

Key Points
Mice with inactivation of NOX2, globally or in macrophages, spontaneously acquire an activated alveolar macrophage phenotype by adulthood.
Epigenetic and transcriptional profiles of NOX2-deleted AMs are influence by their niche and primed to generate inflammation.

Abstract
The leukocyte NADPH oxidase 2 (NOX2) plays a key role in pathogen killing and immunoregulation. Genetic defects in NOX2 result in chronic granulomatous disease (CGD), associated with microbial infections and inflammatory disorders, often involving the lung. Alveolar macrophages (AMs) are the predominant immune cell in the airways at steady state, and limiting their activation is important, given the constant exposure to inhaled materials, yet the importance of NOX2 in this process is not well understood. In this study, we showed a previously undescribed role for NOX2 in maintaining lung homeostasis by suppressing AM activation, in CGD mice or mice with selective loss of NOX2 preferentially in macrophages. AMs lacking NOX2 had increased cytokine responses to Toll-like receptor-2 (TLR2) and TLR4 stimulation ex vivo. Moreover, between 4 and 12 week of age, mice with global NOX2 deletion developed an activated CD11bhigh subset of AMs with epigenetic and transcriptional profiles reflecting immune activation compared with WT AMs. The presence of CD11bhigh AMs in CGD mice correlated with an increased number of alveolar neutrophils and proinflammatory cytokines at steady state and increased lung inflammation after insults. Moreover, deletion of NOX2 preferentially in macrophages was sufficient for mice to develop an activated CD11bhigh AM subset and accompanying proinflammatory sequelae. In addition, we showed that the altered resident macrophage transcriptional profile in the absence of NOX2 is tissue specific, as those changes were not seen in resident peritoneal macrophages. Thus, these data demonstrate that the absence of NOX2 in alveolar macrophages leads to their proinflammatory remodeling and dysregulates alveolar homeostasis.
Introduction
The phagocyte NADPH oxidase 2 (NOX2) generates superoxide, the precursor to reactive oxygen species (ROS) that play essential roles in host defense and immunoregulation.1 Inherited defects in the NOX2 enzyme complex lead to chronic granulomatous disease (CGD), characterized by recurrent, severe bacterial and fungal infections, and aberrant inflammation.2 CGD results from inactivating recessive mutations in any of several genes for different subunits of NOX2, including X-linked CYBB, which encodes a membrane flavocytochrome, and autosomal genes for CYBBA, NCF1, NCF2, or NCF4, which encode critical regulatory subunits, or CYBC1, an endoplasmic reticulum protein important for CYBB biosynthesis.2 Manifestations of CGD typically involve barrier sites, frequently the lung.1,3,4 Indeed, a chronic lung disease afflicts many young adult patients with CGD and is not always a sequela of prior pneumonias but may instead reflect a noninfectious inflammatory response.3-8 Sterile lung inflammation is also reported in CGD mice.9,10 These observations raise the question of whether NOX2 modulates immune responses within the lungs to maintain homeostasis under basal conditions.
In the lung, residential alveolar macrophages (AMs), which are long lived, self-renewing cells derived from fetal monocyte precursors, constitute ≈95% of immune cells in the airways at steady state.11,12 Tissue macrophages are essential as both a first line of host defense and for conducting tissue-specific functions under homeostatic and inflammatory conditions.13-16 Their ability to adapt to specific tissue niches and maintain homeostasis is governed by signals in the local microenvironment as well as cell-intrinsic epigenetic and transcriptional remodeling.16-22 AMs are strategically located on the luminal side of the alveoli, where they continually sample the environment and respond accordingly.11,23,24 To limit collateral tissue damage, AMs maintain a relatively high threshold for inflammatory activation by inhaled materials, unless potentially infectious.11,24 Molecular mechanisms that prevent de novo polarization in hyperreactive states are incompletely understood.
We investigated the cell-intrinsic role of NOX2 in AMs, as studied in mice with either global or cell type–restricted deletion of NOX2. NOX2-deficient mice spontaneously developed AMs that were epigenetically, transcriptionally, and phenotypically distinct from wild-type (WT) AMs, including a proinflammatory CD11bhigh AM population. These changes became evident as the mice reached adulthood, coinciding with increased alveolar neutrophils and inflammatory mediators, independent of host or environmental microbes. Moreover, NOX2-deleted AMs exhibited heightened activation ex vivo, and mice with either global or macrophage deletion of NOX2 displayed enhanced lung inflammation after insults. Thus, NOX2 is essential for modulating AM responses that maintain pulmonary homeostasis.
Methods
Details of experimental procedures are provided in the supplemental Methods (available on the Blood Web site).
Mice
Experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, under protocols approved by the Institutional Animal Care and Use Committee at Washington University in St. Louis, and the Animal Care Committee of the University of Louisville or the Hospital for Sick Children. WT C57BL/6J mice were purchased from The Jackson Laboratory or were bred in-house. X-linked CGD25 (CybbKO), Ncf2−/−26 (Ncf2KO), Ncf2fl/fl, and Ncf2fl/fl LysMWT/Cre (Ncf2LysMCre) mice were bred in-house. CybbKOIfnar1KO mice27 were bred in the Animal Care Facility of the Hospital for Sick Children. The mice were maintained under specific-pathogen–free conditions, which included monitoring for pathogenic bacteria and viruses, or, for germ-free CybbKO mice, under gnotobiotic conditions at the University of Louisville Gnotobiotic Mouse Facility. Both male and female mice were studied at the indicated ages.
In vivo studies
Bronchoalveolar lavage (BAL), lung cell suspensions, and lung histology were analyzed28 in naive mice. The mice were also challenged with intranasal zymosan (1 μg/g), and lung inflammation was analyzed after 18 hours.28 In other studies, the mice were challenged with intraperitoneal zymosan (0.05 mg/g) and peritoneal, and lung inflammation was analyzed after 48 hours.28-31
Macrophage stimulation ex vivo
AMs and resident peritoneal macrophages (RPMs) isolated from naïve mice were plated in 96-well tissue culture plates. After adhesion, cells were treated with UltraPure lipopolysaccharide (LPS; Escherichia coli, 10 ng/mL; Invivogen,) or Pam3csk4 (100 ng/mL; Invivogen) for 4 hours at 37°C. Gene expression was studied by quantitative reverse transcription-polymerase chain reaction (qRT-PCR).
Macrophage sorting for transcriptional and epigenetic analysis
RNA sequencing (RNA-seq) and assay for transposase accessible chromatin sequencing (ATAC-seq) were performed on BAL AMs (CD45+Ly6G−CD11c+Sigle F+) sorted on the basis of CD11b expression. RNA microarray analysis was performed on RPMs sorted as B220−CD115+F4/80highMHC class II− and B220-F4/80lowMHC class IIhigh.
Statistics
Unpaired Student t tests were used to compare 2 groups, and 1-way analysis of variance, followed by Tukey’s post hoc test, was used to compare among multiple groups. For nonparametric data, unpaired, 2-tailed Mann-Whitney tests were used. Data were analyzed and graphed with Prism 5 (GraphPad Software, Inc).
Results
Mice with global deletion of NOX2 spontaneously develop lung inflammation and a population of CD11bhigh AMs
To investigate mechanisms underlying the spontaneous development of lung inflammation in CGD mice in the absence of infection, we used CGD mice with inactivating mutations in Cybb (CybbKO mice25) or Ncf2 (Ncf2KO mice26) and also developed mice with LysM-Cre–mediated deletion of Ncf2 (Ncf2LysMCre mice).
As a starting point, we examined the inflammatory status of the lungs at 4 and 12 weeks of age in WT and CybbKO mice. BAL samples from 4-week-old WT and CybbKO mice had similar total leukocyte counts with few neutrophils (Figure 1A). However, these were significantly increased in 12-week-old CybbKO mice (Figure 1A). The number of BAL AMs and eosinophils was similar in both genotypes (Figure 1B). Ncf2KO CGD mice showed comparable findings (data not shown). Multiplex analysis of BAL from 12-week-old CybbKO mice showed significantly higher levels of tumor necrosis factor-α (TNF-α), interferon-α (IFN-α), interleukin-1β (IL-1β), IL-18, and chemokines, along with reduced IL-10 than the WT BAL analysis (supplemental Figure 2A). BAL IL-1β and CXCL2 levels measured by enzyme-linked immunosorbent assay were similar in 4-week-old WT and CybbKO mice, but were increased in 12-week-old CybbKO mice (Figure 1C). Together, these results set a timeline for the spontaneous development of an inflammatory alveolar state in CGD mice from 4 to 12 weeks after birth.
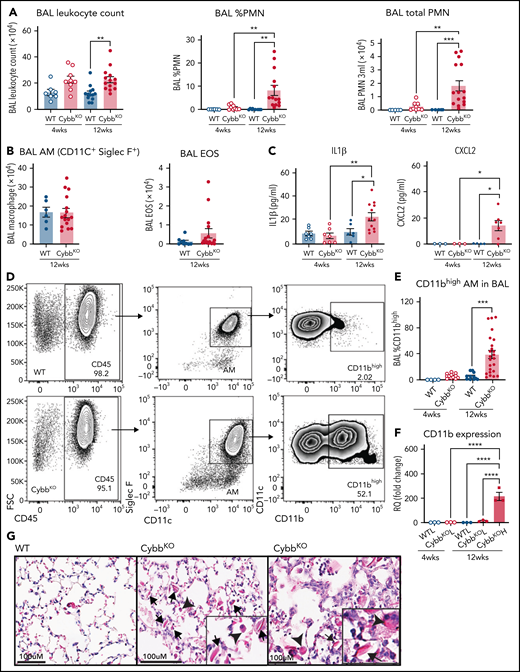
Mice with global deletion of NOX2 develop lung inflammation and a subset of CD11bhigh AMs. (A) BAL leukocyte count, flow cytometric analysis of the percentage of polymorphonuclear leukocytes (PMNs) and total PMNs in 3 mL BAL from 4- and 12-week-old WT and CybbKO male and female mice (n ≥ 7 in each group). (B) Flow cytometric analysis of total AMs (CD45+Siglec F+CD11c+ cells) and total eosinophils (EOS) (CD45+Siglec F+CD11c− cells) from 3 mL of BAL (n ≥ 7). (C) ELISA of IL-1β and CXCL2 in BAL (n ≥ 3 in each group). (D) Gating strategy to identify CD11b+ AMs in the BAL (CD45+SIglec F+ CD11c+CD11bhigh). (E) Percentage of AMs that are CD11bhigh in BAL samples from different groups (n ≥ 4). (F) CD11b expression by qRT-PCR in flow-sorted AMs of different groups. WTL, WT CD11blow AMs; CybbKO L, CybbKO CD11blow AMs; and CybbKO H, CybbKO CD11bhigh AMs (n = 3). (G) Lung sections from 12-week-old WT and CybbKO mice, as indicated, stained with hematoxylin-eosin showing intracellular (arrowhead) and extracellular eosinophilic crystals (arrow) in CybbKO mice. The rightmost panel shows lung tissue from a CybbKO mouse with small infiltrates adjacent to some of the crystals. Scale bars, 100 μm. Representative images from ≥5 mice of each genotype. Bar graph data are expressed as mean ± standard error of the mean. The Student t test was performed for samples distributed in 2 groups. *P < .05; **P < .01; ***P < .001. One-way analysis of variance was performed followed by Tukey’s post hoc analysis comparing multiple groups. Data represent at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001. ELISA, enzyme linked immunosorbent assay.
Mice with global deletion of NOX2 develop lung inflammation and a subset of CD11bhigh AMs. (A) BAL leukocyte count, flow cytometric analysis of the percentage of polymorphonuclear leukocytes (PMNs) and total PMNs in 3 mL BAL from 4- and 12-week-old WT and CybbKO male and female mice (n ≥ 7 in each group). (B) Flow cytometric analysis of total AMs (CD45+Siglec F+CD11c+ cells) and total eosinophils (EOS) (CD45+Siglec F+CD11c− cells) from 3 mL of BAL (n ≥ 7). (C) ELISA of IL-1β and CXCL2 in BAL (n ≥ 3 in each group). (D) Gating strategy to identify CD11b+ AMs in the BAL (CD45+SIglec F+ CD11c+CD11bhigh). (E) Percentage of AMs that are CD11bhigh in BAL samples from different groups (n ≥ 4). (F) CD11b expression by qRT-PCR in flow-sorted AMs of different groups. WTL, WT CD11blow AMs; CybbKO L, CybbKO CD11blow AMs; and CybbKO H, CybbKO CD11bhigh AMs (n = 3). (G) Lung sections from 12-week-old WT and CybbKO mice, as indicated, stained with hematoxylin-eosin showing intracellular (arrowhead) and extracellular eosinophilic crystals (arrow) in CybbKO mice. The rightmost panel shows lung tissue from a CybbKO mouse with small infiltrates adjacent to some of the crystals. Scale bars, 100 μm. Representative images from ≥5 mice of each genotype. Bar graph data are expressed as mean ± standard error of the mean. The Student t test was performed for samples distributed in 2 groups. *P < .05; **P < .01; ***P < .001. One-way analysis of variance was performed followed by Tukey’s post hoc analysis comparing multiple groups. Data represent at least 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001. ELISA, enzyme linked immunosorbent assay.
Next, we examined AM surface markers by flow cytometry to evaluate whether they changed as lungs became inflamed in CybbKO mice. Although AMs with a conventional marker phenotype (CD45+CD11chighSiglec FhighCD11blow32 ) were present, we identified an additional CD11chighSiglec FhighCD11bhigh population in BAL and lung tissue of 12-week-old CybbKO mice, representing ∼40% of the total AMs (Figure 1D-E; supplemental Figure 1A-B). This CD11bhigh subset, which had increased CD11b transcription (Figure 1F), was virtually absent in 4-week-old CybbKO and WT mice (Figure 1E). At homeostasis, AMs both self-renew in situ and are slowly replaced in adulthood by bone marrow–derived precursors.33 CD11chighSiglec FhighCD11bhigh “activated” macrophages in the BAL, which were derived from resident AMs, have been identified in various inflammatory settings, monocytes, or both.34-36 Expression of the monocyte marker CCR2 was not increased on CD11bhighAMs in CybbKO mice (supplemental Figure 1C-D), suggesting that these were not derived from very recently emigrated monocytes.37CybbKO AMs showed similar levels of annexin V staining and bromodeoxyuridine incorporation relative to WT AMs, even within the CD11bhigh population (supplemental Figure1E-F). Thus, 12-week-old CybbKO mice develop an additional population of AMs that are CD11bhigh, which could be derived from either resident AMs or bone marrow, but this process is not associated with increased apoptosis or turnover of AMs.
We also observed eosin-staining YM1 crystals within alveoli and AMs of 12-week-old CybbKO mice, accompanied in some mice by patchy intra-alveolar infiltrates (Figure 1G; supplemental Figure 2B), similar to NCF1-deleted CGD mice.9,10 YM1 is a secreted chitinaselike protein (CLP) that easily forms crystals, often seen in the lungs of mice with various inflammatory conditions.9,10,38,39 Notably, transcripts for CLPs CHIL1, CHIL3, CHIL4, and proteins related to their secretion were elevated in CD11bhigh AMs from 12-week-old CybbKO mice, consistent with an activated AM phenotype (supplemental Figure 2C).
Thus, by the time CGD mice reach adulthood, absence of NOX2 activity leads to spontaneous development of an activated CD11bhigh AM subset and an increase in alveolar neutrophils, proinflammatory cytokines, and YM1 crystals, all consistent with the dysregulation of alveolar homeostasis.
Deletion of NOX2 preferentially in macrophages is sufficient for an inflammatory lung phenotype
We next tested whether inactivating NOX2 primarily in macrophages is sufficient to cause the low-grade alveolar inflammation observed in germline loss-of-function mice. We crossed mice with a conditional Ncf2 loss-of-function allele (Ncf2fl/fl) to LysMCre mice40 (supplemental Figure 3A). In the resulting Ncf2LysMCre mice, ∼95% of AMs or RPMs lacked NOX2 enzyme activity (supplemental Figure 3B-C), and NCF2 protein was not detected in RPM lysates (supplemental Figure 3D-E). However, NCF2 expression was only partially reduced in Ncf2LysMCre neutrophils (supplemental Figure 3D-E). Peripheral blood neutrophils from Ncf2LysMCre mice had either partial or near normal NOX2 activity by flow cytometry assay and superoxide production by quantitative cytochrome c reduction assay was ≈50% of Ncf2fl/fl(supplemental Figure 3F-G). Ncf2LysMCre monocytes harvested from marrow and peripheral blood had normal NCF2 expression and NOX2 activity (supplemental Figure 3D-E,H).
Although complete loss of NOX2 activity was restricted to macrophages, Ncf2LysMCre mice developed inflammatory changes in the lungs similar to CGD mice. BAL from naive Ncf2LysMCre mice at 12 week of age showed increased total leukocytes, neutrophils, and proinflammatory cytokines (Figure 2A-B) and a population of CD11chighSiglec FhighCD11bhigh AMs (Figure 2C). Ncf2LysMCre mice also had increased transcripts for YM1 (supplemental Figure 2D) and crystals in alveoli and within AMs (Figure 2D; supplemental Figure 2B). Both Ncf2LysMCre and CybbKO mice continued to exhibit CD11bhigh AMs after 1 year of age, but Ncf2LysMCre mice did not develop the focal alveolar infiltrates often seen in CybbKO mice, which also were sometimes larger in aging CybbKO mice (supplemental Figure 3I-K). We conclude that, although infiltrate formation most likely requires cross talk with other leukocytes entirely lacking NOX2, deletion of Ncf2 restricted to macrophages is sufficient to produce an inflammatory alveolar environment in Ncf2LysMCre mice.
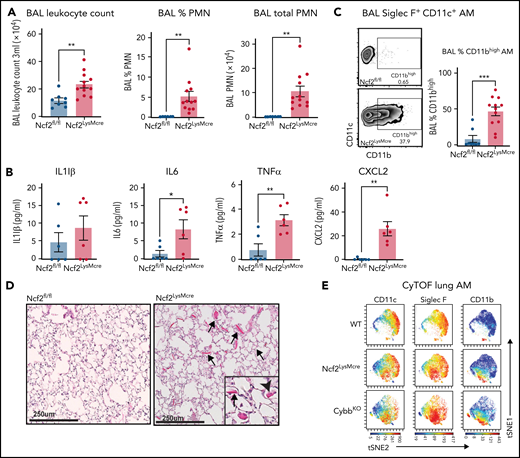
Mice with LysMCre-mediated deletion of NOX2 exhibit an inflammatory lung phenotype. (A) BAL leukocyte count, flow-cytometric analysis of the percentage of polymorphonuclear leukocytes (PMNs), and total PMNs in 3 mL BAL from 12-week-old Ncf2fl/fl and Ncf2LysMCre mice (n ≥ 7). (B) ELISA analysis of BAL samples from 12-week-old mice (n ≥ 6). (C) Flow cytometric analysis of CD11c+CD11bhigh macrophages from the CD45+Siglec F+ CD11c+ gate and the percentage that are CD11bhigh in BAL samples of 12-week-old mice (n ≥ 8). (D) Lung sections from12-week-old Ncf2fl/fl and Ncf2LysMcre mice stained with hematoxylin-eosin showing intracellular (arrowhead) and extracellular (arrow) eosinophilic crystals. Bars represent 250 μm. Representative images from more than 6 samples per genotype. (E) tSNE plot from CyTOF data of CD45+CD11c+Siglec F+ AMs from lung tissue harvested from 12-week-old WT, CybbKO and Ncf2LysMcre mice showing populations expressing CD11c, Siglec F, and CD11b. Representative plots from ≥4 samples in each group. Bar graph data are expressed as the mean ± standard error of the mean. The Student t test was performed for samples distributed in 2 groups. Data represented the results of at least 2 independent experiments. *P < .05; **P < .01; ***P < .001. CyTOF, cytometry by time of flight; t-SNE, t-distributed stochastic neighbor embedding.
Mice with LysMCre-mediated deletion of NOX2 exhibit an inflammatory lung phenotype. (A) BAL leukocyte count, flow-cytometric analysis of the percentage of polymorphonuclear leukocytes (PMNs), and total PMNs in 3 mL BAL from 12-week-old Ncf2fl/fl and Ncf2LysMCre mice (n ≥ 7). (B) ELISA analysis of BAL samples from 12-week-old mice (n ≥ 6). (C) Flow cytometric analysis of CD11c+CD11bhigh macrophages from the CD45+Siglec F+ CD11c+ gate and the percentage that are CD11bhigh in BAL samples of 12-week-old mice (n ≥ 8). (D) Lung sections from12-week-old Ncf2fl/fl and Ncf2LysMcre mice stained with hematoxylin-eosin showing intracellular (arrowhead) and extracellular (arrow) eosinophilic crystals. Bars represent 250 μm. Representative images from more than 6 samples per genotype. (E) tSNE plot from CyTOF data of CD45+CD11c+Siglec F+ AMs from lung tissue harvested from 12-week-old WT, CybbKO and Ncf2LysMcre mice showing populations expressing CD11c, Siglec F, and CD11b. Representative plots from ≥4 samples in each group. Bar graph data are expressed as the mean ± standard error of the mean. The Student t test was performed for samples distributed in 2 groups. Data represented the results of at least 2 independent experiments. *P < .05; **P < .01; ***P < .001. CyTOF, cytometry by time of flight; t-SNE, t-distributed stochastic neighbor embedding.
We also studied female Cybb carrier mice, where approximately half of the leukocytes were NOX2+, and half lacked NOX2 because of X-linked inactivation. Interestingly, ≈10% AMs from Cybb carriers harbored intracellular YM1 crystals by electron microscopy (supplemental Figure 2B), which could be a sign of increased AM activation; however, this fraction is much lower than that for CybbKO and Ncf2LysMCre AMs, where ≈65% contain YM1 crystals detected by electron microscopy. Rare neutrophils were also seen in carrier BAL samples. Notably, carrier females did not develop evidence of lung inflammation by histology, CD11bhigh AMs or increased BAL proinflammatory cytokines, even after 1 year of age (not shown), which suggests that there may be a threshold number of NOX2− cells needed to promote spontaneous inflammatory changes in the alveolar environment.
Cytometry by time of flight for cell surface expression of canonical AM markers confirmed the presence of a CD11bhigh population of CD11chighSiglec F high AMs in Ncf2LysMCre and CybbKO mice that was absent in WT mice (Figure 2E; supplemental Figure 4A-B). There was a modest decrease in CD64 in CD11bhigh AMs, of uncertain biologic significance, along with a modest increase in CX3CR1. However, characteristic expression of other markers (F4/80, Siglec F, CD274, CD206, and MHCII [I-A/I-E]) (supplemental Figure 4B) were unaltered. Additional cytometry by time of flight results showed that Ncf2LysMCre and CybbKO mice had small but significant decreases in lung B lymphocytes and natural killer cells compared with WT, but the percentages of alveolar and interstitial macrophages, dendritic cells, monocytes, eosinophils, and T lymphocytes were similar among all 3 genotypes (supplemental Figure 4C).
Transcriptomic profiles of AMs but not RPMs from NOX2-deleted mice showed an inflammatory signature
To gain insights into how CD11bhigh AMs from NOX2-deficient mice differ from other AMs, we profiled gene expression of sorted AMs using RNA-seq. Multidimensional scaling analysis showed that AMs from 4-week-old WT and CybbKO mice share a similar gene expression signature (Figure 3A). However, in 12-week-old mice, the transcriptome of CybbKO AMs diverged from WT AMs, with differences between CD11blow and CD11bhighCybbKO AMs. Differences are also depicted in a heat map showing the top 500 altered genes among various groups (Figure 3B) and n Venn diagrams (supplemental Figure 5A). We used gene set enrichment analysis (GSEA) of Hallmark gene sets41 to interrogate the 2 most divergent groups: WT CD11blow AMs and CybbKO CD11bhigh AMs from 12-week-old mice. The inflammatory response gene set is among the most significantly different, along with IFN-α and IFN-γ responses, EMT and KRAS signaling (Figure 3C). The inflammatory response was the only gene set significantly altered between CD11blow and CD11bhigh AMs from 12-week-old CybbKO mice and also approached significance for CD11blow AMs from 12-week-old CybbKO and WT mice (Figure 3D). Although no significant differences in GSEA were identified between CD11blow AMs from CybbKO mice at the 2 ages, many of the upregulated genes in the 12-week-old samples are related to inflammation (supplemental Figure 5B). Relative expression of various activation-associated genes in AMs from 12-week-old mice are shown in Figure 3E. IL1a, IL1b, and Chil3 expression were verified among various groups by qRT-PCR (Figure 3F). The skewed transcriptome in NOX2-deleted AMs did not correlate with polarization to either classic (M1) or alternative (M2) macrophage activation (Figure 3G), typical of diversity seen in vivo.16 Finally, AMs sorted from Ncf2LysMCre mice showed increased expression of IL1a, IL1b, Ccl4, Mx1, and Isg15, as compared with Ncf2fl/fl mice, especially in CD11bhigh AMs (Figure 3H). In aggregate, these results indicate that the AM transcriptome becomes skewed toward a more proinflammatory profile as mice with either global or macrophage deletion of NOX2 reach adulthood, with these changes most pronounced in the CD11bhigh AM population.
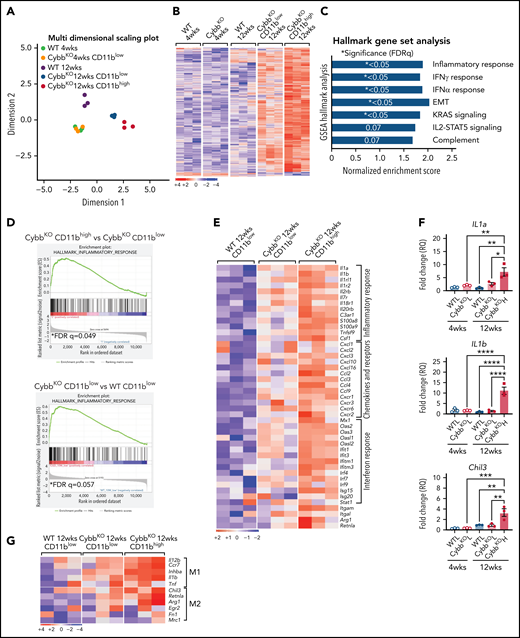
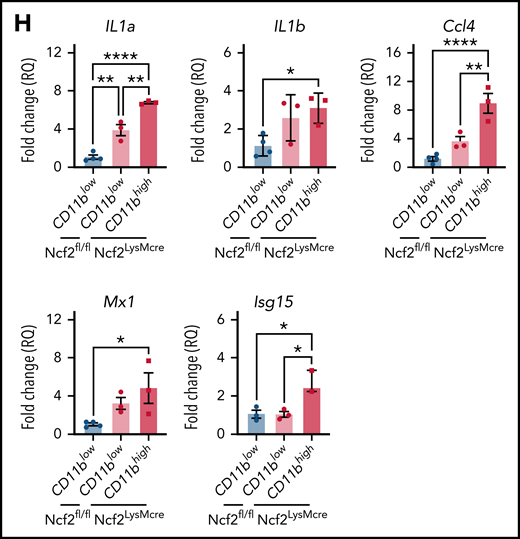
NOX2-deleted CD11bhigh AMs acquire an inflammatory transcriptional signature. (A) Multidimensional scaling plot of RNA-seq transcriptomes in 2 different dimensions showing the differences in gene signatures among the indicated groups of samples of AMs sorted from 4- and 12-week-old mice (n = 3 independent samples in each group). (B) Heat maps showing relative expression of the top 500 altered genes in AMs in the different groups, as indicated (n = 3 independent samples in each group). (C) Hallmark gene sets with significant changes in GSEA between CybbKO CD11bhigh and WT CD11blow AMs from 12-week-old mice. False-discovery rate (FDR)q <0.05 was considered significant (n = 3 independent samples for each group). (D) GSEA plot showing differences in the hallmark inflammatory response gene sets between AMs from 12-week-old CybbKO CD11bhigh and CybbKO CD11blow, as well as CybbKO CD11blow and WT CD11blow mice, as indicated. FDRq <0.05 considered significant (n = 3 independent samples for each group). (E) Heat map from RNA-seq data showing relative expression of selected genes in CybbKO and WT AMs from 12-week-old mice (n = 3 independent samples for each group). (F) qRT-PCR of gene expression in flow-sorted AMs from WT and CybbKO mice of different ages for selected genes. WTL, WT CD11blow AM; CybbKO L, CybbKO CD11blow AM; and CybbKO H, CybbKO CD11bhigh AM (n = 3 samples for each group). (G) Heat map from AM RNA-seq data for selected M1 and M2 genes, as indicated. (H) qRT-PCR for expression of selected genes in flow sorted AMs, as indicated, from Ncf2fl/fl and Ncf2LysMcre mice (n ≥ 3 samples for each group). Adjusted P values: *P < .05; **P < .01; ***P < .001. Bar graph data are expressed as the mean ± standard error of the mean. One-way analysis of variance was performed, followed by Tukey’s post hoc analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001. FDR, false-discovery rate.
NOX2-deleted CD11bhigh AMs acquire an inflammatory transcriptional signature. (A) Multidimensional scaling plot of RNA-seq transcriptomes in 2 different dimensions showing the differences in gene signatures among the indicated groups of samples of AMs sorted from 4- and 12-week-old mice (n = 3 independent samples in each group). (B) Heat maps showing relative expression of the top 500 altered genes in AMs in the different groups, as indicated (n = 3 independent samples in each group). (C) Hallmark gene sets with significant changes in GSEA between CybbKO CD11bhigh and WT CD11blow AMs from 12-week-old mice. False-discovery rate (FDR)q <0.05 was considered significant (n = 3 independent samples for each group). (D) GSEA plot showing differences in the hallmark inflammatory response gene sets between AMs from 12-week-old CybbKO CD11bhigh and CybbKO CD11blow, as well as CybbKO CD11blow and WT CD11blow mice, as indicated. FDRq <0.05 considered significant (n = 3 independent samples for each group). (E) Heat map from RNA-seq data showing relative expression of selected genes in CybbKO and WT AMs from 12-week-old mice (n = 3 independent samples for each group). (F) qRT-PCR of gene expression in flow-sorted AMs from WT and CybbKO mice of different ages for selected genes. WTL, WT CD11blow AM; CybbKO L, CybbKO CD11blow AM; and CybbKO H, CybbKO CD11bhigh AM (n = 3 samples for each group). (G) Heat map from AM RNA-seq data for selected M1 and M2 genes, as indicated. (H) qRT-PCR for expression of selected genes in flow sorted AMs, as indicated, from Ncf2fl/fl and Ncf2LysMcre mice (n ≥ 3 samples for each group). Adjusted P values: *P < .05; **P < .01; ***P < .001. Bar graph data are expressed as the mean ± standard error of the mean. One-way analysis of variance was performed, followed by Tukey’s post hoc analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001. FDR, false-discovery rate.
To investigate whether there is tissue specificity to resident macrophage remodeling with age in CGD mice, we analyzed gene expression in RPMs by RNA microarray. We sorted the 2 groups of RPMs (B220−CD115+F4/80highMHCII− and B220−CD115+F4/80lowMHCIIhigh) from naive 12-week-old WT and CybbKO mice. Although the 2 subtypes of RPMs were shown to be clearly different from each other by principal component analysis, WT and CybbKO macrophages were similar, and within each RPM subgroup, expression of hallmark gene sets was similar between WT and CybbKO (supplemental Figure 5C-D). These results show that the impact of NOX2 deletion on the remodeling of the resident macrophage transcriptome at homeostasis is influenced by the tissue niche.
Chromatin profiles reveal epigenetic changes in CD11bhigh AMs in CybbKO mice
Epigenetic modifications of the enhancer landscape and their plasticity play an important role in regulating macrophage gene expression.15,22 To interrogate differences in the epigenome of WT and CGD AMs, we performed ATAC-seq. A multidimensional scaling plot showed distinct differences among groups by genotype and age (Figure 4A). AMs from 4-week-old WT and CybbKO mice and 12-week-old WT mice had similar ATAC-seq profiles, but AMs from 12-week-old CybbKO mice were distinct, particularly the CD11bhigh subpopulation (Figure 4A). CD11bhigh AMs had a higher number of ATAC-seq peaks compared with CD11blowAMs from 12-week-old mice of either genotype, as did AMs from 12-week-old CybbKO mice compared with those from younger CybbKO mice (supplemental Figure 5E). Analysis of aggregate peak heights showed increased chromatin accessibility in CybbKO AMs compared with WT, with CD11bhigh AMs from 12-week-old CybbKO mice having the greatest difference (Figure 4B). These results indicate that the AM epigenome in NOX2-deficient mice changes between 4 and 12 weeks after birth and acquires a more open chromatin state compared with WT AMs.
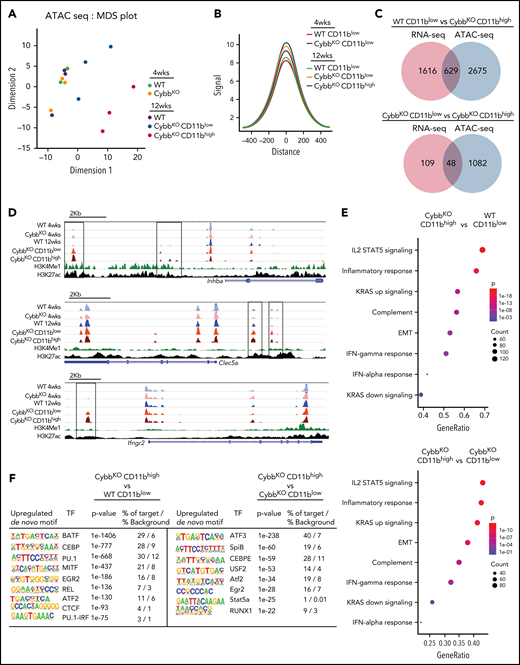
NOX2-deleted AMs acquire epigenetic alterations. (A) An MDS plot of AM ATAC-seq peak signals in 2 different dimensions showing differences among the indicated groups of mice (n = 3 independent samples in each group). (B) Histogram comparing aggregate ATAC-seq peak heights around TSSs (±500 kb) in various groups for the genes with significant differences between CybbKO CD11bhigh AMs and WT CD11blow AMs from 12-week-old mice. (C) Venn diagram showing the overlay of genes with significant alterations in both RNA-seq and ATAC-seq (TSS, ±500 kb) among various groups. (D) Representative WUSTL epigenome browser tracks displaying normalized ATAC-seq peak signal distribution in selected genes from AMs sampled from the indicated groups of 4- and 12-week-old WT and CybbKO mice along with CHIP-seq data for H3K4Me1 and H3K27ac marks from Lavin et al.58 Boxes indicate ATAC-seq peaks that are significantly different in 12-week-old CybbKO samples compared with 12-week-old WT samples. (E) Hallmark gene set analysis for genes with altered ATAC-seq peak signals between CybbKO CD11bhigh and WT CD11blow as well as CybbKO CD11bhigh and CybbKOCD11blowAMs. (F) HOMER de novo motif enrichment analysis for transcription factor binding sites in open chromatin ±500 kb from the TSS, as identified in ATAC-seq, as analyzed in AMs sampled from 12-week-old mice (n = 3 independent samples in each group). The percentage of motif occurrence in peaks vs background is also shown. CHIP, chromatin immunoprecipitation; HOMER, hypergeometric optimization of motif enrichment; MDS, multidimensional scaling; TSS, transcription start site.
NOX2-deleted AMs acquire epigenetic alterations. (A) An MDS plot of AM ATAC-seq peak signals in 2 different dimensions showing differences among the indicated groups of mice (n = 3 independent samples in each group). (B) Histogram comparing aggregate ATAC-seq peak heights around TSSs (±500 kb) in various groups for the genes with significant differences between CybbKO CD11bhigh AMs and WT CD11blow AMs from 12-week-old mice. (C) Venn diagram showing the overlay of genes with significant alterations in both RNA-seq and ATAC-seq (TSS, ±500 kb) among various groups. (D) Representative WUSTL epigenome browser tracks displaying normalized ATAC-seq peak signal distribution in selected genes from AMs sampled from the indicated groups of 4- and 12-week-old WT and CybbKO mice along with CHIP-seq data for H3K4Me1 and H3K27ac marks from Lavin et al.58 Boxes indicate ATAC-seq peaks that are significantly different in 12-week-old CybbKO samples compared with 12-week-old WT samples. (E) Hallmark gene set analysis for genes with altered ATAC-seq peak signals between CybbKO CD11bhigh and WT CD11blow as well as CybbKO CD11bhigh and CybbKOCD11blowAMs. (F) HOMER de novo motif enrichment analysis for transcription factor binding sites in open chromatin ±500 kb from the TSS, as identified in ATAC-seq, as analyzed in AMs sampled from 12-week-old mice (n = 3 independent samples in each group). The percentage of motif occurrence in peaks vs background is also shown. CHIP, chromatin immunoprecipitation; HOMER, hypergeometric optimization of motif enrichment; MDS, multidimensional scaling; TSS, transcription start site.
We next explored possible implications of increased chromatin accessibility in CybbKO AMs for gene expression. Many of the genes with increased RNA expression in CybbKO CD11bhigh AMs showed increased accessibility (Figure 4C-D), which could contribute to their increased transcription. We also investigated the association of differential ATAC-seq signals with GSEA, focusing on gene sets with significant differences (Figure 3C). Six of 8 of these sets showed significant difference in chromatin accessibility in CybbKO CD11bhigh AMs compared with either WT AMs or CybbKO CD11blow AMs from 12-week-old mice (Figure 4E). HOMER (hypergeometric optimization of motif enrichment) analysis showed that the transcription factor motifs most significantly enriched in open chromatin regions in CD11bhigh AMs included those capable of binding to members of the basic helix-loop-helix leucine family (ATF, BATF, USF2, and MITF) or to transcription factors that play prominent roles in myeloid lineage determination and are enriched in macrophage enhancer regions (Pu.1, CEBP, and RUNX; Figure 4F).21,22,42 NFKB/REL and Stat binding sites were also enriched in CybbkoCD11bhigh AMs. The increased accessibility of myeloid lineage enhancers and of the other motifs are consistent with the upregulated proinflammatory gene expression in NOX2-deleted CD11bhigh AMs.
Development of lung inflammation in Cybbko mice does not require host microbiota or the type I IFN receptor
As ongoing exposure to either environmental or host-associated microbes could contribute to the development of alveolar inflammation, we examined the impact of raising CybbKO mice under germ-free conditions. Although there was a small but significant decrease in BAL neutrophils, CD11bhigh AMs were not reduced compared with CybbKO mice maintained in specific-pathogen–free housing (supplemental Figure 6A). Germ-free CybbKO mice also had eosinophilic AM and YM1 crystals within alveoli (supplemental Figure 6B). Although mice could still be exposed to sterile microbe–derived substances, such as in food or bedding, the presence of host microbiota or microbes in the environment are not necessary for NOX2-deleted animals to develop altered alveolar homeostasis.
Enhanced leukocyte expression of type I IFN response genes can be induced by viral or bacterial molecular patterns, as well as other inflammatory stimuli. Increased IFN response genes are reported in humans who lack functional NOX2 and in CGD mice, even in germ-free conditions, which may reflect exposure to nonpathogenic viruses or to sterile proinflammatory molecules.43 Our results showed a significant upregulation in IFN-α response genes in NOX2-deleted CD11bhigh AMs (Figure 3C,E,H) and higher BAL levels of IFN-α (supplemental Figure 2A). To test whether type I IFN drives lung inflammation in NOX2-deleted mice, we studied Ifnar1−/−CybbKO (DKO) mice.27 Lungs of DKO mice displayed inflammatory changes, including increased BAL neutrophils and YM1 crystals (Figure 5A-B). Transcripts for Chil3 (YM1) and proinflammatory cytokines IL-1β, CCL2, and CCL3 were significantly increased in BAL macrophages from both CybbKO and DKO mice compared with littermates that retained a WT Cybb allele (Figure 5C). Thus, IFNAR1 signaling is not necessary for CybbKO mice to develop spontaneous lung inflammation.
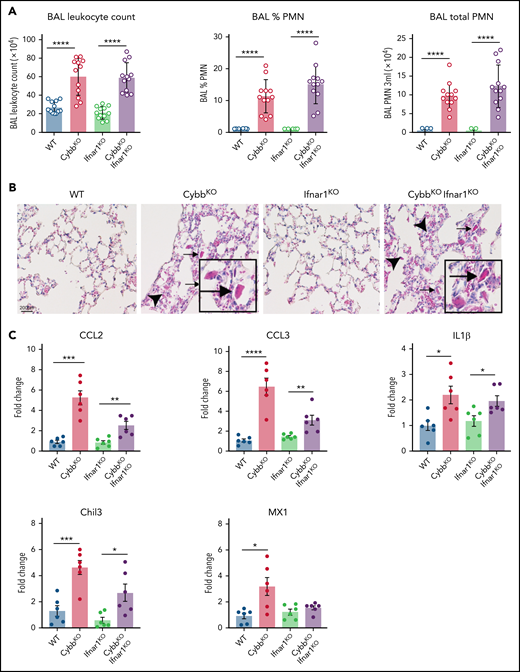
Type I IFN signaling is not necessary for NOX2-deleted mice to develop spontaneous lung inflammation and activated AMs. (A) BAL leukocyte count, percentage of polymorphonuclear leukocytes (PMNs), and total PMNs in 3 mL of BAL from WT, CybbKO, Ifnar1KO, and CybbKOIfnar1KO mice (n = 12 in each group). (B) Hematoxylin-eosin–stained lung tissues showing the presence of eosinophilic macrophages (arrowhead) and extracellular crystals (arrow) in CybbKO and CybbKOIfnar1KO mice. Scale bar, 200 μm and the scale is same for all panels. Representative image from more than 4 samples in each group. (C) qRT-PCR analysis of gene expression in BAL cells isolated from WT, CybbKO, Ifnar1KO, and CybbKOIfnar1KO mice (n = 6 in each group). Bar graph data are expressed as the mean ± standard error of the mean. Experiments were repeated at least twice and the Student t test was performed for samples distributed between 2 groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Type I IFN signaling is not necessary for NOX2-deleted mice to develop spontaneous lung inflammation and activated AMs. (A) BAL leukocyte count, percentage of polymorphonuclear leukocytes (PMNs), and total PMNs in 3 mL of BAL from WT, CybbKO, Ifnar1KO, and CybbKOIfnar1KO mice (n = 12 in each group). (B) Hematoxylin-eosin–stained lung tissues showing the presence of eosinophilic macrophages (arrowhead) and extracellular crystals (arrow) in CybbKO and CybbKOIfnar1KO mice. Scale bar, 200 μm and the scale is same for all panels. Representative image from more than 4 samples in each group. (C) qRT-PCR analysis of gene expression in BAL cells isolated from WT, CybbKO, Ifnar1KO, and CybbKOIfnar1KO mice (n = 6 in each group). Bar graph data are expressed as the mean ± standard error of the mean. Experiments were repeated at least twice and the Student t test was performed for samples distributed between 2 groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
NOX2− AMs, but not RPMs, have increased responses to TLR agonists
We reasoned that the proinflammatory changes in AMs lacking NOX2 may be associated with a cell-intrinsic hyperreactivity to inhaled materials in the environment or debris that AMs continuously manage during homeostasis. To approach this question, we used agonists of Toll-like receptor-2 (TLR2) and TLR4, which can detect DAMPS and exogenous substances in addition to bacterial and fungal cell walls.44-46 We stimulated AMs isolated from Ncf2KO and WT mice of different ages with either Pam3CSK4 or LPS and analyzed gene expression. In AMs harvested from 4-week-old mice, LPS induced significantly higher Il1b, Ccl3, and Ccl4 expression in Ncf2KO AMs compared with Ncf2fl/fl (Figure 6A). Hence, AM reactivity was elevated at a young age, when basal transcriptional and ATAC-seq profiles are similar to WT mice. Ncf2KO AMs from 12-week-old mice additionally showed significantly increased responses to Pam3CSK4 compared with WT AMs, as well as to LPS (Figure 6B). Notably, AMs from 12-week-old Ncf2LysMCre mice were also hyperresponsive to Pam3CSK4 or LPS (Figure 6C). Finally, to investigate whether the macrophage tissue niche influenced this hyperresponsive phenotype, we performed similar studies on RPMs from 12-week-old WT and CybbKO mice, but there were no differences between genotypes after stimulation with either Pam3CSK4 or LPS (supplemental Figure 7). In aggregate, these results show the importance of AM NOX2 activity for limiting their reactivity, and that cell-extrinsic factors related to their tissue niche are also important.
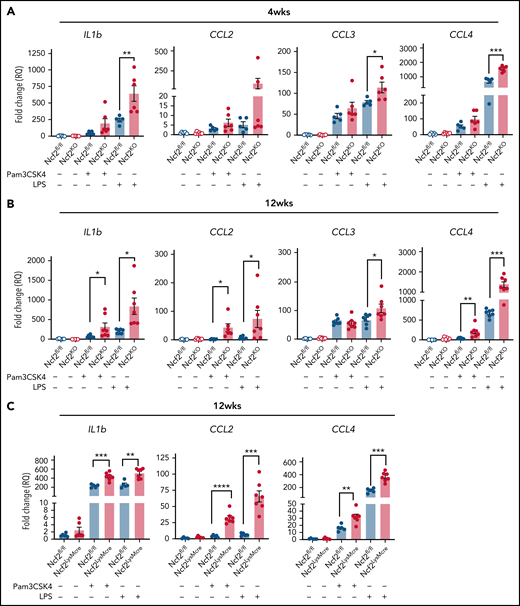
AMs from mice with global or LysMCre-mediated deletion of NOX2 have increased responses to TLR stimulation. (A-B) AMs isolated from BAL of Ncf2fl/fl and Ncf2KO mice were stimulated with Pam3CSK4 or LPS, and gene expression was assayed by qRT-PCR in 4-week-old (A) and 12-week-old (B) mice. (C) AMs isolated from BAL of 12-week-old Ncf2fl/fl and Ncf2LysMCre mice were stimulated with Pam3CSK4 or LPS, and gene expression was assayed by qRT-PCR. Bar graph data are expressed as the mean ± standard error of the mean. Experiments were repeated at least twice. The Student t test was performed for samples distributed between 2 groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
AMs from mice with global or LysMCre-mediated deletion of NOX2 have increased responses to TLR stimulation. (A-B) AMs isolated from BAL of Ncf2fl/fl and Ncf2KO mice were stimulated with Pam3CSK4 or LPS, and gene expression was assayed by qRT-PCR in 4-week-old (A) and 12-week-old (B) mice. (C) AMs isolated from BAL of 12-week-old Ncf2fl/fl and Ncf2LysMCre mice were stimulated with Pam3CSK4 or LPS, and gene expression was assayed by qRT-PCR. Bar graph data are expressed as the mean ± standard error of the mean. Experiments were repeated at least twice. The Student t test was performed for samples distributed between 2 groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Increased lung inflammation after remote tissue inflammation in NOX2-inactivated mice correlates with proinflammatory remodeling of AMs
We next examined whether the age-dependent priming of AMs lacking NOX2 that develops under basal conditions leads to amplified responses to inflammatory insults. We first examined lung inflammation induced by sterile fungal cell walls, which elicit increased neutrophilic infiltration in adult CGD mice.28,47,48 At 18 h after lung instillation of zymosan, both 4- and 12-week-old CybbKO mice displayed a significantly greater number of BAL neutrophils compared with WT mice (Figure 7A-B). Thus, the hyperinflammatory response of NOX2-inactivated mice to direct lung challenge with zymosan is age independent. We next investigated responses of mice at different ages to indirect lung injury induced by remote tissue inflammation (Figure 7D). We modified a peritoneal zymosan-induced systemic inflammatory response syndrome model,29,30,49,50 using a ≈10-fold lower dose of zymosan to avoid mortality. This dose elicited significantly greater neutrophilic inflammation in the peritoneal cavity of CybbKO compared with WT mice at both 4 and 12 wk of age (supplemental Figure 8A-B). However, only 12-week-old CybbKO mice had a significantly increased number of BAL neutrophils, focal lung neutrophil infiltrates, and elevated BAL CXCL2 at 48 hours after induction of peritonitis, whereas neither 4-week-old CybbKO nor WT mice developed evidence of lung inflammation (Figure 7D-H).
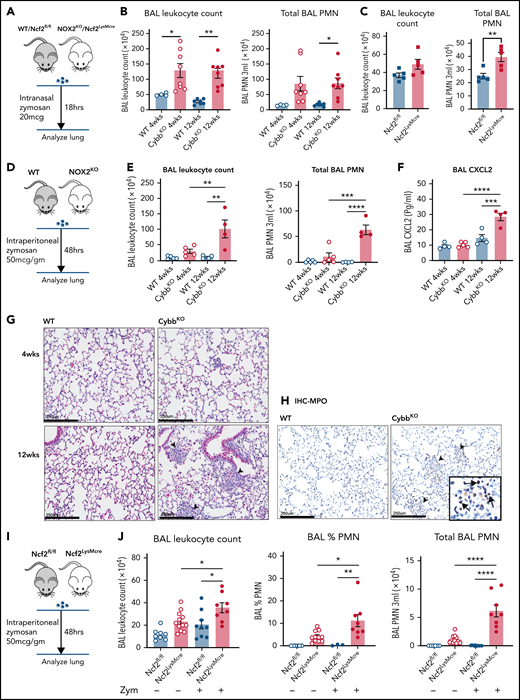
Lung inflammation induced by zymosan challenge or remote tissue inflammation in mice with global or LysMCre-mediated deletion of NOX2. (A) Schema for zymosan-induced lung inflammation in WT or Ncf2fl/fl and CybbKO (NOX2KO) or Ncf2LysMcre mice. (B) BAL leukocyte counts and total polymorphonuclear leukocytes (PMNs), as identified by microscopy in 3 mL of BAL from WT and CybbKO mice of different ages after intranasal administration of zymosan (n ≥ 4 mice in each group). (C) BAL leukocyte counts and total PMNs as identified by microscopy in 3 mL of BAL from Ncf2fl/fl and Ncf2LysMcre mice with different ages following intranasal administration of zymosan (n ≥ 5 mice in each group). (D) Schema for analysis of lung inflammation after intraperitoneal administration of zymosan in WT and CybbKO mice. (E) BAL leukocytes, and total PMNs in 3 mL of BAL from the indicated groups of WT and CybbKO mice of different ages after intraperitoneal zymosan (n ≥ 4 mice in each group). (F) BAL CXCL2 levels in samples from the indicated groups after intraperitoneal zymosan (n ≥ 4 mice in each group). (G) Hematoxylin-eosin staining of lung tissues from WT and CybbKO mice after intraperitoneal zymosan. Representative image from 1 of 4 samples in each group. Arrowheads, focal neutrophil infiltrates. Scale bar, 250 μm. (H) Representative images of immunohistochemistry for MPO to identify lung PMNs after intraperitoneal zymosan. Arrows, MPO+cells. Representative image from 4 samples of each group. Scale bars, 250 μm. (I) Schema for analysis of lung inflammation after intraperitoneal zymosan in 12-week-old Ncf2fl/fl and Ncf2LysMCre mice. (J) BAL leukocyte counts, percentage of PMNs by microscopy, and total PMNs in 3 mL of BAL from naive (data also shown in Figure 2A) or zymosan-challenged Ncf2fl/fl and Ncf2LysMCre mice. Bar graph data are expressed as the mean ± standard error of the mean. Experiments were repeated at least twice. The Student t test was performed to compare results between each genotype for each age group in panel B. *P < .05; **P < .01. One-way analysis of variance was performed followed by Tukey’s post hoc analysis for data (E-F,J). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Lung inflammation induced by zymosan challenge or remote tissue inflammation in mice with global or LysMCre-mediated deletion of NOX2. (A) Schema for zymosan-induced lung inflammation in WT or Ncf2fl/fl and CybbKO (NOX2KO) or Ncf2LysMcre mice. (B) BAL leukocyte counts and total polymorphonuclear leukocytes (PMNs), as identified by microscopy in 3 mL of BAL from WT and CybbKO mice of different ages after intranasal administration of zymosan (n ≥ 4 mice in each group). (C) BAL leukocyte counts and total PMNs as identified by microscopy in 3 mL of BAL from Ncf2fl/fl and Ncf2LysMcre mice with different ages following intranasal administration of zymosan (n ≥ 5 mice in each group). (D) Schema for analysis of lung inflammation after intraperitoneal administration of zymosan in WT and CybbKO mice. (E) BAL leukocytes, and total PMNs in 3 mL of BAL from the indicated groups of WT and CybbKO mice of different ages after intraperitoneal zymosan (n ≥ 4 mice in each group). (F) BAL CXCL2 levels in samples from the indicated groups after intraperitoneal zymosan (n ≥ 4 mice in each group). (G) Hematoxylin-eosin staining of lung tissues from WT and CybbKO mice after intraperitoneal zymosan. Representative image from 1 of 4 samples in each group. Arrowheads, focal neutrophil infiltrates. Scale bar, 250 μm. (H) Representative images of immunohistochemistry for MPO to identify lung PMNs after intraperitoneal zymosan. Arrows, MPO+cells. Representative image from 4 samples of each group. Scale bars, 250 μm. (I) Schema for analysis of lung inflammation after intraperitoneal zymosan in 12-week-old Ncf2fl/fl and Ncf2LysMCre mice. (J) BAL leukocyte counts, percentage of PMNs by microscopy, and total PMNs in 3 mL of BAL from naive (data also shown in Figure 2A) or zymosan-challenged Ncf2fl/fl and Ncf2LysMCre mice. Bar graph data are expressed as the mean ± standard error of the mean. Experiments were repeated at least twice. The Student t test was performed to compare results between each genotype for each age group in panel B. *P < .05; **P < .01. One-way analysis of variance was performed followed by Tukey’s post hoc analysis for data (E-F,J). *P < .05; **P < .01; ***P < .001; ****P < .0001.
We next investigated whether LysMCre-mediated deletion of NOX2 activity resulted in altered responses to inflammatory challenges. Twelve-week-old Ncf2LysMCre mice had a significantly increased number of BAL neutrophils after lung instillation of zymosan compared with Ncf2fl/fl littermates, although the number of neutrophils was about twofold less than that in CGD mice (Figure 7B-C). Zymosan-induced peritoneal inflammation was similar in Ncf2fl/fl and Ncf2LysMCre mice (supplemental Figure 8C-D), indicating that deletion of NOX2 preferentially in macrophages does not enhance the peritoneal response to zymosan. However, the peritoneal inflammation was still accompanied by pulmonary inflammation in Ncf2LysMCre mice at 48 h, with increased BAL leukocytes and neutrophils compared with baseline or to Ncf2fl/fl controls (Figure 7I). Inflammatory changes in the BAL were milder compared with CGD mice, which most likely reflects the more modest inflammatory response to peritoneal zymosan in Ncf2LysMCre mice.
Collectively, these results show that the proinflammatory remodeling of lung AMs and dysregulated alveolar homeostasis in mice with either global or macrophage NOX2 inactivation can result in increased lung inflammation triggered by distant tissue inflammation.
Discussion
Dysregulated inflammatory responses provoked by even noninfectious stimuli is a hallmark of the inherited deficiency of NOX2. In this study, we identified a pivotal anti-inflammatory role of NOX2 in AMs in maintaining alveolar homeostasis. Utilizing genetic models, we showed that mice with NOX2-deleted AMs spontaneously acquire changes in the transcriptome and epigenome over the first few months of life. This remodeling is skewed toward increased chromatin accessibility of motifs for activating transcription factors and expression of genes associated with the inflammatory response, particularly in an activated AM subset distinguished by increased CD11b expression. This is accompanied by low-level inflammatory changes in the alveoli at baseline, including increased proinflammatory cytokines and YM1 and the presence of neutrophils. These changes are also associated with increased inflammation evoked by distant tissue inflammation. Remarkably, although NOX2 is also highly expressed in neutrophils and monocytes, loss of NOX2 activity restricted to AMs is sufficient to elicit similar findings.
Our data show that AM NOX2 attenuates inflammation during homeostasis, which also sets a threshold for responses to insults. Multiple contributing factors are most likely involved in this NOX2-dependent remodeling process. Given their anatomic location as airway sentinels, we speculate that the changes in NOX2-inactivated mice are, at least in part, linked to dysregulated AM activation triggered by ongoing exposure to inhaled materials and clearance of debris that leads to remodeling of the basal AM transcriptome and epigenome. Although our studies using germ-free mice eliminated the involvement of living microorganisms, NOX2 limited cell-intrinsic AM activation to ligands for TLR2 and TLR4, which are receptors that can detect sterile microbial ligands, DAMPs, and exogenous substances.44-46 This hyperresponsiveness could, in part, reflect impaired redox-mediated damping of NFκB transcriptional activity reported for NOX2-deleted lung macrophages.51 Amplified type I IFN-initiated signaling is proposed to contribute to dysregulated inflammation in NOX2 deficiency.43 However, our studies of IFNAR1-deleted CGD mice indicate that the enhanced expression of IFN-α signature genes in CD11bhigh AMs lacking NOX2 is a downstream consequence rather than a driver of proinflammatory changes in the lung. In addition, the altered resident macrophage phenotype in the absence of NOX2 was tissue specific, as these changes were seen in AMs but not in RPMs, which confirms and substantially extends prior work.31,50 Underlying mechanisms for this tissue specificity remain to be clarified, but may reflect epigenetic and transcriptional features of AMs that enable them to otherwise maintain a relatively tolerogenic state in their tissue niche. Finally, epithelial cells in the airways and alveoli produce and release hydrogen peroxide via the NADPH oxidases DUOX1 and DUOX2,52 but this process does not complement the loss of NOX2. We speculate that because deficiency of intracellular ROS is the likely culprit in deregulating AMs lacking NOX2, DUOX1/2-derived cell-extrinsic ROS are not effective because of the limitations on the amounts and/or availability for regulating intracellular macrophage processes.
NOX2-deleted AMs underwent epigenetic changes in the enhancer landscape, including increased accessibility of motifs for myeloid lineage enhancers and other transcription factors that increase responses of associated immune genes to stimulation. These acquired changes in NOX2-deleted AMs could be considered a type of learned or trained immunity, a facet of innate immunity that has attracted much recent attention.21,53 Epigenetic and functional alterations in AMs have previously been described after lung infection, either in residential AMs or in recently emigrated monocyte-derived AMs, although the underlying mechanisms that guide their reprograming remain incompletely understood.12,34,37,54-57 These changes are associated with either increased immunoreactivity of AMs or suppressive effects, depending on the inciting insult. In contrast, the changes in the epigenome of NOX2-deleted AMs develop under basal conditions in the first months of life.
This study provides new insights into the impact of NOX2 on macrophage function and the pathologic consequences of its absence for lung homeostasis and potentially other organs, which has clinical relevance for patients with CGD. Inflammatory lung disease is a poorly understood but serious complication that develops in many adult patients with CGD and can lead to reduced diffusion capacity and progressive hypoxia.3-8 Although difficult to treat, according to anecdotal reports, the inflammatory lung disease can be reversed by allogeneic hematopoietic stem cell transplantation.3 Its pathogenesis is poorly understood but does not always appear to be a consequence of prior lung infections. Computed tomography findings are variable and include interstitial or micronodular infiltrates.3-5 Biopsy studies in patients without intercurrent infections are very limited, but have shown variable findings of neutrophil infiltrates, bronchiolitis, or fibrosis.5 It is unknown whether any features of the lung disease that develops in NOX2-deleted mice overlap those in patients with CGD. However, we hypothesize that loss of NOX leads to hyperinflammatory resident AMs and an alveolar environment that predisposes to lung inflammation in patients with CGD as a consequence of direct or indirect events. This could affect outcomes, even with lung pathogens not associated with increased microbial virulence per se in CGD, such as viruses, Streptococcus pneumonia, or mycoplasma, and it could induce secondary lung inflammation with remote infections or injury. Whether the use of IFN-γ, which boosts phagocyte function and is currently used to reduce the incidence of infections in CGD,1 or the use of agents blocking IL-1 signaling could reduce inflammatory lung disease in CGD may be of interest for future study.
In summary, our results identify a new role for NOX2 to modulate alveolar homeostasis by regulating sentinel AMs and suppressing remodeling of their epigenome and transcriptome to a more proinflammatory state. Both cell-intrinsic factors and their anatomic niche contribute to the acquisition of these changes in the absence of NOX2. This information may shed light on the pathogenesis of chronic lung inflammation that can develop in patients with CGD and may be helpful for the development of treatments to promote protective lung immune responses in general.
Acknowledgments
The authors thank Hongjie Gu for assistance with statistical analysis, Steve Van Dyken and Gwen Randolph for helpful discussions, Jeanette Pingel for early work in generating the NCF2 f/f mice, and Tina McGrath for assistance with manuscript preparation; the Washington University in St. Louis (WUSTL) Genome Technology Core (in part funded by an award from the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital) and the WUSTL Mouse Embryonic Stem Cell Core (affiliated with the Siteman Cancer Center at the time the work was performed), for providing embryonic stem cell culture services; the Mouse Genetics Core at Washington University St. Louis School of Medicine for their support with animal production and care; the Andrew M. and Jane M. Bursky Center for Human Immunology and Immunotherapy Programs (CHiiPs); and the Molecular Microbiology Imaging Core.
This study was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant R01HL140837 (M.C.D.), National Institute of Dental and Craniofacial Research grant R01 DE082896 (J.B.), and National Institute of General Medical Sciences grant GM125504; (J.B.); the Children's Discovery Institute (CDI) of Washington University, and St. Louis Children’s Hospital (M.C.D.); a Leukemia and Lymphoma Scholar award (J.A.M.); and the Canadian Institutes of Health Research FDN 154329 (J.H.B). J.H.B. holds the Pitblado Chair in Cell Biology, and infrastructure for the J.H.B. laboratory was provided by a John Evans Leadership Fund grant from the Canadian Foundation for Innovation and the Ontario Innovation Trust.
Authorship
Contribution: S.B., R.A.I., J.D.R.M., W.L.B., J.J.A., J.H.B., J.B., J.A.M., and M.C.D designed the experiments; S.B., R.A.I., J.D.R.M., Y.L., G.H., W.L.B., J.J.A., and J.B. performed the experiments; S.B., R.A.I., W.Y., J.D.R.M., G.H., W.L..B, J.J.A, J.B., J.A.M., and M.C.D. analyzed the data; S.B., R.A.I., W.Y., Y.L., W.L.B., J.J.A., J.H.B., J.B., J.A.M., and M.C.D. interpreted the data; S.B., R.A.I., J.H.B., J.B., J.A.M., and M.C.D. contributed to writing the manuscript; and S.B. and M.C.D. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mary C. Dinauer, Washington University School of Medicine, PO Box 8208, 660 S Euclid Ave, St. Louis, MO 63110; e-mail: mdinauer@wustl.edu.
Data for RNA microarray, RNA-seq, and ATAC-seq were deposited and available in GEO (accession number GSE198778).
Requests for data sharing may be submitted to Mary C. Dinauer (mdinauer@wustl.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal