis, signaling, and differentiation in primary CML CD34+ cells. (A) Experimental outline of primary CML sample analyses. Both Lv-MS4A3-OE and Lv-dox-shMS4A3 carry GFP to mark transduced cells. GFP- cells are untransduced internal assay controls in the same samples, receiving the same treatment and analyses. Untransduced cells in both groups are combined for analysis. BIT supplement (BIT9500) contains BSA, human insulin, and human transferrin-Fe. StemSpan (CC100) is a cytokine cocktail for HSC and progenitor expansion. (B,C) Flow cytometry analysis of GM-CSF-induced CD116 endocytosis in CML CD34+ cells (n = 3). (D-E) Intracellular phospho-kinase staining of CML CD34+ cell cultures after GM-CSF and IL-3 stimulation for 30 minutes (n = 6). (F) Relative abundance of CD34+ and CD34- cells in the single cytokine pulsing differentiation assay (n = 3). (G) Colony formation of CML CD34+ cells with or without GM-CSF/IL-3-induced differentiation (n = 3). (H) Illustration of the nanoparticle targeting strategy. (I) Size distribution of the manufactured MS4A3 nanoparticles. Sizes are calculated from a standard curve made by a series of microbeads of known sizes. (J) Contour plot of MS4A3 nanoparticles (EGFP+) carrying CD34 ligand, CD62L. (K) Delivery and retention of MS4A3-EGFP by CD62L-coated nanoparticles, as tested in CD34+ Kasumi-1 cells. (L) Experimental design for testing the effect of nanoparticle-based MS4A3 delivery. (M,N) Proportions of various cell types in stem cell cultures with indicated treatments. (O-P) Flow cytometry detection of CD34 level on groups of cells after indicated treatments. (Q-R) Colony-forming capability of stem cells after indicated treatments.
We next analyzed signaling downstream of βc receptors with intracellular phospho-kinase staining and flow cytometry on CD34+ CML LSPCs. MS4A3 overexpression increased pERK and pSTAT5Y694 induced by GM-CSF&IL-3, while MS4A3 KD reduced GM-CSF&IL-3-induced pERK, but had sample-specific effects on pSTAT5Y694 (Figure 7D-E). To test the MS4A3-associated differential sensitivities to βc receptor signaling for functional relevance, we pulsed CML CD34+ cells with GM-CSF or IL-3 for 2 days, without additional cytokines (Figure 7A). MS4A3 overexpression increased CD34- cells following GM-CSF/IL-3 stimulation, while MS4A3 KD preserved CD34+ LSPCs (Figure 7F). Following 48 hours of cytokine pulsing, equal numbers of viable cells were placed in semisolid media supplemented with StemSpan. Differences in clonogenicity between cells expressing high vs low MS4A3 levels were greatly enhanced by 2 days of GM-CSF/IL-3 stimulation (Figure 7G). These data suggest that low MS4A3 maintains clonogenic capacity by reducing response to GM-CSF/IL-3, and that differences in MS4A3 expression regulate susceptibility to differentiating signals. This may contribute to the increasing granulopoietic left shift upon progression from CP-CML (reduced MS4A3) to BP-CML (very low MS4A3).
MS4A3 protein-loaded nanoparticles promote differentiation and reduce clonogenicity of CML CD34+ cells
We manufactured a prototype MS4A3-loaded liposomal nanoparticle with a median size of ∼150 nm (Figure 7H-I), using CD62L coating for targeted delivery to CD34+ cells (Figure 7J). After 1 dose of nanoparticle administration in CD34+ Kasumi-1 cells, ∼11% of cells took up and retained MS4A3-EGFP for 24 hours and ∼6% for 48 hours (Figure 7K). CD34+ cells (MS4A3low) from 2 TKI-resistant AP-CML patients were cultured with GM-CSF&IL-3 and treated with 2 doses of nanoparticles ± MS4A3 payload (Figure 7L). MS4A3 nanoparticles reduced CD34+CD38− cells, but increased CD34+CD38+ and CD34−CD38+ cells (Figure 7M-N). Following MS4A3 uptake, cells showed reduced CD34 expression, suggesting differentiation (Figure 7O-P). MS4A3 nanoparticles reduced colony formation compared with MS4A3-free nanoparticles (Figure 7Q-R), mimicking the results of overexpression (Figure 2A-B). These data with prototype MS4A3 nanoparticles provide proof of concept that therapeutic delivery of MS4A3 to CML LSPCs may be feasible.
Discussion
Here we identify MS4A3 as a regulator of βc receptor endocytosis that promotes differentiation and TKI sensitivity in CML by enhancing response to IL-3 and GM-CSF. The unique rheostat-like functionality of MS4A3 in CML represents a unifying mechanism connecting BCR-ABL1-independent primary TKI resistance, LSPC persistence, and BP transformation. Downregulation of MS4A3 during CML progression is initially triggered by BCR-ABL1 kinase activity but subsequently proceeds in a kinase-independent manner. An early event in MS4A3 suppression, detected in CML CD34+ cells, compared with normal CD34+ cells, appears to be elevated methylation of MS4A3 promoter. Similarly, Ko et al recently reported hypermethylation of the MS4A3 promoter in BP-CML CD34+ cells compared with normal controls.65 Previous work using Scl-tTA+;TRE-BCR-ABL1+ mice showed that BCR-ABL1-induced DNA hypermethylation is not completely reversible after BCR-ABL1 shutoff,66 potentially explaining why TKIs do not restore Ms4a3 expression. Hypomethylating agents promote differentiation of CML cells in vitro and in vivo,66-68 and our data suggest that reexpression of MS4A3 may contribute to these effects. A second level of MS4A3 regulation is EZH2, which is essential for LSC survival and TKI resistance.69,70 EZH2 inhibitors increased MS4A3 expression in case of high H3K27me3 near the MS4A3 locus. This and a recent report of decreased PRC2 activity in BP-CML suggest that EZH2 regulation of MS4A3 is variable.65 A third MS4A3-suppressing mechanism in CML is a MECOM-C/EBPε axis. MECOM upregulation is caused by chromosome 3q26 rearrangements in a minority of cases, with the majority unexplained.57,58 TP53 can suppress MECOM in CML by inhibiting β-catenin/LEF1/TCF,71 suggesting that reduced TP53 activity during progression may increase MECOM and reduce MS4A3, resulting in impaired differentiation. This could explain the enhanced LSC potential of BP-CML GMPs.20
Contrary to a previous report,61 we find no evidence for a cell cycle regulatory role of MS4A3. Instead, we show for the first time that MS4A3 promotes differentiation by regulating βc receptor endocytosis and signaling. Our finding that MS4A3 depletion impairs differentiation of CML CD34+ cells ex vivo and reduces GMPs in vivo implicates MS4A3 as a direct differentiation regulator rather than a mere marker, fitting the broader concept of a functional tumor suppressor in CML. As cycling CML LSPCs40 are sensitive to TKIs,72 survival of LSPCs in TKIs reflects their ability to prevent differentiation by reducing cytokine responsiveness.
βc cytokines, especially IL-3, are critical to the proliferation of primary CML cells via paracrine and autocrine mechanisms.73,74BCR-ABL1 was reported to promote differentiation in Scl-tTA+;TRE-BCR-ABL1+ mice,19 reducing LT/ST-LSCs. We now show that Ms4a3 depletion attenuates this effect, probably through inhibition of IL-3/GM-CSF signaling. As MS4A3 suppression becomes uncoupled from BCR-ABL1 kinase, this mechanism will persist in the presence of TKIs. Additional cytokines are likely involved in maintaining CML hematopoiesis in balance. For instance, paracrine IL-6 redirects MPPs with B-cell lymphoid potential to the myeloid lineage.27 CML CD34+ cells are responsive to SCF,75 but KIT is low in CD34+CD38- cells, rendering primitive cells insensitive to KIT inhibition.76 Similarly, LSCs persisting in CML patients are KITlow or KIT-.77 Therefore, LSPCs reduce responsiveness to certain cytokines to avoid differentiation and remain TKI-resistant.
Receptor endocytosis is often mistaken as the termination of cytokine signaling. However, endosomes with residing signaling complexes can enhance signaling.62,63 We demonstrate that βc cytokines require receptor endocytosis for maximal downstream signaling and that MS4A3 enhances this process, reminiscent of a recent report implicating MS4A4 as a positive regulator of KIT endocytosis and signaling in mast cells.47 Our laboratory is exploring additional membrane proteins with MS4A3-dependent endocytosis.
MS4A3 enhances myeloid differentiation in CML but is not required for maintaining normal hematopoiesis at steady state. Evolutionarily, MS4A3 gene orthologs emerged only in mammals, much later than myeloid hematopoiesis or the essential cytokine M-CSF (supplemental Figure 9), which may explain why MS4A3 depletion has no obvious hematopoietic phenotype in nonleukemic mice. This surprising specificity for CML may reflect BCR-ABL1-driven aberrant cytoskeletal remodeling and vesicular trafficking.78,79 Of interest, MS4A3 KD increased membrane-bound CRKL (Figure 5G), a binding partner and substrate of BCR-ABL1 kinase in CML neutrophils.80,81 The hyperactive endocytic machinery in CML79 may increase the sensitivity of LSPCs to GM-CSF/IL-3 differentiating signals, creating pressure to reduce endocytosis to avoid exhaustion. Our study suggests that rescuing MS4A3 expression by hypomethylating agents, MS4A3-loaded targeted nanoparticles, or in some cases EZH2 inhibitors, may promote differentiation and confer responsiveness to TKIs, providing a rationale for combination therapies for eradicating CML LSPCs.
Acknowledgments
M.W.D. acknowledges funds from the National Institutes of Health (NIH), National Cancer Institute through awards R01CA178397 and R01CA257602-01, and P30CA042014. A.M.E. was supported by a NIH T32 training grant (CA093247), followed by a Career Development Award for postdoctoral fellows (5090-12) from the Leukemia & Lymphoma Society (LLS), followed by a Fellow Scholar Award from the American Society of Hematology (ASH). A.M.E. also acknowledges support from the NIH Loan Repayment Program. D.Y. is supported by the International Award from the Lady Tata Memorial Trust following the Special Fellow Award from the Leukemia & Lymphoma Society. J.S.K. was supported by the Special Fellow Award from the LLS. This work was funded in part by the University of Utah Flow Cytometry Core Facility and the National Cancer Institute through award 5P30CA042014-24 awarded to the Huntsman Cancer Institute and the National Center for Research Resources of the NIH under award 1S10RR026802-01. Research reported in this publication used the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at The University of Utah Huntsman Cancer Institute, supported by the National Cancer Institute of the NIH under award P30CA042014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The Ms4a3tm1.1(KOMP)Vlcg mouse strain used for this research project was generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (www.komp.org). NIH grants to Velocigene at Regeneron Inc (U01HG004085) and the CSD Consortium (U01HG004080) funded the generation of gene-targeted ES cells for 8500 genes in the KOMP Program and archived and distributed by the KOMP Repository at UC Davis and the Children’s Hospital Oakland Research Institute (CHORI) (U42RR024244). The authors thank Clinton C. Mason at The University of Utah for help with computational data analysis. The authors also thank Kristin Dahlin, Huntsman Cancer Institute, for help with illustrations. The authors acknowledge Cell Imaging Core at The University of Utah for use of confocal microscopy equipment.
Authorship
H.Z., A.M.E., A.F., and A.D.P. planned and performed experiments, analyzed data, interpreted results, and wrote the manuscript; J.A., A.S., A.D.P., B.H., D.Y., S.I., M.S.Z., A.A., H.M.R., J.S.K., J.W.T., K.C.B., J.M.V., J.G., and A.D.B. performed experiments; J.-Y.H., J.M.V., J.G., S.K.M., and S.V. performed computational data analysis; P.M.C. processed patient samples; D.L.S., V.G.O., J.P.R., K.E.V., and B.J.D. provided relevant data for preparation of the manuscript; and M.W.D. supervised the study, planned experiments, interpreted results, and wrote the manuscript.
Conflict-of-interest disclosure: M.W.D. reports research funding or paid advisory board membership for the following companies: Blueprint, Pfizer, Novartis, Takeda, Incyte, Fusion Pharma, Medscape, and Ascentage Pharma. None of these relationships pose any conflict of interest with the present manuscript. The remaining authors declare no competing financial interests.
Correspondence: Michael W. Deininger, 8727 W Watertown Plank Rd, Milwaukee, WI 53226; e-mail: mdeininger@versiti.org.
Data have been uploaded to National Center for Biotechnology Information Gene Expression Omnibus (accession number GSE188241).
Requests for data sharing may be submitted to M.W.D. (mdeininger@versiti.org)
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal