

Key Points
PHF6 interacts with NurD, SWI/SNF and ISWI factors, the replication machinery, and DNA repair proteins.
PHF6 associates with heterochromatin at satellite DNA and protects genomic fragile sites from DNA damage–induced genetic instability.

Abstract
The Plant Homeodomain 6 gene (PHF6) encodes a nucleolar and chromatin-associated leukemia tumor suppressor with proposed roles in transcription regulation. However, specific molecular mechanisms controlled by PHF6 remain rudimentarily understood. Here we show that PHF6 engages multiple nucleosome remodeling protein complexes, including nucleosome remodeling and deacetylase, SWI/SNF and ISWI factors, the replication machinery and DNA repair proteins. Moreover, after DNA damage, PHF6 localizes to sites of DNA injury, and its loss impairs the resolution of DNA breaks, with consequent accumulation of single- and double-strand DNA lesions. Native chromatin immunoprecipitation sequencing analyses show that PHF6 specifically associates with difficult-to-replicate heterochromatin at satellite DNA regions enriched in histone H3 lysine 9 trimethyl marks, and single-molecule locus-specific analyses identify PHF6 as an important regulator of genomic stability at fragile sites. These results extend our understanding of the molecular mechanisms controlling hematopoietic stem cell homeostasis and leukemia transformation by placing PHF6 at the crossroads of chromatin remodeling, replicative fork dynamics, and DNA repair.
Introduction
Originally identified as the causative gene of Börjeson-Forssman-Lehmann syndrome, an X-linked neurodevelopmental disorder,1 the Plant Homeodomain 6 gene (PHF6) functions as an epigenetic regulator of long-term self-renewal in hematopoietic stem cells frequently mutated in T-cell acute lymphoblastic leukemia (T-ALL), in T-myeloid mixed lineage tumors, and, albeit less frequently, in acute myeloid leukemia and myelodysplastic syndromes.2-5 Functionally, PHF6 localizes to the nucleolus and interacts with the PAF1 transcription elongation complex6 implicated in the control of RNA polymerase I activity and ribosomal DNA (rDNA) transcription and with UBF,7 a transcriptional activator in the RNA polymerase I pre-initiation complex; these actions support a role for PHF6 in the control of ribosome biogenesis. Moreover, PHF6 associates with the nucleosome remodeling and deacetylase (NuRD) complex,8 a major chromatin regulator controlling nucleosome positioning and transcription with important roles in development, genome integrity, and cell cycle progression.9,10 Finally, early and recent work on the characterization of factors involved in the clearance of γ-H2AX after DNA damage revealed that suppression of PHF6 expression can impair the clearance of this DNA damage–associated mark.11,12 Consistently, PHF6 inactivation results in γ-H2AX accumulation,3 indicating a potential link between PHF6 function and maintenance of genomic integrity.3,12
Mechanistically, increased self-renewal in the hematopoietic stem cell compartment seems to be a major effector contributing to leukemia development after PHF6 loss.13-15 Thus, genetic inactivation of Phf6 primes hematopoietic stem cells to transformation by oncogenic NOTCH1 in mice, and secondary loss of Phf6 in NOTCH1-induced T-ALL increases the numbers of self-renewing leukemia-initiating cells.13 Consistently, PHF6 mutations are recurrently found in clonal hematopoiesis associated with aging16 and in clonal hematopoiesis developing in patients with aplastic anemia as they recover from bone marrow failure.17 In agreement, loss of PHF6 is frequently an early initiating event in leukemia transformation.13,18 Molecularly, Phf6 inactivation seems to favor increased chromatin accessibility in hematopoietic stem cells and leads to the upregulation of JAK-STAT target genes.13 In addition, loss of Phf6 causes the upregulation of expression of gene sets linked with increased leukemia stem cell activity. However, the molecular mechanisms underlying its tumor suppressor activity remain unknown. To bridge this gap, we sought to gain further insight into the molecular functions of PHF6 by analyzing the composition of PHF6-associated protein complexes isolated by tandem affinity purification. Our results uncovered a broader and largely unanticipated role of PHF6 in chromatin regulation in association not only with the NuRD complex but also with the SWI/SNF machinery and implicate PHF6 in the control of replication fork dynamics and DNA repair specifically at difficult-to-replicate satellite DNA sites.
Methods
Isolation of PHF6 protein complexes by tandem affinity purification
We harvested HEK293T and Jurkat cells (2000 million cells) stably expressing PHF6-FLAG-HA and green fluorescent protein (GFP) and empty vector control GFP-expressing cells, and we then extracted the cytoplasmic fraction by incubation in 20 volumes of Cytosol Hypotonic Buffer A (10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES] pH 7.9, 10 mM potassium chloride, 1.5 mM magnesium chloride [MgCl2], 0.1 mM EDTA) supplemented with protease (11697498001; Sigma-Aldrich) and phosphatase (4906845001; Sigma-Aldrich) inhibitors for 15 minutes on ice, with occasional vortexing. We added 0.1% NP40 and isolated cell nuclei by centrifugation at 100g for 15 minutes. Nuclear pellets were washed once in Cytosol Hypotonic Buffer A and then resuspended in 5 volumes of Nuclear Extraction Buffer C (20 mM HEPES, pH 7.9, 400 mM sodium chloride [NaCl], 1.5 mM MgCl2, 0.4% Triton X-100, 1 mM EDTA) supplemented with protease and phosphatase inhibitors. After incubation on ice for 60 minutes, with frequent vortexing, nuclear extracts were centrifuged at 2000g for 30 minutes, and we collected the high-salt nuclear fraction supernatants and adjusted to reduce the NaCl concentration to 150 to 200 mM by adding Equilibration Buffer (20 mM HEPES, pH 7.9, 10% glycerol, 1 mM EDTA) supplemented with protease and phosphatase inhibitors. To immunoprecipitate PHF6-Flag-HA–containing protein complexes, nuclear extracts were incubated with anti–Flag M2 beads (M8823; Sigma-Aldrich) overnight at 4°C. Beads were washed 3 times with phosphate-buffered saline supplemented with protease and phosphatase inhibitors at 4°C, and eluted protein complexes by overnight incubation in 150 mM NaCl Equilibration Buffer containing 1 mg/mL Flag peptide (F3290; Sigma-Aldrich). We performed a second round of immunoprecipitation with anti-HA beads (26182; Thermo Fisher Scientific) as before and eluted with HA peptide (I2149; Sigma-Aldrich). Pulled down proteins were analyzed by using mass spectrometry at the Taplin Biological Mass Spectrometry Facility.
PHF6 native chromatin immunoprecipitation
We harvested 20 million cells per condition and resuspended at 10 million cells per milliliter of 0.3% Triton X-100/phosphate-buffered saline supplemented with protease and phosphatase inhibitors to isolate nuclei. Nuclei were resuspended in 250 µL of EX100 buffer (10 mM HEPES [pH 7.6], 100 mM NaCl, 1.5 mM MgCl2, 0.5 mM EGTA, 10% glycerol, 0.2 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol) and added 1.5 U/µL of micrococcal nuclease for 20 minutes at room temperature. The reaction was stopped by adding EGTA to a final concentration of 10 mM, resuspended nuclear pellets in EX100 buffer and incubated overnight at 4°C with PHF6 (HPA001023; Sigma-Aldrich) or immunoglobulin G (C15400001-15; Diagenode) antibodies. We used 75 µL of equilibrated magnetic beads (16-662; Sigma-Aldrich) in EX-100 buffer for 3 hours at 4°C. Samples were washed twice with Wash Buffer 1 (10 mM Tris pH 7.5, 1 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 0,1% sodium deoxycholate, 1% Triton X-100/H2O), once with Wash Buffer 2 (10 mM Tris pH 7.5, 1 mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 150 mM NaCl/H2O) and once with 1X TE + 0.2% Triton X-100. The samples were resuspended in 100 µL of 1X TE, eluted DNA in 10% SDS + 20 mg/mL proteinase K for 1 hour at 65°C and resuspended in 100 µL of TE + 0.5 M NaCl. We eluted and purified DNA from input and immunoprecipitation samples by using phenol:chloroform extraction followed by ethanol precipitation, resuspended it in water, and quantified it by UV absorbance in a NanoDrop spectrophotometer. The Diagenode MicroPlex Kit for Illumina platforms (C05010012; Diagenode) was used, following manufacturer’s instructions for library preparation. The libraries were quantified by using the KAPA Library Quantification Kit for Illumina Platforms (KR0405; Kappa Biosystems) and AMPure XP (A63880; Beckman Coulter) for library purification. Amplicon pools were sequenced in an Illumina NextSeq500/550. Data are available in the Gene Expression Omnibus database (accession number: GSE152292).
Single-molecule analysis of replicated DNA
Single-molecule analysis of replicated DNA (SMARD) was conducted by using a procedure reported previously19,20 and described in detail in the supplemental Methods (available on the Blood Web site).
Statistical analyses
Statistical analysis was performed by using GraphPad Prism version 5.0 (GraphPad Software). We assumed normality and equal distribution of variance between groups, and results of P < .05 by a Student's t test were considered as statistically significant. Replication fiber analyses were conducted on blinded images.
Results
PHF6 prominently interacts with nucleosome remodeling protein complexes, the replication machinery, and DNA repair factors
To gain insight regarding the molecular mechanisms engaged in the tumor suppressor activity of PHF6 in human leukemia and general functions controlled by PHF6, we performed mass spectrometry analysis of PHF6-HA-FLAG protein complexes isolated from Jurkat, a PHF6 wild-type T-ALL cell line, and in HEK293T cells, a human fetal kidney–derived cell line. These analyses revealed fundamentally overlapping protein interactions in support of a general role for PHF6 in cellular homeostasis. Across both data sets, 85 PHF6-associated factors were identified (Figure 1; supplemental Table 1), inclusive of known PHF6-interacting proteins such as multiple members of the NuRD complex (CHD4, HDAC1, HDAC2, RBBP4, RBBP7, MBD2, MTA2, MBD3, and GATAD2B). In addition, PHF6 also associated with the SWI/SNF family of chromatin remodelers in our tandem affinity purification profiling (SMARCA4, SMARCB1, SMARCC1, SMARCC2, and SMARCE1), as well as by coimmunoprecipitation and western blot analyses (SMARCA4/BRG1, ARID2, BRD7, SMARCA5/SNF2H, BCL11B, WDR5, ASH2L, HCFC1, and SETD1A) (supplemental Figure 1).These findings suggest a broader and more complex role of PHF6 in chromatin remodeling and nucleosome repositioning than previously recognized.21
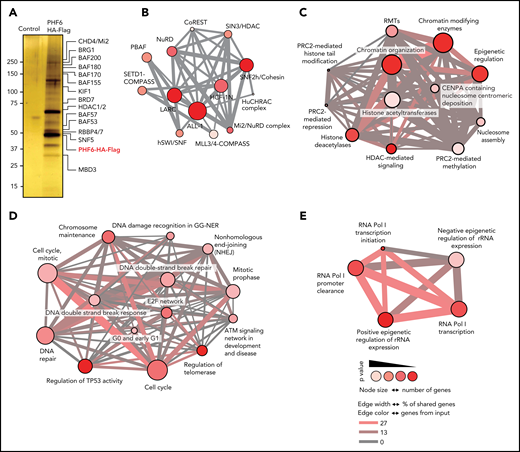
PHF6 associates with protein complexes involved in chromatin regulation and DNA repair. (A) Purified proteins after tandem affinity purification visualized by silver staining. Molecular weight marker is indicated on the left. (B) ConsensusPathDB over-representation analysis of protein complexes. (C) ConsensusPathDB over-representation analysis showing enrichment in chromatin remodeling pathways. (D) ConsensusPathDB over-representation analysis showing enrichment in DNA repair pathways. (E) ConsensusPathDB over-representation analysis showing enrichment in rRNA expression regulation pathways. ALL-1, leukemia acute lymphocytic susceptibility to 1; BAF, BRG1/BRM-associated factor; BRD7, bromodomain-containing protein 7; BRG1, brahma-related gene 1; CHD4, chromodomain helicase DNA binding protein 4; COMPASS, complex proteins associated with Set1; CoREST, RE1-silencing transcription factor corepressor; GG-NER, global-genome nucleotide excision repair; HCF1N, host cell factor 1; HDAC1/2, histone deacetylase 1/2; hSWI/SNF, human SWItch/sucrose nonfermentable; HuCHRAC, human chromatin accessibility complex; KIF1, kinesin family member 1a; LARC, LCR-associated remodeling complex; MBD3, methyl-CpG binding domain protein 3; MLL3/4, mixed-lineage leukemia-like protein 3/4; NuRD, nucleosome remodeling and deacetylase; PBAF, polybromo Brg1-associated factor; PRC2, polycomb repressive complex 2; RBBP4/7, retinoblastoma-binding protein 4/7; RMT, histone arginine methylase; RNA Pol I, RNA polymerase I; SETD1, SET domain containing 1A; SIN3, Swi-independent 3; SNF2h, sucrose nonfermenting 2 homolog; SNF5, switching defective/sucrose nonfermenting subunit 5.
PHF6 associates with protein complexes involved in chromatin regulation and DNA repair. (A) Purified proteins after tandem affinity purification visualized by silver staining. Molecular weight marker is indicated on the left. (B) ConsensusPathDB over-representation analysis of protein complexes. (C) ConsensusPathDB over-representation analysis showing enrichment in chromatin remodeling pathways. (D) ConsensusPathDB over-representation analysis showing enrichment in DNA repair pathways. (E) ConsensusPathDB over-representation analysis showing enrichment in rRNA expression regulation pathways. ALL-1, leukemia acute lymphocytic susceptibility to 1; BAF, BRG1/BRM-associated factor; BRD7, bromodomain-containing protein 7; BRG1, brahma-related gene 1; CHD4, chromodomain helicase DNA binding protein 4; COMPASS, complex proteins associated with Set1; CoREST, RE1-silencing transcription factor corepressor; GG-NER, global-genome nucleotide excision repair; HCF1N, host cell factor 1; HDAC1/2, histone deacetylase 1/2; hSWI/SNF, human SWItch/sucrose nonfermentable; HuCHRAC, human chromatin accessibility complex; KIF1, kinesin family member 1a; LARC, LCR-associated remodeling complex; MBD3, methyl-CpG binding domain protein 3; MLL3/4, mixed-lineage leukemia-like protein 3/4; NuRD, nucleosome remodeling and deacetylase; PBAF, polybromo Brg1-associated factor; PRC2, polycomb repressive complex 2; RBBP4/7, retinoblastoma-binding protein 4/7; RMT, histone arginine methylase; RNA Pol I, RNA polymerase I; SETD1, SET domain containing 1A; SIN3, Swi-independent 3; SNF2h, sucrose nonfermenting 2 homolog; SNF5, switching defective/sucrose nonfermenting subunit 5.
Indeed, functional annotation of PHF6 protein complexes revealed striking enrichment in factors involving chromatin organization (P = 2.18−16) and epigenetic regulation of gene expression (P = 2.23−15) (Figure 1; supplemental Table 2). Moreover, and in agreement with the proposed roles of PHF6 in chromatin remodeling and control of ribosomal RNA (rRNA) transcription, PHF6-associated factors prominently included epigenetic regulators involved in control of rRNA expression (P = 5.13−16) and proteins involved in RNA polymerase I–mediated transcriptional regulation (P = 6.82−14). Finally, the PHF6 interactome also included numerous factors involved in chromosome maintenance (P = 3.1−5) and DNA repair (P = 3.21−4) as well as proteins controlling cell cycle and DNA synthesis (P = 4.80−5).
PHF6 is a DNA repair factor recruited to double-strand breaks
PHF6 inactivation can lead to accumulation of the γ-H2AX DNA damage marker,3,11,12 increased replication–transcription conflicts at rDNA sites,7 and delayed DNA repair.12 To explore the relationship between PHF6 and the DNA damage response, we tested whether PHF6 could be recruited to DNA damage sites. Notably, immunofluorescence analysis revealed that PHF6 protein relocated to laser-generated stripes marked by γ-H2AX 5 minutes after micro-irradiation, pointing to a role of PHF6 as an early repair factor (Figure 2A). Similarly, immunofluorescence (Figure 2B) and chromatin immunoprecipitation (ChIP) (Figure 2C) assays documented the recruitment of PHF6 to a single double-strand DNA break generated by the I-SceI restriction enzyme in U2OS cells that faded away when moving away from the break site, indicating a specific binding of PHF6 to the break site.
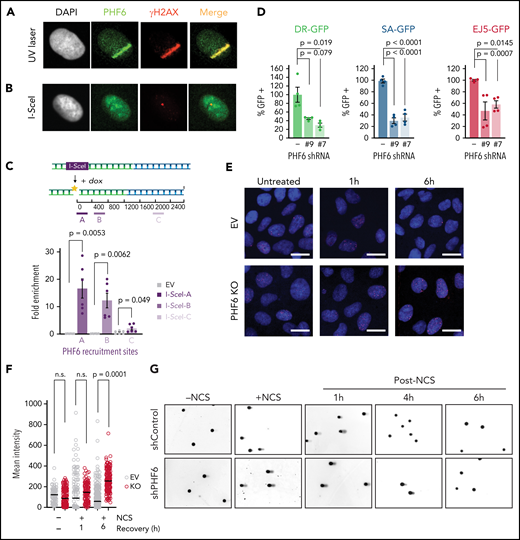
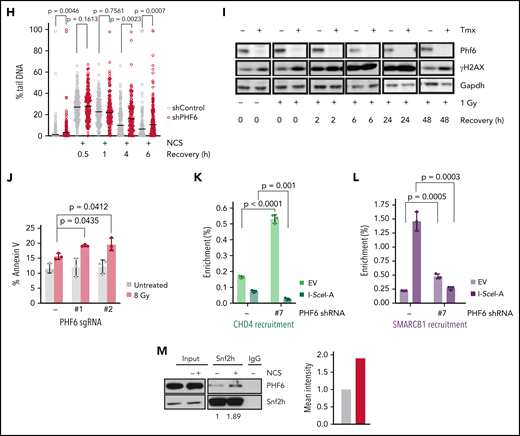
PHF6 is recruited to the vicinity of DNA breaks for efficient DNA repair. (A) Representative confocal images showing PHF6 colocalization with γ-H2AX after UV micro-irradiation in U2OS cells. (B) Representative confocal images showing PHF6 colocalizing with γ-H2AX in a single double-strand break (DSB) induced by I-SceI expression in U2OS-DR GFP cells. (C) Upper panel, schematics of DSB induction after doxycycline and PHF6 recruitment to the vicinity of a DSB (region A) and to 2 different regions away from the DSB (regions B and C). Lower panel, quantification of ChIP assay showing PHF6 recruitment to the vicinity of the I-SceI DSB site in U2OS-DR-GFP cells in 3 different genomic regions (regions A-C) in 2 independent experiments. Bar graphs represent mean ± standard error of the mean. EV (empty-vector, not I-SceI-induced DSB control). (D) GFP percentage measured by flow cytometry in U2OS cells expressing 2 different short hairpin RNAs (shRNAs) targeting PHF6 or control shRNA containing integrated reporters to measure DNA repair efficiency through homologous recombination (U2OS-DR-GFP), single-strand annealing (SA-GFP), or non-homologous end-joining (EJ5-GFP). The percentage of GFP-positive cells is plotted as percent relative to the control cells. Data are representative of 4 independent experiments. Bar graphs represent mean ± standard error of the mean. (E) Representative images of Rad51 foci (red) obtained after 1- or 6-hour recovery from neocarzinostatin (NCS) treatment in control (EV, control sgRNA) and PHF6-knockout cells. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 25 μM. (F) Quantification of the intensity of Rad51 foci per cell in control (EV, control sgRNA) and PHF6-knockout U2OS cells. Between 100 and 200 cells were analyzed per condition. Statistical analysis was conducted by using a nonparametric Mann-Whitney test. Data are representative of 2 independent experiments. (G) Representative alkaline comet images performed in untreated U2OS cells or after NCS treatment (100 ng/mL) and recovery for 1, 4, or 6 hours in cells infected with a control shRNA (shControl) or a PHF6-targeting shRNA (shPHF6). (H) Dot plot showing individual percentages of comet tail DNA. The median value of >70 nuclei per experimental condition is indicated. Statistical analysis was conducted by using the Mann-Whitney test. Data are representative of 2 independent experiments. (I) Western blot showing the presence of phosphorylated γ-H2AX after recovery from irradiation (1 Gy) for the indicated times in PHF6 control or knock-out primary T-ALL cells. Gapdh is shown as loading control. Tmx, tamoxifen. (J) Analysis of apoptosis upon irradiation at 8 Gy in U2OS infected with control single guide RNA (sgRNA) or sgRNA#1/sgRNA#2. Bar graphs represent mean ± standard deviation (SD). (K) Quantification of ChIP assay showing CHD4 recruitment to the vicinity of the I-SceI DSB site in U2OS-DR-GFP cells (region A) in the presence (PHF6 shRNA '-') or absence of PHF6 (PHF6 shRNA '#7'). Data are representative of 3 independent experiments. EV (empty-vector, not I-SceI-induced DSB control). Bar graphs represent mean ± SD. (L) Quantification of ChIP assay showing SMARCB1 recruitment to the vicinity of the I-SceI DSB site in U2OS-DR-GFP cells (region A) in the presence (PHF6 shRNA '-') or absence of PHF6 (PHF6 shRNA '#7'). Data are representative of 3 independent experiments. EV (empty-vector, not I-SceI-induced DSB control). Bar graphs represent mean ± SD. (M) Left, western blot confirming endogenous PHF6 interaction by immunoprecipitation with SNF2H before and after treatment with 100 ng/mL NCS. Right: quantification of Phf6 levels normalized to Phf6 input ± NCS. All P values in the graphics were assessed by using a 2-tailed, unpaired Student’s t test. IgG, immunoglobulin G; n.s., not significant.
PHF6 is recruited to the vicinity of DNA breaks for efficient DNA repair. (A) Representative confocal images showing PHF6 colocalization with γ-H2AX after UV micro-irradiation in U2OS cells. (B) Representative confocal images showing PHF6 colocalizing with γ-H2AX in a single double-strand break (DSB) induced by I-SceI expression in U2OS-DR GFP cells. (C) Upper panel, schematics of DSB induction after doxycycline and PHF6 recruitment to the vicinity of a DSB (region A) and to 2 different regions away from the DSB (regions B and C). Lower panel, quantification of ChIP assay showing PHF6 recruitment to the vicinity of the I-SceI DSB site in U2OS-DR-GFP cells in 3 different genomic regions (regions A-C) in 2 independent experiments. Bar graphs represent mean ± standard error of the mean. EV (empty-vector, not I-SceI-induced DSB control). (D) GFP percentage measured by flow cytometry in U2OS cells expressing 2 different short hairpin RNAs (shRNAs) targeting PHF6 or control shRNA containing integrated reporters to measure DNA repair efficiency through homologous recombination (U2OS-DR-GFP), single-strand annealing (SA-GFP), or non-homologous end-joining (EJ5-GFP). The percentage of GFP-positive cells is plotted as percent relative to the control cells. Data are representative of 4 independent experiments. Bar graphs represent mean ± standard error of the mean. (E) Representative images of Rad51 foci (red) obtained after 1- or 6-hour recovery from neocarzinostatin (NCS) treatment in control (EV, control sgRNA) and PHF6-knockout cells. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 25 μM. (F) Quantification of the intensity of Rad51 foci per cell in control (EV, control sgRNA) and PHF6-knockout U2OS cells. Between 100 and 200 cells were analyzed per condition. Statistical analysis was conducted by using a nonparametric Mann-Whitney test. Data are representative of 2 independent experiments. (G) Representative alkaline comet images performed in untreated U2OS cells or after NCS treatment (100 ng/mL) and recovery for 1, 4, or 6 hours in cells infected with a control shRNA (shControl) or a PHF6-targeting shRNA (shPHF6). (H) Dot plot showing individual percentages of comet tail DNA. The median value of >70 nuclei per experimental condition is indicated. Statistical analysis was conducted by using the Mann-Whitney test. Data are representative of 2 independent experiments. (I) Western blot showing the presence of phosphorylated γ-H2AX after recovery from irradiation (1 Gy) for the indicated times in PHF6 control or knock-out primary T-ALL cells. Gapdh is shown as loading control. Tmx, tamoxifen. (J) Analysis of apoptosis upon irradiation at 8 Gy in U2OS infected with control single guide RNA (sgRNA) or sgRNA#1/sgRNA#2. Bar graphs represent mean ± standard deviation (SD). (K) Quantification of ChIP assay showing CHD4 recruitment to the vicinity of the I-SceI DSB site in U2OS-DR-GFP cells (region A) in the presence (PHF6 shRNA '-') or absence of PHF6 (PHF6 shRNA '#7'). Data are representative of 3 independent experiments. EV (empty-vector, not I-SceI-induced DSB control). Bar graphs represent mean ± SD. (L) Quantification of ChIP assay showing SMARCB1 recruitment to the vicinity of the I-SceI DSB site in U2OS-DR-GFP cells (region A) in the presence (PHF6 shRNA '-') or absence of PHF6 (PHF6 shRNA '#7'). Data are representative of 3 independent experiments. EV (empty-vector, not I-SceI-induced DSB control). Bar graphs represent mean ± SD. (M) Left, western blot confirming endogenous PHF6 interaction by immunoprecipitation with SNF2H before and after treatment with 100 ng/mL NCS. Right: quantification of Phf6 levels normalized to Phf6 input ± NCS. All P values in the graphics were assessed by using a 2-tailed, unpaired Student’s t test. IgG, immunoglobulin G; n.s., not significant.
We next assessed the impact of PHF6 knockdown on the efficacy of homologous-recombination, single-strand annealing and non-homologous end-joining DNA repair pathways with specific GFP reporters22 in U2OS cells. In these analyses, PHF6 knockdown resulted in a highly significant decrease in both homologous-recombination and single-strand annealing double-strand break repair compared with control cells and a more moderate, yet significant, reduction in the efficiency of non-homologous end-joining (Figure 2D; supplemental Figure 2A-B), which implicates PHF6 in the resolution of single- and double-strand breaks. To gain mechanistic insights on the different repair pathways in which PHF6 was involved, we transiently treated PHF6 knockdown and control cells with neocarzinostatin, a radiomimetic DNA-damaging agent, and performed immunofluorescence assays of different players in recombination-dependent or recombination-independent repair. We first addressed the level and nuclear localization of BRCA1, the main factor promoting resection in homologous recombination repair23 and Rad51, a key player in crossover regulation.24 To evaluate the initial steps of non-homologous end-joining in PHF6-deficient cells, we checked the levels of Ku80, the main double-strand break sensor, and of XRCC4, which promotes the re-ligation of broken ends and serves as an activity readout for the main NHEJ kinase, DNA-PK.25 Although no major differences were observed in the number or intensity of Ku80 or XRCC4 foci (supplemental Figure 2C), a clear persistence of Rad51 foci after 6 hours of recovery was found, indicating a failure in resolving DNA damage downstream resection or at the level of homologous strand search (Figure 2E-F). We did not observe a different intensity of BRCA1 foci, indicating that PHF6 loss does not have an impact on the global levels of BRCA1. However, and as reported before in the case of a low dose of irradiation,26 BRCA1 nuclear export was observed after the induction of DNA damage. Interestingly, PHF6-deficient cells exhibited an earlier cytoplasmic BRCA1 signal soon after treatment, suggesting that PHF6 could be necessary for BRCA1 nuclear retention (supplemental Figure 2D). This premature BRCA1 export in PHF6-deficient cells could contribute to the observed defects in homologous recombination.
Following on these results, we monitored and quantified the resolution of single- and double-strand DNA breaks visualized by alkaline comet assay. PHF6 depletion resulted in delayed resolution of both types of breaks over time (Figure 2G-H), which was in line with the impaired resolution of Rad51 foci, suggesting a functional role for PHF6 in recombination-mediated repair. Consistently, we observed a more rapid and persistent increase in γ-H2AX levels in Phf6-deficient primary NOTCH1-induced leukemic lymphoblasts compared with their isogenic controls (Figure 2I).
Ultimately, the loss of PHF6 led to increased apoptosis in PHF6-deficient cells after gamma irradiation and after a high-dose of hydroxyurea, indicating that PHF6 loss can promote DNA damage–induced apoptosis (Figure 2J; supplemental Figure 2E-I). Importantly, we did not observe increased apoptosis outside highly genotoxic conditions. Specifically, low amounts of replication stress as induced by sustained low-dose hydroxyurea treatment did not result in increased apoptosis in PHF6 knockdown cells (supplemental Figure 2G). The association of PHF6 with both chromatin remodeling complexes and with the replication and DNA repair machinery suggested a potential role for PHF6 in the epigenetic control of DNA repair. To formally test this hypothesis, we evaluated the recruitment of the chromatin remodeling factors CHD4 and SMARCB1 to a single DNA damage site induced by I-SceI in wild-type or PHF6-deficient cells. Interestingly, CHD4 recruitment to a unique DNA damage site was significantly impaired upon PHF6 loss, whereas a similar trend was observed in the recruitment of SMARCB1 (Figure 2K-L; supplemental Figure 2J-K), suggesting that PHF6 creates an adequate chromatin environment needed for its DNA repair functions.
To further investigate the interaction between PHF6-associated epigenetic regulators and the DNA damage response, we evaluated the effects of neocarzinostatin treatment in the interaction of PHF6 with different chromatin remodelers. These experiments revealed an increased PHF6–SMARCA5/SNF2H interaction in response to DNA damage, whereas no changes in the PHF6-bound fraction to NuRD or SWI/SNF chromatin remodelers was observed (Figure 2M; supplemental Figure 2L-M). These results support a potential role for CHD4/PHF6 and SMARCA5/SNF2H-PHF6 complexes in providing an adequate chromatin environment during DNA damage. Interestingly, SMARCA5/SNF2H accumulates on nascent DNA upon replication-associated damage,27 whereas CHD4 loss promotes increased replication fork stability, increasing the chemoresistance of homologous recombination-deficient cells,28 data that position these 2 chromatin remodelers as important responders against replication stress.
To further understand the mechanisms of DNA repair, replication, and PHF6 regulation, we performed mass spectrometry–based phosphoproteomic analysis of PHF6 protein immunoprecipitated from neocarzinostatin-treated HEK293T cells29 and assessed whether PHF6 was a target of the DNA damage response kinases. Five damage-dependent phosphorylation sites were identified in PHF6 (S120, S138, S155, S199, and S204), 3 of which (S120, S199, and S204) were suppressed by treatment with caffeine, an inhibitor of phosphatidylinositol 3-kinase–related kinase kinases (ATR, PRKDC/DNA-PK, and ATM) (supplemental Figure 2N). Furthermore, western blot analysis of HEK293T Flag-HA-PHF6 immunoprecipitates exhibited increased immunoreactivity with an antibody recognizing the ATM/ATR DNA phosphorylation motif after gamma radiation (supplemental Figure 2O). Together, these results implicate PHF6 as a DNA repair factor recruited to double-strand breaks and phosphorylated by DNA repair–signaling kinases. Moreover, the presence of PHF6 in complex with factors involved in the control of cell cycle and DNA synthesis (Figure 1D) suggests a potential role for PHF6 in DNA homeostasis in association with the DNA replication machinery.
PHF6 protects replication fork integrity by regulating the speed of DNA synthesis
To formally evaluate this possibility, we performed DNA fiber analysis to document replication fork dynamics30 in PHF6 wild-type and knockout cells after sequential labeling of nascent DNA with chlorodeoxyuridine and iododeoxyuridine. CUTLL1 PHF6 and Jurkat PHF6 knockout T-ALL leukemia cells showed a significant increase in iododeoxyuridine (green) and chlorodeoxyuridine (red) DNA track lengths compared with isogenic PHF6 wild-type controls (Figure 3A-B; supplemental Figure 3A), indicative of accelerated replication fork progression. In addition, we noted increased replication fork pausing, shown by asymmetric DNA fibers, in PHF6 knockout cells compared with controls (Figure 3C; supplemental Figure 3B-C), which is consistent with increased genomic instability in the context of accelerated DNA replication.31 PHF6 interacts with the UBF transcription factor in the nucleolus, which contributes to downregulate rDNA transcription and to prevent rDNA damage.7 The observed increase in replication fork progression upon PHF6 loss in association with upregulation of rDNA transcription may explain the observation of an increase in collapsed replication forks and double-strand DNA breaks or R-loop DNA–RNA hybrids at rDNA sites after PHF6 inactivation. To determine if the loss of PHF6 could also exacerbate the formation of R-loops in the presence of replication-induced damage, we treated wild-type or PHF6-deficient cells with hydroxyurea alone or hydroxyurea and ATR inhibitor VE-821 during 30 minutes and measured the levels of replication-induced damage after 1 hour of recovery. These experiments revealed an increase in the number of R-loops in PHF6-deficient cells under replication stress which was more pronounced after the inhibition of the ATR-mediated replication stress response, further supporting a role for PHF6 in the resolution of DNA–RNA hybrids (supplemental Figure 3D-E).
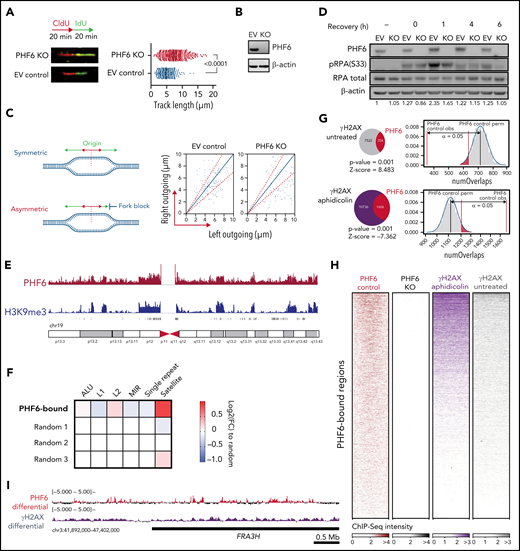
PHF6 protects from replication-associated DNA damage and binds to satellite DNA heterochromatin. (A) Schematic of chlorodeoxyuridine (CldU, red)/iododeoxyuridine (IdU, green) pulse labeling (upper left). Representative images of CldU and IdU replication tracks in Jurkat control (EV, control sgRNA) or PHF6 knockout (KO) cells (bottom left). Fork rate dot plot showing the IdU tract length of individual replication forks in untreated Jurkat cells (right). The median value of >350 tracts per experimental condition is indicated. Statistical analysis was conducted by using the Mann-Whitney test (P < .0001). Data are representative of 2 independent experiments. (B) Western blot showing PHF6 KO in Jurkat cells infected with a control sgRNA (EV) or a single guide RNA targeting the second PHD2 domain of PHF6 (KO). β-actin concentrations are shown as a loading control. (C) Left, scheme of the signals used for quantification of asymmetry analysis of forks moving from a single origin (outgoing forks). Right, scatter diagram of fork symmetry in Jurkat cells. Each dot corresponds to the ratio between the right and the left fork velocities of a pair of outgoing forks belonging to the same replication bubble. The areas outside of the dotted lines include all points whose ratios deviate from the expected theoretical value of 1 ± 0.3, corresponding to forks moving bidirectionally at nearly the same rate. EV control (control sgRNA), PHF6 KO (sgRNA targeting second PHD2 domain of PHF6). Statistical analysis was performed with the Mann-Whitney rank sum test (P < .001). (D) Western blot showing the presence of phosphorylated RPA (pRPA) after recovery from 30 minutes of 1 μM camptothecin treatment. The “–” sign indicates untreated conditions. Both RPA total amount and β-actin concentrations are shown as a loading control as used for the blot quantification shown below each band. EV control (control sgRNA), PHF6 KO (sgRNA targeting second PHD2 domain of PHF6). (E) Chromosome 19 distribution of normalized PHF6 (red track) or H3K9me3 (blue track) ChIP-Seq intensities in Jurkat. (F) Heat map indicating the log2FC enrichment in repetitive regions according to category compared with the average in 3 random subsets. (G) Overlap between PHF6 peaks and γ-H2AX genomic regions in untreated (upper panel) and after aphidicolin treatment (lower panel). The indicated P value and Z scores are the result of permutation testing (n = 1000 trials). PHF6 control obs, refers to observed regions. PHF6 control perm, refers to median expected permutated regions. (H) Normalized ChIP-Seq heat maps of Jurkat PHF6 control and KO and K562 γ-H2AX aphidicolin treated and untreated. PHF6-bound regions (n = 11 528) were scaled to the same length. (I) Differential PHF6 (control/KO cell lines) and γ-H2AX (treated/untreated) ChIP-Seq intensities within the fragile site FRA3H.
PHF6 protects from replication-associated DNA damage and binds to satellite DNA heterochromatin. (A) Schematic of chlorodeoxyuridine (CldU, red)/iododeoxyuridine (IdU, green) pulse labeling (upper left). Representative images of CldU and IdU replication tracks in Jurkat control (EV, control sgRNA) or PHF6 knockout (KO) cells (bottom left). Fork rate dot plot showing the IdU tract length of individual replication forks in untreated Jurkat cells (right). The median value of >350 tracts per experimental condition is indicated. Statistical analysis was conducted by using the Mann-Whitney test (P < .0001). Data are representative of 2 independent experiments. (B) Western blot showing PHF6 KO in Jurkat cells infected with a control sgRNA (EV) or a single guide RNA targeting the second PHD2 domain of PHF6 (KO). β-actin concentrations are shown as a loading control. (C) Left, scheme of the signals used for quantification of asymmetry analysis of forks moving from a single origin (outgoing forks). Right, scatter diagram of fork symmetry in Jurkat cells. Each dot corresponds to the ratio between the right and the left fork velocities of a pair of outgoing forks belonging to the same replication bubble. The areas outside of the dotted lines include all points whose ratios deviate from the expected theoretical value of 1 ± 0.3, corresponding to forks moving bidirectionally at nearly the same rate. EV control (control sgRNA), PHF6 KO (sgRNA targeting second PHD2 domain of PHF6). Statistical analysis was performed with the Mann-Whitney rank sum test (P < .001). (D) Western blot showing the presence of phosphorylated RPA (pRPA) after recovery from 30 minutes of 1 μM camptothecin treatment. The “–” sign indicates untreated conditions. Both RPA total amount and β-actin concentrations are shown as a loading control as used for the blot quantification shown below each band. EV control (control sgRNA), PHF6 KO (sgRNA targeting second PHD2 domain of PHF6). (E) Chromosome 19 distribution of normalized PHF6 (red track) or H3K9me3 (blue track) ChIP-Seq intensities in Jurkat. (F) Heat map indicating the log2FC enrichment in repetitive regions according to category compared with the average in 3 random subsets. (G) Overlap between PHF6 peaks and γ-H2AX genomic regions in untreated (upper panel) and after aphidicolin treatment (lower panel). The indicated P value and Z scores are the result of permutation testing (n = 1000 trials). PHF6 control obs, refers to observed regions. PHF6 control perm, refers to median expected permutated regions. (H) Normalized ChIP-Seq heat maps of Jurkat PHF6 control and KO and K562 γ-H2AX aphidicolin treated and untreated. PHF6-bound regions (n = 11 528) were scaled to the same length. (I) Differential PHF6 (control/KO cell lines) and γ-H2AX (treated/untreated) ChIP-Seq intensities within the fragile site FRA3H.
The single-strand scaffolding protein RPA (replication protein A) is progressively phosphorylated by ATR at Ser33 at replication-associated DNA double-strand breaks to promote DNA repair,32 and it specifically functions as a rate-limiting factor shielding replication forks from collapse.33 To further explore the role of PHF6 in the sensing and resolution of DNA damage, we analyzed the effect of PHF6 inactivation in the induction of RPA Ser33 phosphorylation after treatment with the replication stress–inducing agent camptothecin (Figure 3D). In this setting, PHF6 inactivation resulted in decreased RPA S33 phosphorylation in agreement with a defective replicative stress response. ATR phosphorylation at T1989 is required for ATR activation upon replication-associated DNA double-stranded breaks34 upstream of RPA S33 phosphorylation. Notably, we observed increased ATR T1989 phosphorylation in PHF6 knockout cells under different replication stress conditions (camptothecin and neocarzinostatin treatment) (supplemental Figure 3F-G). Because PHF6 knockout cells display a reduced RPA S33 phosphorylation (with increased ATR pT1989), we concluded that PHF6 is necessary for the phosphorylation of RPA by activated ATR. Together, these results implicate PHF6 in the preservation of replication fork integrity.
To gain a better understanding of the role of PHF6 across the chromatin landscape, ChIP followed by deep sequencing (ChIP-Seq) was performed in native chromatin from T-ALL lymphoblast cells. These analyses revealed that PHF6 was specifically enriched at genomic locations corresponding to histone H3 lysine 9 trimethyl marks (H3K9me3) domains in Jurkat cells but not in PHF6 knockout controls (Figure 3E; supplemental Figure 4A-B). H3K9me3 marks heterochromatic regions, which in the human genome primarily correspond to genomic areas with high copy number tandem repeat sequences, including satellite repeat and transposon regions.35 Analysis of the distribution of PHF6 ChIP-Seq signals across different heterochromatin domains revealed a marked overlap between PHF6 occupancy and satellite DNA regions (Figure 3F), a feature readily noticeable in the large satellite heterochromatin areas of human chromosome 19 (Figure 3E; supplemental Figure 4C). Notably, satellite DNA poses a major challenge for the DNA replication machinery, resulting in increased replication fork stalling, which can lead to chromosomal breaks and rearrangements.36
The functional requirement of PHF6 for efficient DNA repair, its association with chromatin remodeling factors, and its localization to satellite DNA domains support a role in chromatin dynamics during DNA replication and in the maintenance of genomic integrity. Consistently, even though PHF6-containing chromatin in untreated conditions showed limited overlap with γ-H2AX, we observed a high overlap between nontreated PHF6-containing regions and aphidicolin-treated γ-H2AX domains (P = .001); these findings support the idea that PHF6 preoccupies difficult-to-replicate DNA regions, which are revealed upon replication stress (Figure 3G-H). Indeed, PHF6 was markedly enriched at the common fragile site FRA3H, which is induced by aphidicolin in the human genome37 that appears as a hotspot of replicative stress marked by high levels of γ-H2AX after aphidicolin treatment (Figure 3I).
Altogether, these observations indicate that PHF6 is likely recruited to fragile genomic regions, which are highly susceptible to replicative stress–induced DNA damage, where it could facilitate DNA replication and repair to maintain genomic integrity. To test this hypothesis, we analyzed the impact of PHF6 loss at common fragile sites as these hypermutable repetitive loci are highly susceptible to replicative stress–induced DNA damage38 and represent common hotspots for chromosomal rearrangements in human cancer.39 SMARD20 at the FRA16D fragile site (Figure 4A) revealed that replication proceeds predominantly in the 3′ to 5′ direction, with limited fork stalling in PHF6 wild-type cells (Figure 4B), a pattern consistent with that reported in B cells.20 In contrast, analysis of PHF6 knockout cells revealed an increase in the number of stalled replication forks at this locus, as well as extended DNA fragment lengths and abnormal fluorescence in situ hybridization probe patterns indicative of accumulating genomic rearrangements (Figure 4B-C). These results implicate PHF6 in the maintenance of genomic integrity and more specifically in the protection of difficult-to-replicate heterochromatin-associated fragile sites.
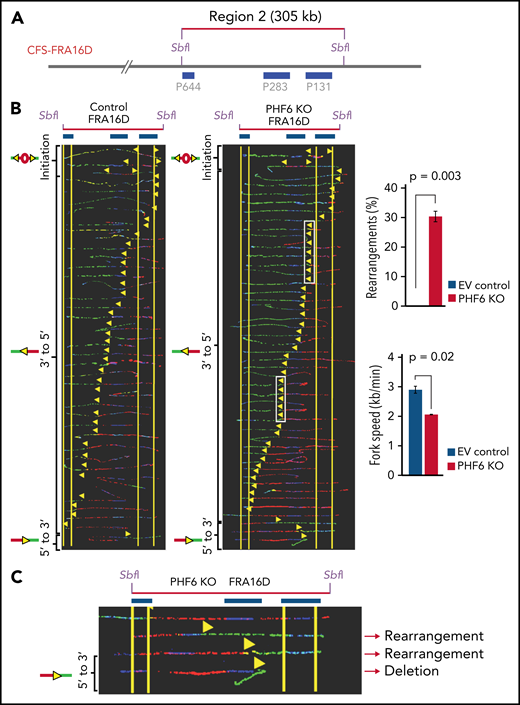
PHF6 prevents replication-associated damage and accumulation of genomic rearrangements at the FRA16D chromosome fragile site. (A) Locus map of CFS-FRA16D SbfI digested segment. The fluorescence in situ hybridization probes that identify the segment are labeled in blue. (B) Aligned photomicrograph images of labeled DNA molecules from Jurkat PHF6 infected with an empty vector control (EV, control sgRNA) or with an sgRNA targeting the second PHD2 domain of PHF6 (PHF6 PHF6 KO). The yellow arrows indicate the sites along the molecules where the iododeoxyuridine transitioned to chlorodeoxyuridine. White rectangles indicate representative sites of replication fork pausing. The molecules are arranged in the following order: molecules with initiation events, molecules with 3′ to 5′ progressing forks, molecules with 5′ to 3′ progressing forks, and molecules with termination events. The quantification in the upper right panel shows the percentage of molecules with rearrangements at CFS-FRA16D in Jurkat PHF6 EV (blue bar) and Jurkat KO (red bar). Error bars represent mean ± standard deviation from data collected from 2 independent experiments. The quantification in the lower right panel shows the replication fork speed at CFS-FRA16D in Jurkat PHF6 EV (blue bar) and Jurkat KO (red bar). Error bars represent mean ± standard deviation from data collected from 2 independent experiments. (C) Close-up of the 5′ to 3′ region of CFS-FRA16D showing aberrant probe patterns in individual DNA molecules.
PHF6 prevents replication-associated damage and accumulation of genomic rearrangements at the FRA16D chromosome fragile site. (A) Locus map of CFS-FRA16D SbfI digested segment. The fluorescence in situ hybridization probes that identify the segment are labeled in blue. (B) Aligned photomicrograph images of labeled DNA molecules from Jurkat PHF6 infected with an empty vector control (EV, control sgRNA) or with an sgRNA targeting the second PHD2 domain of PHF6 (PHF6 PHF6 KO). The yellow arrows indicate the sites along the molecules where the iododeoxyuridine transitioned to chlorodeoxyuridine. White rectangles indicate representative sites of replication fork pausing. The molecules are arranged in the following order: molecules with initiation events, molecules with 3′ to 5′ progressing forks, molecules with 5′ to 3′ progressing forks, and molecules with termination events. The quantification in the upper right panel shows the percentage of molecules with rearrangements at CFS-FRA16D in Jurkat PHF6 EV (blue bar) and Jurkat KO (red bar). Error bars represent mean ± standard deviation from data collected from 2 independent experiments. The quantification in the lower right panel shows the replication fork speed at CFS-FRA16D in Jurkat PHF6 EV (blue bar) and Jurkat KO (red bar). Error bars represent mean ± standard deviation from data collected from 2 independent experiments. (C) Close-up of the 5′ to 3′ region of CFS-FRA16D showing aberrant probe patterns in individual DNA molecules.
Discussion
Nucleosome remodeling complexes play an essential role in creating a dynamic environment in which chromatin can be accessible for DNA replication and repair.38,40-42 However, the mechanisms that coordinate chromatin reorganization with the DNA synthesis and repair machinery and the role of chromatin remodeling DNA–repair interactions in the pathogenesis of cancer remain poorly understood.
Nucleosome displacement in the context of DNA damage and repair may comprise diverse nucleosome remodeling complexes involved both in increasing chromatin accessibility (SWI/SNF) and in favoring a closed chromatin configuration (NuRD). In addition, local mono-ubiquitylation of H2BK120 at double-strand DNA breaks promotes increased chromatin accessibility by SMARCA5 SWI/SNF complexes to facilitate efficient recruitment of factors involved in homologous recombination, in support of a functional interaction between histone marks and nucleosome repositioning in DNA repair.43,44 Interestingly, a recent report proposed a dual role for PHF6 as a epigenetic reader of H2BK12 acetylation and as a epigenetic writer for histone H2BK120 ubiquitination of functional relevance for regulation of trophectodermal gene expression.45 A prominent finding reported here is the increased interaction between PHF6 and SMARCA5 after induction of DNA damage. SMARCA5, the central component of the mammalian ISWI family of chromatin remodelers, is actively involved in regulating chromatin structure and in this role facilitates the efficient recruitment of DNA repair factors.21 After DNA damage, SMARCA5 is actively recruited to break sites through PARP146; in addition, it can be found enriched at active elongating replication forks,47 where it enables replication through highly heterochromatic regions.48 The role of PHF6 in restraining rRNA expression,7 together with our observation of increased replication fork progression, is consistent with the documented increase in collapsed replication forks due to the presence of DNA–RNA hybrids in the nucleolus after PFH6 inactivation. However, the role of PHF6 in the maintenance of genomic integrity does not seem to be restricted to rDNA loci. Indeed, PHF6 is actively recruited to sites of DNA double-strand breaks and associates with numerous factors involved in chromosome maintenance and DNA repair, suggesting a more general role in genomic integrity. Moreover, we observed a particularly prominent overlap between PHF6-preoccupied genomic locations and sites of DNA damage marked by γ-H2AX upon induction of replicative stress. The location of PHF6 at sites of replicative stress–induced DNA damage could well correspond with its proposed role in the maintenance of genomic integrity, as resolution of R-loops and replication fork collapse events involves both active assembly of DNA repair complexes and active chromatin remodeling. However, the association of PHF6 with protein complexes directly involved in controlling cell cycle and DNA synthesis suggests a more direct link with the replication machinery.
The rDNA loci are composed of multiple sequence repeats in tandem, which represents a particular challenge for the replication machinery. However, most highly repetitive sequence domains in the genome correspond to satellite DNA heterochromatin regions marked by the H3K9me3 histone mark.35 Cancer genome studies have shown that mutations accumulate at much higher levels in compact, H3K9me3-rich heterochromatin domains,49 consistent with the slower rates of DNA repair reported in heterochromatin.50,51 Remarkably, we observed a prominent overlap between PHF6 occupancy and satellite DNA domains, arguing for a broader functional role of PHF6 in preventing and resolving replication fork stalling at difficult-to-replicate DNA sites beyond rDNA repeats. This hypothesis is further supported by our single-molecule locus-specific analysis of replication dynamics and genomic integrity at the FRA16D fragile site, which revealed increased numbers of stalled replication forks and accumulating genomic rearrangements. Our results unravel a discrete and distinct role for PHF6 in the maintenance of genomic integrity by limiting replication fork dynamics, particularly in difficult-to-replicate satellite DNA regions but also by active recruitment to sites of double-strand DNA break where it facilitates the resolution of DNA damage. We propose that PHF6 has a local effect at repetitive regions conducive of fork stalling that can result in the observed global increase in replication fork progression and asymmetric forks. Although a functional role for PHF6 in non-homologous end-joining has been recently proposed,12 we observed broader defects in DNA repair implicating PHF6 also in homologous recombination and single-strand annealing. Additionally, its participation in DNA repair in concert with the recruitment of nucleosome remodeling factors required to facilitate access to damage sites suggests a more general role of PHF6 in the resolution of DNA damage. Alternatively, it is also possible that PHF6 participates in different forms of DNA repair via association with distinct chromatin remodeling complexes.
Leukemia-focused and pan-cancer mutational profiling analyses have established a specific tumor suppressor role for PHF6 in the hematopoietic compartment. This activity seems to be functionally linked to an increased stem cell self-renewal and sensitization to NOTCH1-induced transformation.13 However, the identification of a PHF6 function in the control of replicative dynamics and DNA repair suggests that defects in PHF6 could also favor leukemia transformation by accelerating the accumulation of DNA damage with consequent accumulation of secondary genetic alterations in oncogenes and tumor suppressor genes. In an analogous way genes associated with DNA repair, DNA ligase IV and Fanconi DNA repair factor gene mutations result in abrogation of self-renewal in the hematopoietic system and cause bone marrow aplasia while at the same time favoring leukemia development as a result of increased genomic instability.52,53 An intriguing possibility that warrants further studies is that loss of PHF6 could associate with collateral vulnerabilities which could be exploited therapeutically in the treatment of human leukemia.
Acknowledgments
The authors thank Jeremy Stark for the U2OS-EJ5-GFP and U2OS SA-GFP cells.
This work was supported by the National Institutes of Health, National Cancer Institute (R35 CA210065 and R01 CA155743 [A.A.F.]), National Institute on Aging (R01AG077020 [A.A.F.]), and National Cancer Institute grant P30 CA013696 (in support of the Herbert Irving Comprehensive Cancer Center Genomics, Flow Cytometry and Molecular Cytogenetics, Genetically Manipulated Mouse Models, Proteomics and Macromolecular Crystallography, Genomics and High Throughput Screen Shared Resource, and Confocal and Specialized Microscopy Shared Resources). A.W. was supported by a Rally Foundation fellowship. P.V.V. was supported by the Fund for Scientific Research Flanders (postdoctoral fellowship and Odysseus type 2 grant). S.A. was supported by a Leukemia and Lymphoma Society Special Fellowship Award. R.A. was supported by a Leukemia and Lymphoma Society Fellowship Award.
Authorship
Contribution: S.A. and A.C.d.S.A. performed most of the experiments; R.A. and M.B. analyzed the ChIP-Seq data; S.G. analyzed the mass spectrometry data; T.A. performed immunostainings on laser stripes and I-SceI lesions; A.B.-G., A.S., and A.M. performed SMARD; A.W., E.P., and P.V.V. performed research; T.S.G. provided technical assistance with experiments; and A.A.F. conceived and designed the project, analyzed data with J.G. and A.M., and wrote the manuscript with S.A. and A.C.d.S.A.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.C.d.S.A. is Biogen Inc, Cambridge, MA.
The current affiliation for A.W. is Calico Life Sciences, South San Francisco, CA.
Correspondence: Adolfo A. Ferrando, 1130 St. Nicholas Ave, ICRC 402, New York, NY 10032; e-mail: af2196@columbia.edu; or Silvia Alvarez, 1130 St. Nicholas Ave, ICRC 402, New York, NY 10032; e-mail: sa3358@columbia.edu.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE152292).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal