

Key Points
CDX2 cis-deregulation and UBTF::ATXN7L3 fusion driven by focal deletions define a novel subtype of B-ALL.
CDX2/UBTF::ATXN7L3 is a high-risk B-ALL subtype in young adults, which warrants improved therapeutic strategies.

Abstract
Oncogenic alterations underlying B-cell acute lymphoblastic leukemia (B-ALL) in adults remain incompletely elucidated. To uncover novel oncogenic drivers, we performed RNA sequencing and whole-genome analyses in a large cohort of unresolved B-ALL. We identified a novel subtype characterized by a distinct gene expression signature and the unique association of 2 genomic microdeletions. The 17q21.31 microdeletion resulted in a UBTF::ATXN7L3 fusion transcript encoding a chimeric protein. The 13q12.2 deletion resulted in monoallelic ectopic expression of the homeobox transcription factor CDX2, located 138 kb in cis from the deletion. Using 4C-sequencing and CRISPR interference experiments, we elucidated the mechanism of CDX2 cis-deregulation, involving PAN3 enhancer hijacking. CDX2/UBTF ALL (n = 26) harbored a distinct pattern of additional alterations including 1q gain and CXCR4 activating mutations. Within adult patients with Ph− B-ALL enrolled in GRAALL trials, patients with CDX2/UBTF ALL (n = 17/723, 2.4%) were young (median age, 31 years) and dramatically enriched in females (male/female ratio, 0.2, P = .002). They commonly presented with a pro-B phenotype ALL and moderate blast cell infiltration. They had poor response to treatment including a higher risk of failure to first induction course (19% vs 3%, P = .017) and higher post-induction minimal residual disease (MRD) levels (MRD ≥ 10−4, 93% vs 46%, P < .001). This early resistance to treatment translated into a significantly higher cumulative incidence of relapse (75.0% vs 32.4%, P = .004) in univariate and multivariate analyses. In conclusion, we discovered a novel B-ALL entity defined by the unique combination of CDX2 cis-deregulation and UBTF::ATXN7L3 fusion, representing a high-risk disease in young adults.
Introduction
Acute leukemia is a paradigm for the oncogenic role of deregulated transcription factors (TF).1 Most acute lymphoblastic leukemias (ALL) involve the activation of an aberrant transcriptional program as the initiating oncogenic event, which can be caused by inappropriate expression of a genuine TF or expression of a corrupted TF. Both situations occur mainly by chromosomal translocations, involving either immunoglobulin or T-cell receptor regulatory sequences that deregulate expression of the targeted gene or fusion genes encoding chimeric proteins that harbor altered transcriptional properties.
B-cell precursor ALL (B-ALL) can be subclassified according to recurrent cytogenetic abnormalities that drive specific transcriptional programs and are associated with distinct clinical presentation, response to treatment, and prognosis.2,3 Consequently, cytogenetic and/or molecular characterization at diagnosis is the cornerstone of risk-adapted treatment stratification in children, along with minimal residual disease assessment.4 This is the result of extensive annotation of large cohorts of patients and prognostic correlations in the context of clinical trials. In adults, with the exception of Philadelphia chromosome (Ph)-positive ALL that benefit from distinct treatment protocols including tyrosine kinase inhibitors, few genomic abnormalities are widely recognized as useful for treatment stratification.4 This is partly related to the poorer knowledge of genomic abnormalities defining clinically relevant subgroups of patients, as compared with B-ALL in children.
In order to improve our understanding of adult B-ALL, we performed transcriptome profiling of a large cohort of adult B-ALL harboring no known cytogenetic abnormalities. We identified a novel subtype of B-ALL, elucidated its underlying genomic alterations, and described its clinical features. This novel B-ALL entity affects young women, is characterized by poor treatment outcome, and relies on the unique combination of 2 genomic alterations resulting in CDX2 cis-deregulation and UBTF::ATXN7L3 fusion.
Methods
Patients and samples
Patients with Ph-negative B-ALL treated in the GRAALL-2005,5 GRAALL-2014, and EWALL-INO trials (clinicaltrials.gov numbers NCT00327678, NCT02617004, and NCT03249870, respectively) were included. Screening for recurrent cytogenetic aberrations was performed by standard cytogenetic procedures in local laboratories. Targeted molecular analyses were performed centrally, including screening for recurrent fusion transcripts by reverse transcription multiplex ligation-dependent probe amplification,6 screening for ERG and IKZF1 intragenic deletions by breakpoint-specific PCR,7,8 and quantitative reverse transcription PCR (qRT-PCR) for CRLF2 and EPOR transcripts. All cases negative for the major classifying chromosomal abnormalities (ie, KMT2A rearrangements, TCF3::PBX1, high hyperdiploidy, low hypodiploidy, and ETV6::RUNX1) or for BCR::ABL1–like alteration or ERG intragenic deletion were subjected to RNA sequencing (RNA-seq). Blast percentage was assessed by cytometry analysis of mononuclear cells subjected to nucleic acid extractions, and cases with <20% blasts were excluded. The studied cohort had higher white blood cells (WBC) and cumulative incidence of relapse (CIR) as compared with nonstudied patients (supplemental Table 1). Additional cases allocated to the major adult B-ALL subtypes were included in the RNA-seq cohort (ie, KMT2A-rearranged, low hypodiploidy and high hyperdiploidy; n = 16, 10, and 8 cases, respectively). Nine additional cases referred to our laboratory for molecular investigation in the context of patient care and for whom a CDX2/UBTF ALL was identified, including 3 relapses, were included in the cohort for characterization of CDX2/UBTF ALL. All patients or relatives gave informed consent according to the Declaration of Helsinki, and the study was approved by the local ethics committee. A flowchart describing the study cohort is provided in supplemental Figure 1.
High-throughput sequencing
RNA-seq libraries were prepared using the Truseq stranded mRNA library preparation kit (Illumina, San Diego, CA), and 2x150bp paired-end sequencing was performed using a NextSeq 500 (Illumina). Raw, unfiltered RNA-seq reads were aligned to human reference genome hg19 using STAR 2.5.2b.9 Fusions transcripts were detected using FusionCatcher 0.99.7c β.10 For UBTF::ATXN7L3 fusion, visual inspection of each BAM file was done because it could be occasionally overlooked by bioinformatic algorithms. Details of gene expression analysis and mutation detection are provided in supplemental Methods.
Whole-genome long-read sequencing was performed using the PacBio platform. High-molecular-weight DNA was extracted and purified using the MagAttract HMW DNA kit (Qiagen). DNA shearing was performed using g-TUBE (Covaris) with a targeted 20 kb fragment size, and fragments below 5 kb were removed using AMPure PB beads at 3.1X. Library preparation was conducted according to the manufacturer protocol (SMRTbell Express Template Prep Kit 2.0; Pacific Biosciences), and samples were processed using sequencing primer v2 and Sequel II Binding Kit 2.0. Samples were sequenced on a Sequel II instrument in consensus circular sequencing (CCS) mode using 2 SMRTcells per sample. CCS sequencing reads were generated using SMRTlink software and aligned against the human genome (hg38) using pbmm2 of PacBio SMRTtools v.8.0. Structural variant calling was performed using SMRT Analysis. Structural variants of a minimum of 10 bp were retained if covered by at least 2 CCS reads.
Experimental procedures regarding RNA-DNA fluorescent in situ hybridization (FISH), chromatin immunoprecipitation sequencing (ChIP-seq), Assay for Transposase-Accessible Chromatin with highthroughput sequencing (ATAC-seq), 4C-seq,CRISPR interference (CRISPRi), targeted next-generation sequencing (NGS), and array-CGH are provided in supplemental Methods.
Results
A novel B-ALL subtype characterized by a unique gene expression signature and 2 distinct genomic alterations pointing to candidate oncogenic drivers: CDX2 gene and UBTF::ATXN7L3 fusion
We performed comprehensive transcriptome and genome analyses of a cohort of 302 B-ALL cases, most of which were negative for the common classifying cytogenetic aberrations, formerly named “B-other” (268/302, 89%). By combining 2 unsupervised methods, weighted gene coexpression network analysis and hierarchical bi-clustering, several clusters were identified (supplemental Figure 2; supplemental Tables 2-6). Aggregation with genomic and fusion transcript data enabled us to allocate major clusters to previously described subtypes within B-other ALL, namely Ph-like,11,12 DUX4/ERG,7,13-15 ZNF384,13 MEF2D,16 and PAX5 P80R17 as visualized on t-distributed stochastic neighbor embedding analysis (Figure 1A). In addition, we identified a novel cluster comprising 23 B-other cases defined by a unique transcriptional signature.
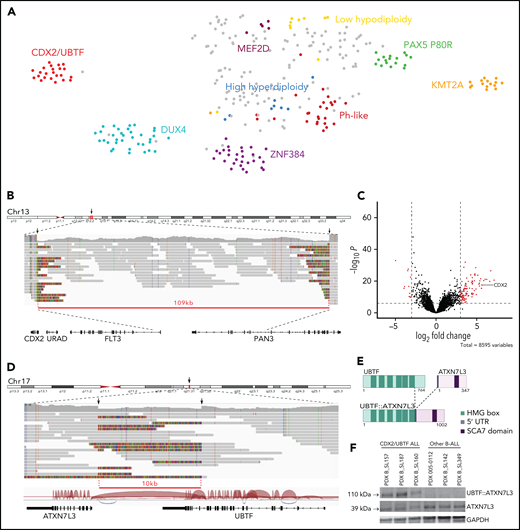
Identification of a novel gene expression B-ALL cluster characterized by CDX2 high expression and UBTF::ATX7NL3 fusion. (A) Gene expression profiling of 302 B-ALL cases shown in a 2-dimensional tSNE plot. This analysis was performed with 235 selected genes listed in supplemental Table 6. Major B-ALL subtypes are highlighted in different colors, and gray dots indicate other B-ALL. A novel cluster of cases with distinct gene expression profiling was identified, thereafter named CDX2/UBTF ALL. (B) WGS long-reads visualized on IGV software in 1 CDX2/UBTF case (B_SL160) showing a deletion at 13q12.2 locus close to the CDX2 gene. The red line underlines the deleted region. (C) Volcano plot of differentially expressed genes between CDX2/UBTF ALL (n = 23) and other B-ALL cases (n = 279). Genes with fold change greater than 3 and -log10 P value >7 are shown in red. CDX2 is highlighted as 1 of the 10 most highly expressed genes in the novel cluster. (D) WGS long-reads and RNA-seq splice junctions visualized on IGV software in 1 CDX2/UBTF case (B_SL160) showing a deletion at 17q21.31 locus involving the UBTF and ATXN7L3 genes. The red line underlines the deleted region. (E) Scheme of the predicted UBTF::ATXN7L3 chimeric protein. The resulting in-frame fusion transcript incorporated 60 nucleotides upstream the canonical translation start codon of ATXN7L3. (F) Western-blot of CDX2/UBTF cases using ATXN7L3 antibody shows detection of normal ATXN7L3 protein (39 kDa) in all samples and another protein at a size corresponding to the predicted UBTF::ATXN7L3 chimeric protein (110 kDa) in CDX2/UBTF cases only. Western blot performed on PDX from 3 CDX2/UBTF cases (B_SL157, B_SL187, and B_SL160), 2 PAX5 P80R cases (005-0112 and B_SL142), and 1 KMT2A-r case (B_SL349). IGV, integrative genomics viewer; PDX, patient-derived xenograft; tSNE, t-distributed stochastic neighbor embedding; WGS, whole-genome sequencing.
Identification of a novel gene expression B-ALL cluster characterized by CDX2 high expression and UBTF::ATX7NL3 fusion. (A) Gene expression profiling of 302 B-ALL cases shown in a 2-dimensional tSNE plot. This analysis was performed with 235 selected genes listed in supplemental Table 6. Major B-ALL subtypes are highlighted in different colors, and gray dots indicate other B-ALL. A novel cluster of cases with distinct gene expression profiling was identified, thereafter named CDX2/UBTF ALL. (B) WGS long-reads visualized on IGV software in 1 CDX2/UBTF case (B_SL160) showing a deletion at 13q12.2 locus close to the CDX2 gene. The red line underlines the deleted region. (C) Volcano plot of differentially expressed genes between CDX2/UBTF ALL (n = 23) and other B-ALL cases (n = 279). Genes with fold change greater than 3 and -log10 P value >7 are shown in red. CDX2 is highlighted as 1 of the 10 most highly expressed genes in the novel cluster. (D) WGS long-reads and RNA-seq splice junctions visualized on IGV software in 1 CDX2/UBTF case (B_SL160) showing a deletion at 17q21.31 locus involving the UBTF and ATXN7L3 genes. The red line underlines the deleted region. (E) Scheme of the predicted UBTF::ATXN7L3 chimeric protein. The resulting in-frame fusion transcript incorporated 60 nucleotides upstream the canonical translation start codon of ATXN7L3. (F) Western-blot of CDX2/UBTF cases using ATXN7L3 antibody shows detection of normal ATXN7L3 protein (39 kDa) in all samples and another protein at a size corresponding to the predicted UBTF::ATXN7L3 chimeric protein (110 kDa) in CDX2/UBTF cases only. Western blot performed on PDX from 3 CDX2/UBTF cases (B_SL157, B_SL187, and B_SL160), 2 PAX5 P80R cases (005-0112 and B_SL142), and 1 KMT2A-r case (B_SL349). IGV, integrative genomics viewer; PDX, patient-derived xenograft; tSNE, t-distributed stochastic neighbor embedding; WGS, whole-genome sequencing.
To investigate the molecular basis of this novel B-ALL subtype, we performed long-read WGS in 2 cases. Integrated analysis of long-read WGS and RNA-seq data uncovered 2 relevant somatic structural variants, both observed in the 2 cases. First, short deletions were observed at 13q12.2 locus (83 and 109 kb) (Figure 1B). Strikingly, those were located 138 kb upstream of the CDX2 gene, one of the most highly expressed genes in the novel cluster (Figure 1C; supplemental Table 7). Second, short deletions (10 kb) at 17q21.31 involved the UBTF and ATXN7L3 genes, with the loss of the 4 last exons of UBTF (exons 18-21) and intergenic sequence, bringing the 5′ portion of UBTF closer to the adjacent gene ATXN7L3 (Figure 1D). This resulted in an in-frame UBTF::ATXN7L3 fusion transcript detected in RNA-seq data and confirmed by qRT-PCR (Figure 1D-E).
CDX2 is a member of the caudal-type family (Cdx) of homeodomain TFs. It plays essential roles in development during early stages of embryogenesis, including primitive hematopoiesis.18,19 By contrast, CDX2 is not expressed in hematopoietic cells in postnatal life at any stage of differentiation20 but is restricted to the gut, where it is critical for homeostasis and behaves as a tumor suppressor in colorectal cancer.21 Intriguingly, ectopic CDX2 expression has been reported in most myeloid and lymphoblastic acute leukemias,20,22 suggesting a role in leukemogenesis. Further works have shown that ectopic Cdx2 is indeed able to induce acute myeloid leukemia (AML) and myelodysplastic syndrome in mice.23-25 To understand the relevance of our finding with respect to these observations, we analyzed CDX2 expression in a panel of AML, T-, and B-ALL and normal blood and bone marrow samples (supplemental Figure 3). Strikingly, CDX2 expression in B-ALL cases from the novel cluster was 1000-fold higher than in all other leukemia cases, suggesting a major driving role in this subtype.
UBTF (upstream binding transcription factor) is expressed ubiquitously and encodes a HMG-box DNA-binding protein involved in ribosome biogenesis. ATXN7L3 is a component of the histone deubiquitination module of the SAGA complex, which activates transcription by chromatin remodeling.26 UBTF was previously found involved in oncogenic fusions with ETS4 in prostate cancer27 and with MAML3 in neuroendocrine tumors,28 where it promotes expression of its partner genes. In agreement with this model, ATXN7L3 was slightly but significantly more expressed in cases from the novel cluster (supplemental Figure 4). In addition, tandem duplication of UBTF was recently reported in pediatric AML.29,30 The predicted UBTF::ATXN7L3 fusion protein retained the major part of UBTF, including all high mobility group box domains and the entire ATXN7L3. Western blot analysis confirmed the presence of an ATXN7L3-containing protein at the expected size in cases harboring the alteration (Figure 1F). Importantly, UBTF::ATXN7L3 fusion transcripts were found in each case of the cluster and were absent in all but 1 other B-ALL (Figure 2), suggesting a role as a codriver in this subtype, further named CDX2/UBTF.
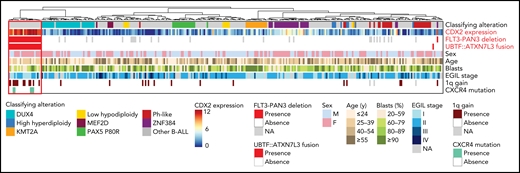
Summary oncoplot highlighting the main features of CDX2/UBTF ALL within the whole RNA-seq cohort. Classifying alterations, sex, age, blast percentage, and immunophenotypic classification according to European Group for the Immunological Classification of Leukaemia (EGIL); 1q gain and CXCR4 mutations are indicated. CDX2 expression is indicated in color scale from RNA-seq data. FLT3-PAN3 deletion corresponds to the 13q12.2 deletion involving FLT3 and PAN3 promoters and exon 1 (excluding 13q12.2 deletion sparing FLT3). UBTF::ATXN7L3 fusion indicates the presence of the fusion transcript as detected on RNA-seq data.
Summary oncoplot highlighting the main features of CDX2/UBTF ALL within the whole RNA-seq cohort. Classifying alterations, sex, age, blast percentage, and immunophenotypic classification according to European Group for the Immunological Classification of Leukaemia (EGIL); 1q gain and CXCR4 mutations are indicated. CDX2 expression is indicated in color scale from RNA-seq data. FLT3-PAN3 deletion corresponds to the 13q12.2 deletion involving FLT3 and PAN3 promoters and exon 1 (excluding 13q12.2 deletion sparing FLT3). UBTF::ATXN7L3 fusion indicates the presence of the fusion transcript as detected on RNA-seq data.
13q12.2 deletion drives cis-deregulation of CDX2 expression
Using array-CGH and/or targeted NGS, focal 13q12.2 deletions were identified in every CDX2/UBTF case (Figure 3A; supplemental Table 8). Fine mapping of those deletions using capture-based sequencing of the genomic region identified a common minimal deleted region of 41 kb length, which included the intergenic region between 2 contiguous genes, FLT3 and PAN3, and the promoter and first exon of both genes. CDX2/UBTF cases had lower expression of FLT3 and PAN3, consistent with gene dosage (Figure 3B). Interestingly, our data also revealed a correlated expression of FLT3 and CDX2 in all the other, non-CDX2/UBTF, B-ALL (Figure 3C), suggesting coregulation of the 2 genes within the locus.
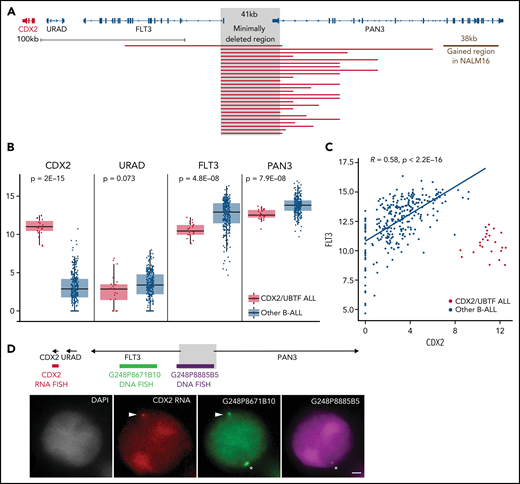
The 13q12.2 microdeletion upstream CDX2 is associated with deregulation of CDX2 in cis. (A) Capture-based sequencing of 13q12.2 locus identified focal deletion upstream of CDX2 in all CDX2/UBTF cases. The gray zone indicates the minimally deleted region. (B) Expression analysis of CDX2, URAD, FLT3, and PAN3 in CDX2/UBTF cases as compared with other B-ALL. Gene expression was quantified by RNA-seq (log2 of counts normalized using DESeq2’s median of ratios), and comparison analysis was performed by the Wilcoxon rank-sum test. (C) Dot-plot of expression data of CDX2 and FLT3 genes based on RNA-seq data for the whole cohort (n = 302) and Spearman's rank correlation analysis of non-CDX2/UBTF ALL. (D) Representative experiment (B_SL157) of sequential RNA-DNA FISH showing CDX2 primary transcripts (red signal) expressed from the allele located on the chromosome 13 exhibiting the genomic deletion (green G248P8671B10 signal and missing purple G248P8885B5 signal), shown with arrowheads. Wild-type chromosome 13 (green G248P8671B10 and purple G248P8885B5 signals) is shown by asterisks. DNA counterstained with DAPI (gray). Scale bar: 1 µm. DAPI, 4′,6-diamidino-2-phenylindole.
The 13q12.2 microdeletion upstream CDX2 is associated with deregulation of CDX2 in cis. (A) Capture-based sequencing of 13q12.2 locus identified focal deletion upstream of CDX2 in all CDX2/UBTF cases. The gray zone indicates the minimally deleted region. (B) Expression analysis of CDX2, URAD, FLT3, and PAN3 in CDX2/UBTF cases as compared with other B-ALL. Gene expression was quantified by RNA-seq (log2 of counts normalized using DESeq2’s median of ratios), and comparison analysis was performed by the Wilcoxon rank-sum test. (C) Dot-plot of expression data of CDX2 and FLT3 genes based on RNA-seq data for the whole cohort (n = 302) and Spearman's rank correlation analysis of non-CDX2/UBTF ALL. (D) Representative experiment (B_SL157) of sequential RNA-DNA FISH showing CDX2 primary transcripts (red signal) expressed from the allele located on the chromosome 13 exhibiting the genomic deletion (green G248P8671B10 signal and missing purple G248P8885B5 signal), shown with arrowheads. Wild-type chromosome 13 (green G248P8671B10 and purple G248P8885B5 signals) is shown by asterisks. DNA counterstained with DAPI (gray). Scale bar: 1 µm. DAPI, 4′,6-diamidino-2-phenylindole.
By screening additional patients’ samples and B-ALL cell lines for CDX2 expression, we identified 3 patients and the NALM16 cell line as having high CDX2 expression (supplemental Figure 5). The 3 patients presented 13q12.2 deletion and UBTF::ATXN7L3 fusion and were thus included in the CDX2/UBTF group. NALM16 harbored a short duplication (38 kb) at 13q12.2 within the PAN3 gene but no UBTF::ATXN7L3 fusion (Figure 3A).
Analysis of breakpoint sequences revealed recombination signal sequence–like motifs and nontemplated sequence nucleotides at junction (supplemental Figure 6), indicating that the deletion most likely occurred through off-target cleavage by the RAG1/2 recombinase in a differentiating B-cell precursor. Such deletions usually occur in regions of open chromatin around active genes.31 To understand if those deletions were causative of CDX2 deregulated expression rather than merely related to active transcription of the locus, we analyzed allele-specific expression of CDX2 using single-nucleotide polymorphisms located in its untranslated regions. In every CDX2/UBTF case with informative single-nucleotide polymorphisms (n = 10 cases), a clear mono-allelic expression pattern was observed (supplemental Table 9). We performed sequential RNA and DNA FISH on PDX of CDX2/UBTF ALL combining a probe to visualize the CDX2 nascent transcripts (RNA FISH part) and 2 probes mapping the deleted and nondeleted regions at 13q12.2 (DNA FISH part). RNA-DNA FISH showed that CDX2 was uniquely transcribed from the allele in cis from the deletion (Figure 3D; supplemental Table 10), demonstrating that CDX2 deregulated expression was mediated by the genomic deletion in cis.
Recurrent 13q12.2 deletions were recently reported in B-ALL and shown to deregulate FLT3.32 However, those deletions were different because they spared the FLT3 gene. In our cohort, we identified 10 cases having such deletions, which did not cluster with CDX2/UBTF ALL but spread across other B-ALL. Indeed, our expression data show distinct transcriptional effects on the locus, drastic deregulation of CDX2 expression on one hand vs moderate FLT3 increase on the other hand (supplemental Figure 7). Of note, 1 case had a deletion involving FLT3 with high CDX2 expression but no UBTF::ATXN7L3 fusion, and it did not cluster with CDX2/UBTF cases.
13q12 deletion alters 3D DNA topology with hijacking of a PAN3 enhancer
Using ChIP-seq for histone modifications, we mapped cis-regulatory elements within the 13q12.2 locus in 2 PDX of CDX2/UBTF cases and in NALM16 and control cell lines (Figure 4A). H3K4me3 and H3K27ac peaks were observed at promoter regions of CDX2, FLT3, and PAN3 in CDX2/UBTF ALL and NALM16, indicating active promoters. In addition, a strong H3K27ac peak was observed within the PAN3 gene, indicative of an active enhancer. Importantly, this element is spared in both types of 13q12.2 deletions, and Yang et al32 showed that it drives cis-deregulation of FLT3. In addition, the short duplication found in NALM16 encompasses this region, suggesting that this enhancer may be also involved in CDX2 deregulation. We performed 4C-seq on NALM16 and control cell lines using the CDX2 promoter as the viewpoint (Figure 4B). In addition to a conserved interaction downstream CDX2 seen in all cell lines, an interaction between the CDX2 promoter and the PAN3 enhancer region was observed in NALM16 only. Finally, we designed a CRISPRi experiment using single-guide RNAs targeting the PAN3 enhancer, coexpressed with a transcriptional repressor domain fused to catalytically dead CRISPR-Cas9 (Figure 4C). Upon PAN3 enhancer repression in NALM16, we observed marked reduction of CDX2 expression at the transcript and protein levels (Figure 4D-E). Collectively, these results definitively demonstrate the role of the PAN3 enhancer on CDX2 aberrant expression due to a genomic alteration within the 13q12.2 locus and strongly suggest that CDX2 deregulation in CDX2/UBTF ALL patients is the result of hijacking of the PAN3 enhancer.
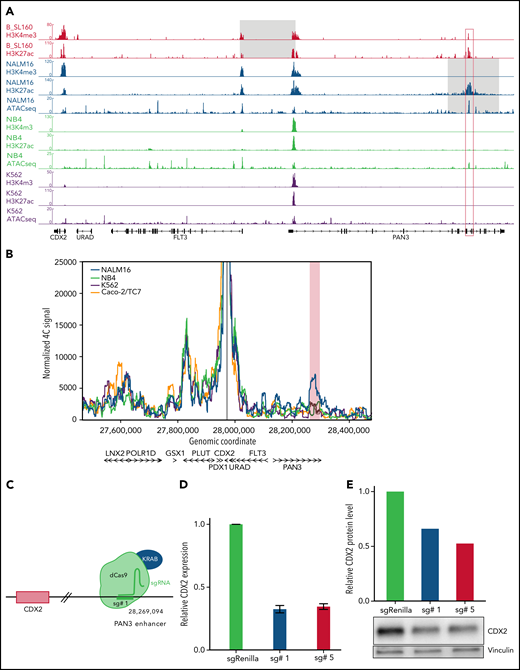
CDX2 cis-deregulation is driven by hijacking of an enhancer located in the PAN3 gene. (A) ChIP-seq signals of histone modifications (H3K4me3 and H3K27ac) of 1 representative CDX2/UBTF case (B_SL160) and ChIP-seq and ATAC-seq signals of NALM16, NB4, and K562 cell lines at the CDX2-PAN3 locus. CDX2 expression levels of cell lines are provided in supplemental Figure 5. The gray zone on CDX2/UBTF ALL and NALM16 traces indicate respectively the minimally deleted region and the duplicated region. The red box corresponds to the position of the enhancer identified in PAN3. (B) 4C-seq normalized signals of NALM16, NB4, K562, and Caco-2/TC7 cell lines with CDX2 promoter as view point. The red zone highlights the NALM16-specific called interaction (C) CRISPRi experimental scheme. In order to inactivate the enhancer in PAN3, we used the catalytically inactive CRISPR-associated protein 9 (dCas9) fused to the repressor KRAB with specific single-guide RNA. Representative example of a CRISPRi experiment showing CDX2 expression decrease in NALM16 after CRISPRi targeting the PAN3 enhancer, measured by qRT-PCR (D), and western blot (E).
CDX2 cis-deregulation is driven by hijacking of an enhancer located in the PAN3 gene. (A) ChIP-seq signals of histone modifications (H3K4me3 and H3K27ac) of 1 representative CDX2/UBTF case (B_SL160) and ChIP-seq and ATAC-seq signals of NALM16, NB4, and K562 cell lines at the CDX2-PAN3 locus. CDX2 expression levels of cell lines are provided in supplemental Figure 5. The gray zone on CDX2/UBTF ALL and NALM16 traces indicate respectively the minimally deleted region and the duplicated region. The red box corresponds to the position of the enhancer identified in PAN3. (B) 4C-seq normalized signals of NALM16, NB4, K562, and Caco-2/TC7 cell lines with CDX2 promoter as view point. The red zone highlights the NALM16-specific called interaction (C) CRISPRi experimental scheme. In order to inactivate the enhancer in PAN3, we used the catalytically inactive CRISPR-associated protein 9 (dCas9) fused to the repressor KRAB with specific single-guide RNA. Representative example of a CRISPRi experiment showing CDX2 expression decrease in NALM16 after CRISPRi targeting the PAN3 enhancer, measured by qRT-PCR (D), and western blot (E).
Gene expression signature of CDX2/UBTF ALL
To explore the deregulated gene expression in CDX2/UBTF ALL, we analyzed the core set of genes most differentially expressed (Figure 5A; supplemental Table 7). This signature comprised a small number of genes highly expressed in CDX2/UBTF ALL and, to a lesser extent, in most other B-ALL, like CD34 (cluster 4), and also genes shared with some subtypes, like MEIS1, also expressed in KMT2A-rearranged cases (cluster 3). Of note, some HOX genes were also moderately expressed in CDX2/UBTF ALL, in agreement with the role of CDX2 as an upstream factor (supplemental Figure 8).33,34 A small group of genes had much lower expression in CDX2/UBTF ALL, like CD9 (cluster 5). The vast majority of the signature comprised genes not expressed in other B-ALL and ectopic with regard to hematopoietic cells (annotated “not expressed in CD34+”), a number of which being expressed in brain or blood vessels. A limited number of CDX2 direct target genes in the context of colon cancer (Caco-2/TC7 cell line) were identified, including CD9, CDH4, NR6A1, PLD5, and SPON1. GSEA showed enrichment in genes regulated by the polycomb repressive complex 2 (PRC2) in embryonic stem cells, expressed in cord blood–derived hematopoietic stem cells, and related to homophilic cell adhesion (Figure 5B). GSEA also showed negative enrichment in genes related to cell cycle and mTOR signaling, suggestive of a lower metabolic state.
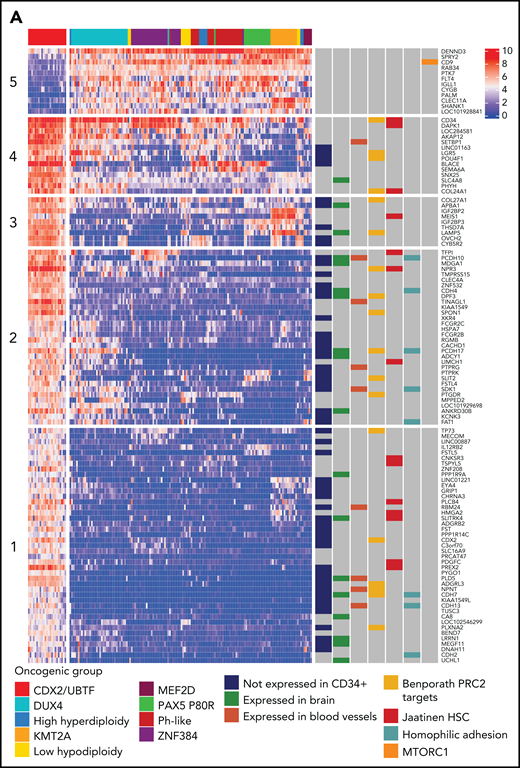
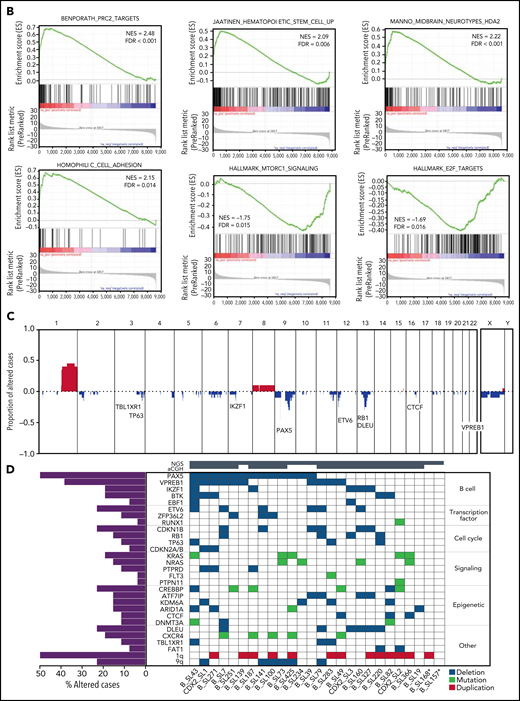
Transcriptional signature and genomic landscape of CDX2/UBTF ALL. (A) Heatmap representing biclustering of gene expression from the major subtypes in the RNA-seq cohort (excluding unclassified cases). Genes were selected after comparison between expression means (fold change >3, P < 10−7) resulting in a list of 110 genes (supplemental Table 7). Annotations on the right are derived from selected gene set enrichment analyses (GSEA) shown in (B) and expression data from The Human Cell Atlas Bone Marrow Single-Cell Interactive Web Portal (http://www.altanalyze.org/ICGS/HCA/splash.php) and the TSEA atlas (genetics.wustl.edu/jdlab/tsea/) (B) Selected GSEA for genes expressed in CDX2/UBTF ALL as compared with other cases, showing significant enrichment in genes regulated by the Polycomb repressive complex 2 (PRC2), genes expressed in cord blood–derived CD133+ cells enriched in hematopoietic stem cells, genes involved in homophilic cell-cell adhesion, and genes expressed in a subset of embryonic neurons. The differential gene list is negatively enriched in genes upregulated through mTORC1 signaling and E2F target genes. GSEA core genes are listed in supplemental Table 11. (C) Copy-number alterations in CDX2/UBTF ALL as determined by array-CGH analysis (n = 20). (D) Heatmap of recurrent alterations in CDX2/UBTF cases as determined by array-CGH and targeted sequencing (n = 26), with frequencies of altered cases indicated on the left. *Cases with low blast infiltration (ie, <30%), with possibly focal copy-number alterations overlooked. TSEA, tissue specific expression analysis.
Transcriptional signature and genomic landscape of CDX2/UBTF ALL. (A) Heatmap representing biclustering of gene expression from the major subtypes in the RNA-seq cohort (excluding unclassified cases). Genes were selected after comparison between expression means (fold change >3, P < 10−7) resulting in a list of 110 genes (supplemental Table 7). Annotations on the right are derived from selected gene set enrichment analyses (GSEA) shown in (B) and expression data from The Human Cell Atlas Bone Marrow Single-Cell Interactive Web Portal (http://www.altanalyze.org/ICGS/HCA/splash.php) and the TSEA atlas (genetics.wustl.edu/jdlab/tsea/) (B) Selected GSEA for genes expressed in CDX2/UBTF ALL as compared with other cases, showing significant enrichment in genes regulated by the Polycomb repressive complex 2 (PRC2), genes expressed in cord blood–derived CD133+ cells enriched in hematopoietic stem cells, genes involved in homophilic cell-cell adhesion, and genes expressed in a subset of embryonic neurons. The differential gene list is negatively enriched in genes upregulated through mTORC1 signaling and E2F target genes. GSEA core genes are listed in supplemental Table 11. (C) Copy-number alterations in CDX2/UBTF ALL as determined by array-CGH analysis (n = 20). (D) Heatmap of recurrent alterations in CDX2/UBTF cases as determined by array-CGH and targeted sequencing (n = 26), with frequencies of altered cases indicated on the left. *Cases with low blast infiltration (ie, <30%), with possibly focal copy-number alterations overlooked. TSEA, tissue specific expression analysis.
Genomic landscape of CDX2/UBTF ALL
We characterized additional alterations using array-CGH (n = 20, Figure 5C) and targeted sequencing of a custom panel of genes recurrently altered in B-ALL (n = 26, Figure 5D). A number of were recurrent in CDX2/UBTF ALL, the most frequent being duplication of the 1q arm, seen in 50% (13/26) of patients, often related to unbalanced chromosomal translocations as shown by cytogenetic data (supplemental Table 12). Other nonfocal copy-number alterations involved loss of 6q (n = 6), 9q (n = 6), 13q including DLEU (n = 6) and RB1 (n = 4), and 3q including TBL1XR1 (n = 3) and TP63 (n = 3). Focal deletions involving PAX5 5′ or gene promoter were particularly frequent (n = 13/26, 50%); other genes targeted by recurrent deletions were IKZF1 (n = 5/26, 19%), VPREB1, ETV6, CDKN1B, KDM6A, CTCF, and PTPRD.
Targeted sequencing showed frequent mutations of RAS pathway and CREBBP genes (27% and 19%, respectively) (Figure 5D; supplemental Tables 13-14). In addition, RNA-seq analysis for variant calling uncovered recurrent mutations of CXCR4, further confirmed by Sanger sequencing and present in 5 cases (22%). Interestingly, CXCR4 mutations were uniquely detected in CDX2/UBTF ALL. Those mutations are predicted to truncate the C-terminus, like described in WHIM syndrome and in Waldenström macroglobulinemia and shown to confer gain-of-function.35-37
Clinical presentation and outcome of CDX2/UBTF ALL
The 26 CDX2/UBTF ALL patients included 20 females, demonstrating highly skewed sex ratio (male/female ratio, 0.3), and comprised 4 adolescents and 22 adults. No CDX2/UBTF ALL patient was identified in adults aged 55 or older.
The immunophenotype of CDX2/UBTF ALL was frequently classified as pro–B/B-I stage according to EGIL (16/26, 62%), with negative or partial expression of CD10 (n = 22/26, 85%) and negative expression of CD20 (n = 20/22, 91%), whereas CD34 and CD38 were highly expressed (supplemental Table 15).
Seventeen CDX2/UBTF ALL patients were included in the GRAALL-2005/2014 protocols prospectively enrolling adults aged 18 to 59 years (Table 1). Thus, the prevalence of this subtype in adult Ph-negative ALL is 2.4% (17/723). Female predominance was significant in this cohort (83% vs 44% in other B-ALL, P = .002), and patients tended to be younger (median age, 31 years vs 38 years, P = .13). Blast infiltration in the bone marrow was significantly lower in CDX2/UBTF ALL (91% vs 81%, P = .04), and their immunophenotype was skewed to pro–B/B-I EGIL stage (12/17, 71%, P < .001).
Study patient characteristic
| All patients (n = 723) | Other B-ALL (n = 706) | CDX2/UBTF ALL (n = 17) | P value | |
|---|---|---|---|---|
| Patient-related characteristics | ||||
| Median age, y (range) | 38 (18-60) | 38 (18-60) | 31 (19-50) | .13 |
| Gender, M/F (ratio) | 400/323 (1.2) | 397/309 (1.3) | 3/14 (0.2) | .002 |
| Disease-related characteristics | ||||
| Median WBC, G/L (range) | 9 (0-712) | 9 (0-712) | 9 (1-63) | .72 |
| BM blast percentage (range) | 91 (21-100) | 91 (21-100) | 81 (35-98) | .04 |
| CNS involvement | 57/713 (8%) | 57/696 (8%) | 0/17 (0%) | .39 |
| Immunophenotypic classification (EGIL) | <.001 | |||
| Pro-B (I) | 148/622 (24%) | 136/605 (22%) | 12/17 (71%) | |
| Common (II) | 364/622 (59%) | 361/605 (60%) | 3/17 (18%) | |
| Pre-B (III) | 102/622 (16%) | 100/605 (17%) | 2/17 (12%) | |
| Mature (IV) | 8/622 (1%) | 8/605 (1%) | 0/17 (0%) | |
| Response-related characteristics | ||||
| Good PB prednisone response at day 8 | 582/701 (83%) | 574/685 (84%) | 8/16 (50%) | .002 |
| Good BM response at day 15 | 397/668 (59%) | 395/653 (60%) | 2/15 (13%) | <.001 |
| Late CR (achieved after induction 2) | 24/672 (4%) | 21/656 (3%) | 3/16 (19%) | .017 |
| CR (after induction 1 and 2) | 672/723 (93%) | 656/706 (93%) | 16/17 (94%) | 1.0 |
| Postinduction MRD ≥10-4 | 255/542 (47%) | 242/528 (46%) | 13/14 (93%) | <.001 |
| Postinduction MRD ≥10-3 | 151/542 (28%) | 144/528 (27%) | 7/14 (50%) | .073 |
| Postremission outcome | ||||
| Allo-SCT in first CR | 184/672 (27%) | 174/656 (27%) | 10/16 (63%) | .003 |
| 3-y CIR, % (95% CI) | 33.5 (29.8-37.6) | 32.4 (28.7-36.5) | 75.0 (50.6-93.5) | .004 |
| 3-y OS, % (95% CI) | 65.9 (62.0-69.5) | 65.9 (61.9-69.5) | 67.9 (38.8-85.4) | .86 |
| All patients (n = 723) | Other B-ALL (n = 706) | CDX2/UBTF ALL (n = 17) | P value | |
|---|---|---|---|---|
| Patient-related characteristics | ||||
| Median age, y (range) | 38 (18-60) | 38 (18-60) | 31 (19-50) | .13 |
| Gender, M/F (ratio) | 400/323 (1.2) | 397/309 (1.3) | 3/14 (0.2) | .002 |
| Disease-related characteristics | ||||
| Median WBC, G/L (range) | 9 (0-712) | 9 (0-712) | 9 (1-63) | .72 |
| BM blast percentage (range) | 91 (21-100) | 91 (21-100) | 81 (35-98) | .04 |
| CNS involvement | 57/713 (8%) | 57/696 (8%) | 0/17 (0%) | .39 |
| Immunophenotypic classification (EGIL) | <.001 | |||
| Pro-B (I) | 148/622 (24%) | 136/605 (22%) | 12/17 (71%) | |
| Common (II) | 364/622 (59%) | 361/605 (60%) | 3/17 (18%) | |
| Pre-B (III) | 102/622 (16%) | 100/605 (17%) | 2/17 (12%) | |
| Mature (IV) | 8/622 (1%) | 8/605 (1%) | 0/17 (0%) | |
| Response-related characteristics | ||||
| Good PB prednisone response at day 8 | 582/701 (83%) | 574/685 (84%) | 8/16 (50%) | .002 |
| Good BM response at day 15 | 397/668 (59%) | 395/653 (60%) | 2/15 (13%) | <.001 |
| Late CR (achieved after induction 2) | 24/672 (4%) | 21/656 (3%) | 3/16 (19%) | .017 |
| CR (after induction 1 and 2) | 672/723 (93%) | 656/706 (93%) | 16/17 (94%) | 1.0 |
| Postinduction MRD ≥10-4 | 255/542 (47%) | 242/528 (46%) | 13/14 (93%) | <.001 |
| Postinduction MRD ≥10-3 | 151/542 (28%) | 144/528 (27%) | 7/14 (50%) | .073 |
| Postremission outcome | ||||
| Allo-SCT in first CR | 184/672 (27%) | 174/656 (27%) | 10/16 (63%) | .003 |
| 3-y CIR, % (95% CI) | 33.5 (29.8-37.6) | 32.4 (28.7-36.5) | 75.0 (50.6-93.5) | .004 |
| 3-y OS, % (95% CI) | 65.9 (62.0-69.5) | 65.9 (61.9-69.5) | 67.9 (38.8-85.4) | .86 |
For BM blast percentage, data were available for 423 patients. Good PB prednisone response at day 8 is defined by less than 1 G/L blast. Good BM response at day 15 is defined by <5% blast. MRD evaluation was performed by PCR quantification of immunoglobulin or T-cell receptor clono-specific rearrangements according to EuroMRD guidelines.
Allo-SCT, allogeneic stem cell transplantation; BM, bone marrow; CNS, central nervous system; CR, complete remission; EGIL, European Group for the Immunological Characterization of Leukemias; F, female; M, male; MRD, minimal residual disease; OS, overall survival; PB, peripheral blood.
Patients with CDX2/UBTF ALL displayed a slow response to induction phase. Peripheral blood response to steroid prophase at day 8 and bone marrow blast clearance at day 15 (M1 bone marrow) were observed in only 50% (vs 84% in other B-ALL, P = .002) and 13% (vs 60%, P < .001) of CDX2/UBTF cases, respectively. One patient died during induction, and 3 patients achieved a late CR after second induction (19%, vs 3%, P = .017). All but 1 evaluable patient (13/14, 93%) had a high MRD (≥10−4) at CR, as compared with 46% in others (P < .001). Therefore, CDX2/UBTF ALL is a subtype with very poor response to chemotherapy.
Patients with poor response at day 8 or at day 15 were eligible to allo-SCT in GRAALL-2005, whereas patients with high postinduction MRD were eligible in GRAALL-2014. Accordingly, patients with CDX2/UBTF ALL received more frequent allo-SCT (10/16, 63% vs 74/656, 27%, P = .003). The median follow-up of the cohort was 3.4 years. At 3 years, the CIR estimates were 75.0% (95% CI, 50.6-93.5) in CDX2/UBTF ALL patients and 32.4% (95% CI, 28.7-36.5) in other B-ALL (specific hazard ratio, 2.43; 95% CI, 1.32-4.46; P = .004, Figure 6; supplemental Figure 9). CDX2/UBTF ALL was still associated with a significantly higher CIR in multivariate analysis including age, WBC counts, and postinduction MRD as covariates (specific hazard ratio, 2.41; 95% CI, 1.20-4.84; P = .013, Figure 6C). However, the 3-year OS did not differ between CDX2/UBTF ALL (67.9%; 95% CI, 38.8-85.4) and other B-ALL patients (65.9%; 95% CI, 61.9-69.5), which may reflect a favorable response to current salvage procedures in B-ALL.
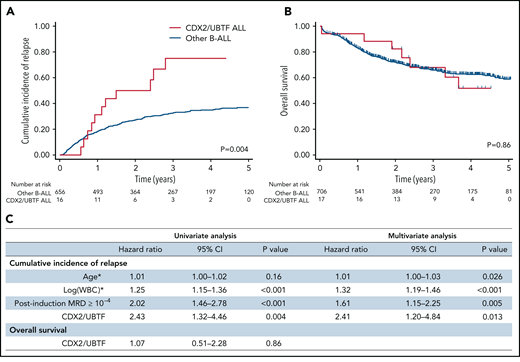
Outcome analyses of CDX2/UBTF ALL patients. Cumulative incidence of relapse (A) and OS (B) Kaplan-Meier curves for B-ALL patients enrolled in GRAALL-2005/2014 trials according to CDX2/UBTF ALL status. (C) Univariate and multivariate outcome analyses for CDX2/UBTF ALL patients. *Continuous variables. WBC, white blood cell count.
Outcome analyses of CDX2/UBTF ALL patients. Cumulative incidence of relapse (A) and OS (B) Kaplan-Meier curves for B-ALL patients enrolled in GRAALL-2005/2014 trials according to CDX2/UBTF ALL status. (C) Univariate and multivariate outcome analyses for CDX2/UBTF ALL patients. *Continuous variables. WBC, white blood cell count.
Discussion
Aiming to elucidate the oncogenic bases of unresolved cases of adult B-ALL, we uncovered a novel leukemia entity with unique genomic and phenotypic features. Most intriguing is the systematic combination of 2 genomic alterations pointing to distinct candidate oncogenic drivers, namely CDX2 and UBTF::ATXN7L3. CDX2 is a homeobox TF well known for its role during embryogenesis and in adult intestinal homeostasis. Inappropriate reactivation of CDX2 has been widely observed in leukemias, and an oncogenic role was proposed.20,22 However, the mechanism of ectopic deregulation has remained an enigma and, even though CDX2 expression in the hematopoietic compartment is able to induce leukemia in mice,23-25 there was no direct evidence of its contributing role in human leukemogenesis. Our study, demonstrating the targeting and strong deregulation of CDX2 by a recurrent alteration in a subset of B-ALL, thus provides definitive support for its oncogenic role in human leukemia. A recent article from Yasuda et al38 reported the CDX2-expressing B-ALL subtype but did not elucidate the mechanism of its deregulation. Interestingly, we uncovered focal alterations within the CDX2-FLT3-PAN3 locus in all CDX2/UBTF ALL patients and in the NALM16 cell line and proved CDX2 cis-deregulation mediated by enhancer hijacking. We also provide evidence that those deletions likely result from RAG-mediated inappropriate recombination events, which are commonly seen in ALL. To be targeted by RAG enzymes, a locus should be in an open chromatin state and, accordingly, translocations and deletions are enriched in B-cell TFs and other genes actively transcribed during B-cell development.31 Because CDX2 is not expressed in normal hematopoietic cells, one can speculate that recombination events close to FLT3 and PAN3 promoters could occur, owing to their active transcriptional state. Hence, FLT3 expression is initiated in multipotent progenitors and plays a key role in the maintenance of lymphoid‐primed multipotent and common lymphoid progenitors. Expression pattern and conditional knockout mouse studies showed that FLT3 expression is maintained and critical at the pro-B stage, where RAG expression is induced.39 Therefore, our data highlight how neighbor genes can be involved in a deleterious loop linking gene expression, chromatin structure, and oncogenic activation, suggesting a mechanistic framework for deregulation of ectopic genes by RAG-mediated alterations.
The functional consequences of both CDX2 deregulation and UBTF::ATXN7L3 chimeric fusion remain to be investigated, as well as the likely oncogenic cooperation suggested by the unique combination of those 2 alterations. CDX2 is known to act as an upstream regulator of HOX genes,33,34 the expression of which was observed in CDX2/UBTF ALL, although at a lesser extent than in KMT2A-rearranged ALL. However, this probably does not recapitulate CDX2-mediated oncogenesis, and identifying the relevant targets of CDX2 in B-ALL will require dedicated work, taking into account its context-dependent function.40,41 Not less complex may be the role of UBTF::ATXN7L3. Upstream binding transcription factor, the product of UBTF, plays a critical role in ribosomal RNA transcription.42 Interestingly, alterations targeting UBTF (ie, internal tandem duplications) were reported as a recurrent lesion in pediatric AML.29,30 Because it is ubiquitously expressed and interacts with DNA as a dimer, its role in the fusion may also fit with the common model for chimeric fusions where the 5′ partner allows deregulated expression and dimerization of the 3′ partner, altering its properties of DNA binding or cofactors recruitment.43
ATXN7L3 is an adaptor protein that orchestrates activities of multiple deubiquitinating enzymes and acts as a global facilitator for histone 2B (H2B) deubiquitination.26 Misregulation of H2B monoubiquitination has been observed in cancers and shown to promote oncogenesis.44 Hence, H2B deubiquitination is required for optimal expression of many target genes of transcription activators.45,46 One may speculate that altered activity of ATXN7L3 factor fused to UBTF might enable CDX2 to drive an aberrant transcriptional program and reveal its oncogenic potential. The core signature of CDX2/UBTF ALL comprises a number of ectopic genes normally expressed in brain or blood vessels, the chromatin state of which in lymphoid progenitors may not allow activation by CDX2 alone. In support of a unique oncogenic cooperation are the 2 outlier cases from our cohort, 1 having only the CDX2 cis-deregulating deletion and the other only the UBTF::ATXN7L3 fusion and neither presenting the typical and complete signature of the subtype. However, functional analyses will be required to decipher the role of these drivers and their possible functional interaction.
By analyzing biological and patients’ characteristics as well as additional genetic alterations of CDX2/UBTF ALL patients, we highlighted several salient features. First, patients were strikingly enriched in females. The basis for this bias is still unknown and deserves investigation. Second, the immunophenotype of leukemic cells was remarkable, with negativity of CD10 and CD20 (pro–B/B-I EGIL stage), a common feature of KMT2A-rearranged ALL but otherwise uncommon. CD34 high expression and CD9 negativity were also noted in both immunophenotypic data and gene expression signature. Third, the pattern of additional genomic alterations exhibited several notable features. They comprised a number of large chromosomal imbalances, including recurrent duplications in 1q arm, also observed by Yasuda et al,38 and deletions in 3q, 6q, 9q, and 13q, which is not common in B-ALL and suggests underlying genomic instability. Of note, CDX2 has been previously involved in inhibition of DNA double-strand break repair activity in intestinal context.47 Focal deletions commonly observed in B-ALL involving IKZF1, PAX5, and ETV6 deletions were also observed in CDX2/UBTF ALL, with a notable high frequency of deletions of PAX5 5′ or promoter regions. CREBBP and RAS pathway alterations were relatively frequent in CDX2/UBTF ALL. Interestingly, this association has been reported in high-hyperdiploid B-ALL and linked to treatment resistance and relapse.48 Finally, we identified recurrent mutations of CXCR4, which had never been reported to date in acute leukemias. CXCR4 encodes a chemokine receptor broadly expressed in hematopoietic cells and critical for their interaction with the bone marrow niche and migration. Mutations observed in CDX2/UBTF ALL were somatically acquired and were seen in a minor leukemia fraction in some patients. Of note, 2 distinct CXCR4 mutations were detected in 2 patients, which suggests specific constraints or cooperation in oncogenesis paths of CDX2/UBTF ALL. Further work is warranted to elucidate the significance of CXCR4 mutations in CDX2/UBTF ALL and, possibly, the relevance of the therapeutic use of a CXCR4 antagonist to mobilize leukemic cells.
Prognosis of CDX2/UBTF ALL was analyzed within the GRAALL-2005 and 2014 trials, comprising 17 cases out of 723 with available molecular and clinical data. CDX2/UBTF ALL patients exhibited criteria of poor response to treatment at every stage of evaluation (ie, response at day 8, day 15, and end of induction). Of note, poor sensitivity to chemotherapy may be related to a low-proliferating condition of these leukemia cells, as suggested by normal WBC counts at diagnosis and negative enrichment in cell cycle genes and mTOR signaling. Three patients failed to reach CR after 1 induction phase, and all but 1 had positive postinduction MRD. Accordingly, a very high relapse rate was observed, beyond those of known poor-risk B-ALL subtypes like KMT2A rearrangement and low hypodiploidy. A poor prognosis was also reported in Yasuda et al, with lower disease-free and overall survival.38 By contrast, OS of CDX2/UBTF ALL was not lower in the present study, likely because of efficient salvage therapies currently available. Overall, our study reveals a novel high-risk B-ALL subtype, which requires novel therapeutic developments.
Acknowledgments
The authors thank the patients and all the GRAALL investigators and biologists who contributed samples and data for this study. The authors thank the Saint-Louis Molecular Hematology laboratory for technical support, especially Agnès Pérus, Hélène Boyer, and Emilie Gaudas, and Loïc Maillard from the research team. The authors thank Pierre Lemaire and Clémentine Chauvel for expert advice on immunophenotypic data. The authors thank Delphine Ndiaye-Lobry and Alexandra da Silva Babinet for the help with A-DNA FISH experiments. They thank the IMAG’IC facility (member of the National Infrastructure France BioImaging [ANR-10-INBS-04]) of Institut Cochin. The authors thank the Platform of Biological Resources from Robert Debré hospital, Assistance Publique-Hôpitaux de Paris, Paris, France. Long-read WGS was performed on the Meary third-generation sequencing platform of Université de Paris. Sequencing of 4C-seq was performed by the GenomEast platform, a member of the “France Génomique” consortium (ANR-10-INBS-0009). UMR-944, EA-3518, UMR-1274, Meary platform, and GRAALL are members of the Institut Carnot OPALE.
This study was supported by grants from Association Laurette Fugain, La Ligue régionale du Haut-Rhin Contre le Cancer (2019-20), and Institut National du Cancer (PLBIO 19-289). M.P. was a recipient of a grant from ITMO Cancer of Aviesan Cancer Control Strategy, on funds administered by INSERM. This work was supported by THEMA, the national center for precision medicine in leukemia.
Authorship
Contribution: M.P. performed experiments, analyzed data, and wrote the manuscript; R.K. and S.G. performed experiments and contributed to data analysis and study design; F.S. analyzed gene expression data and contributed to study design and manuscript writing; J.C., A.G., N.K., L.H., and B.H. performed experiments and analyzed data; S.Q., T.S., and T.Y. performed bioinformatics analyses; L.L. and H.B. contributed to patients’ annotations and diagnostic analyses; A.C. contributed to patient’s sample and annotation; C.L., F.P., and J-N.F. contributed to study design; T.L., M.H., F.H., V.L., and H.D. contributed to patients’ samples and clinical annotations and led the clinical trials; C.D.-D. contributed to data analysis and study design; J.S. contributed to data analysis, study design, and manuscript writing; N.B. contributed to patients’ samples and clinical annotations, led the clinical trials, performed clinical statistical analyses, and contributed to manuscript writing; E.C. designed research, analyzed data, and wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emmanuelle Clappier, Laboratoire d’Hématologie, Hôpital Saint-Louis, 1 Ave Claude Vellefaux, 75010 Paris, France; e-mail: emmanuelle.clappier@aphp.fr.
RNA-seq, WGS, and ChIPseq data files from B-ALL patients have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession numbers E-MTAB-11364, E-MTAB-11400, and E-MTAB-11500. Files can be made available upon comprehensive request after internal approval according to the Saint-Louis Research Institute scientific policy and to French laws.
Send data sharing requests via e-mail to the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal