

Key Points
Disrupting the adult globin promoter alleviates promoter competition and reactivates fetal globin gene expression.

Abstract
The benign condition hereditary persistence of fetal hemoglobin (HPFH) is known to ameliorate symptoms of co-inherited β-hemoglobinopathies, such as sickle cell disease and β-thalassemia. The condition is sometimes associated with point mutations in the fetal globin promoters that disrupt the binding of the repressors BCL11A or ZBTB7A/LRF, which have been extensively studied. HPFH is also associated with a range of deletions within the β-globin locus that all reside downstream of the fetal HBG2 gene. These deletional forms of HPFH are poorly understood and are the focus of this study. Numerous different mechanisms have been proposed to explain how downstream deletions can boost the expression of the fetal globin genes, including the deletion of silencer elements, of genes encoding noncoding RNA, and bringing downstream enhancer elements into proximity with the fetal globin gene promoters. Here we systematically analyze the deletions associated with both HPFH and a related condition known as δβ-thalassemia and propose a unifying mechanism. In all cases where fetal globin is upregulated, the proximal adult β-globin (HBB) promoter is deleted. We use clustered regularly interspaced short palindromic repeats-mediated gene editing to delete or disrupt elements within the promoter and find that virtually all mutations that reduce ΗΒΒ promoter activity result in elevated fetal globin expression. These results fit with previous models where the fetal and adult globin genes compete for the distal locus control region and suggest that targeting the ΗΒΒ promoter might be explored to elevate fetal globin and reduce sickle globin expression as a treatment of β-hemoglobinopathies.
Introduction
Hemoglobin, the abundant oxygen carrying protein in red blood cells, is made up of 2 α-type and 2 β-type subunits. The β-subunits are encoded by a cluster of 5 developmentally regulated genes that depend on a large upstream enhancer, the locus control region (LCR), for their expression. The embryonic ɛ-globin gene (HBE) is expressed early in gestation, followed by 2 fetal γ-globin genes (HBG2 and HBG1). Around birth, the minor adult δ-globin gene (HBD) and major adult β-globin gene (HBB) are upregulated, with reciprocal silencing of fetal HBG genes. This process is referred to as hemoglobin gene switching.1
Individuals with pathogenic mutations in the adult HBB gene suffer from β-hemoglobinopathies such as sickle cell disease (SCD) and β-thalassemia, with symptoms manifesting after birth as the fetal HBG genes are silenced. Rare individuals who co-inherit a benign condition known as hereditary persistence of fetal hemoglobin (HPFH) have few or no symptoms of SCD or β-thalassemia as they maintain fetal γ-globin chain production throughout life and produce sufficient fetal hemoglobin (HbF) (α2γ2) to overcome the effects of defective or missing adult β-globin chains. This clinical observation supports the premise that genetic or pharmacological induction of fetal HBG gene expression postnatally will reduce the symptoms of those suffering from β-hemoglobinopathies. Accordingly, efforts to understand the mechanism of action of natural mutations affecting globin switching have been intense, especially now that it is possible to recapitulate these mutations using genome editing strategies.
HPFH is caused by either point mutations, very small deletions in the proximal promoters of the fetal HBG genes, or large kilobase-scale deletions that lie downstream of the first fetal gene, HBG2 (historically referred to as Gγ). The point mutations either disrupt the binding of transcriptional repressors BCL11A or ZBTB7A/LRF or create de novo binding sites for activators, such as GATA1, KLF1, or TAL1.2-5 The underlying mechanisms of deletional HPFH, on the other hand, are not well understood. At least 13 large deletions ranging from 12.5 kb to more than 80 kb in length are associated with HPFH (Figure 1). In addition, HbF levels have also been found to be elevated in a related clinical condition, termed δβ-thalassemia, that is associated with mild hypochromic, microcytic anemia, and partial amelioration of β-hemoglobinopathies.6 Like HPFH, large deletions downstream of the HBG genes are a hallmark of δβ-thalassemia. Some smaller deletions associated with β0-thalassemia have also been reported to be accompanied by modestly elevated HbF and milder clinical symptoms.7,8
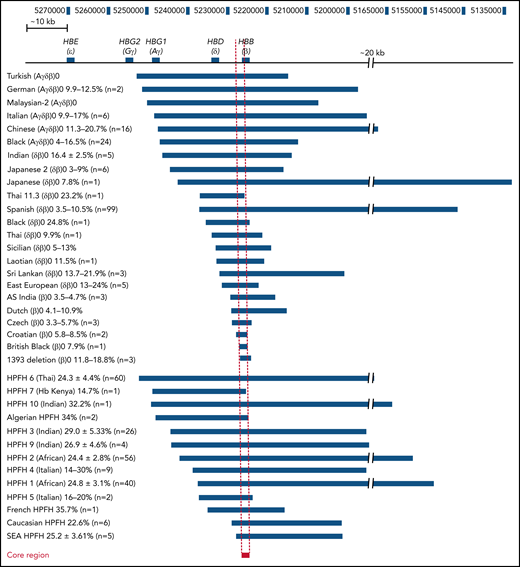
Comparison of the breakpoints of naturally occurring deletions associated with HBG upregulation. Naturally occurring deletions in both thalassemic and HPFH individuals with upregulated HbF levels are shown in relation to their location in the β-globin locus (numbers represent the human chromosome 11 coordinates from the hg38 genome assembly). The name and deletion type are indicated on the left, and the deletion associated with each condition is indicated by the blue bars. The percentage of HbF detected in patients with each condition who are heterozygous for the respective deletion is provided next to the condition name as well as the number of patients that this data were derived from. The core region has been indicated by a red bar with a dashed line indicating the corresponding region in all other deletions and the β-globin locus, which we have defined as the ∼1100 bp region between the 5′ end of the 1393 deletion and the 3′ end of the Croatian deletion (hg38 chromosome 11 coordinates 5 226 452 to 5 227 556). SEA, South East Asian.
Comparison of the breakpoints of naturally occurring deletions associated with HBG upregulation. Naturally occurring deletions in both thalassemic and HPFH individuals with upregulated HbF levels are shown in relation to their location in the β-globin locus (numbers represent the human chromosome 11 coordinates from the hg38 genome assembly). The name and deletion type are indicated on the left, and the deletion associated with each condition is indicated by the blue bars. The percentage of HbF detected in patients with each condition who are heterozygous for the respective deletion is provided next to the condition name as well as the number of patients that this data were derived from. The core region has been indicated by a red bar with a dashed line indicating the corresponding region in all other deletions and the β-globin locus, which we have defined as the ∼1100 bp region between the 5′ end of the 1393 deletion and the 3′ end of the Croatian deletion (hg38 chromosome 11 coordinates 5 226 452 to 5 227 556). SEA, South East Asian.
Several mechanisms have been proposed to explain how large deletions in the extended β-like globin locus may impair globin switching. These include the deletion of silencer elements, deletion of regions encoding various RNAs, and the juxtaposition of enhancer elements adjacent to the HBG gene promoters.9-12 Although all of these events are likely to quantitatively affect the expression of various globin genes, the mechanisms are diverse and do not provide a unifying framework for understanding deletional HPFH.13-16
Here we compiled a list of reported naturally occurring deletions that are accompanied by significantly increased (>2%) postnatal expression of HbF (Figure 1) and identified a core region of approximately 1100 bp that was deleted in these patients. Interestingly, this region included little more than the promoter of the adult HBB gene. This result is consistent with early hypotheses that the HBB gene has a role in deletional HPFH.8,17 We tested the effect of deleting the adult HBB gene promoter in cellular models. Modifications that impaired the activity of the HBB promoter interfered with fetal HBG gene silencing, resulting in increased production of HbF. Deletion or impairment of the HBB promoter also redirected looping of the LCR from the adult HBB gene and to the fetal HBG genes. These results support a model where the adult HBB promoter silences expression of the fetal HBG genes in part by competing for the LCR.8,18 Thus, this study provides a unified model for understanding why HbF levels are elevated in deletional HPFH.19 Our results also suggest that targeting the adult HBB promoter, perhaps in combination with mutations that disrupt repressors in the fetal HBG promoter, may produce synergistic effects for improved β-hemoglobinopathy gene therapy.
Materials and methods
HUDEP-2 cell culture
HUDEP-2 cells20 and HUDEP-2Δɛγδβ/GγAγ cells21 were cultured in StemSpan serum-free expansion medium supplemented with doxycycline (1 µg/mL) (Sigma-Aldrich, Melbourne, Australia), dexamethasone (10−6 M) (Sigma-Aldrich), erythropoietin (EPO) (3 IU/mL) (Peprotech, Sydney, Australia), stem cell factor (SCF) (50 ng/mL) (R&D Systems, Melbourne, Australia) and 1% penicillin-streptomycin-glutamine (PSG) (Gibco-Life Technologies, Sydney, Australia). Media was prepared fresh before each use. Cells were maintained at a concentration of 5 × 105 cells per mL at 37°C with 5% CO2.
Genome editing
For clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) genome editing, the pSpCas9(BB)-2A-GFP (PX458) plasmid was used. PX458 was a gift from F. Zhang (Addgene plasmid #48138).22 Sequences for the single-guide RNA (sgRNA) were designed using the optimized CRISPR online tool provided by the Zhang laboratory of the Massachusetts Institute of Technology, Boston: http://crispr.mit.edu (supplemental Table 1). sgRNAs were cloned into the PX458 plasmid and were subjected to Sanger sequencing to confirm the correct sequence (Australian Genome Research Facility, Melbourne, Australia).
Nucleofection
Cells were transfected for CRISPR-Cas9 gene editing by electroporation using a Neon Transfection kit (Thermo Fisher Scientific, Melbourne, Australia). 5 × 105 cells were used per electroporation and were resuspended in 9 µL of Buffer T (supplied with Neon Transfection System 10 µL Kit). A master mix was added to the cells in T buffer containing the PX458 plasmids and guide/s (0.8 µL of 2 mg/mL PX458 plus sgRNA single guide or a mixture of 0.4 µL of 2 mg/mL PX458 plus sgRNA of each guide if 2 guides were used). For some deletion strategies, a single-stranded oligonucleotide donor template (ssOND) was included to encourage homology directed repair (0.2 µL of 50 µM ssOND) (supplemental Table 2). Nucleofections were performed in triplicate. Each nucleofection involved 1 pulse of 1100 to 1300 V for 20 to 40 milliseconds. Cells were cultured for 48 to 72 hours in normal culture media lacking PSG. Samples were subjected to fluorescence-activated cell sorting on the BD Aria III cell sorter (BD Biosciences, Sydney, Australia) into a pooled population selecting for Kusabira-Orange–positive (live) and GFP+ cells. Seventy-two to 96 hours later, a second round of fluorescence-activated cell sorting was performed, sorting into single-cell populations selecting for Kusabira-Orange–positive and GFP− (Mark Wainwright Analytical Centre, Sydney, Australia).
CD34+ cell genome editing and culture
Circulating granulocyte colony-stimulating factor–mobilized human mononuclear cells were obtained from deidentified healthy adult donors (Key Biologics, Lifeblood, Memphis, TN). CD34+ hematopoietic stem and progenitor cells (HSPCs) were thawed on day 0 into X-VIVO 15 (Lonza, 04-418Q, Morristown, TN) supplemented with 100 ng/mL human SCF, 100 ng/mL human thrombopoietin (TPO), and 100 ng/mL recombinant human Flt3-ligand. HSPCs were nucelofected with Cas9 ribonucleoprotein complex (RNP) 24 hours after thawing; purified recombinant Cas9 protein was obtained from Berkeley Macrolabs (Berkeley, CA). Chemically modified sgRNAs were synthesized by Synthego (Menlo Park, CA). RNPs were formed by incubating 50 pmol of Cas9 and 50 pmol sgRNA in 5 µL of 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Thermo Fisher Scientific, catalog number 15630080), 150 mM NaCl (Thermo Fisher Scientific, catalog number 9759) for 20 to 30 minutes. 2 × 105 CD34+ cells were washed with phosphate-buffered saline (PBS) twice, resuspended in 5 µL T buffer (Thermo Fisher Scientific), mixed with RNP complex, and electroporated with 1600 V, 3 pulses of 10 milliseconds, using a Neon Transfection System (Thermo Fisher Scientific, catalog number MPK5000). As controls, cells were electroporated with Cas9 and nontargeting RNP complex. Twenty-four hours after electroporation, HSPCs were transferred into erythroid differentiation medium. Erythroid differentiation of CD34+ cells was performed using a 3-phase protocol. Phase 1 (days 1 to 7): Iscove's modified Dulbecco's medium with 2% human blood type AB plasma, 3% human AB serum, 1% penicillin/streptomycin, 3 units per mL heparin, 10 µg/mL insulin, 3 units per mL EPO, 200 µg/mL holo-transferrin, 10 ng/mL SCF, and 1 ng/mL interleukin IL-3. Phase 2 (days 8 to 14): phase 1 medium without IL-3. Phase 3 (days 15 to 21): the holo-transferrin concentration was increased to 1 mg/mL and SCF withdrawn.
Gene expression studies
Total RNA was extracted from cells using TRI Reagent (Sigma-Aldrich) and the RNeasy Mini Kit (Qiagen, Melbourne, Australia). Samples were DNase treated with an Ambion DNA-free kit using the manufacturer’s protocol. One microgram of RNA per sample was used for complimentary DNA synthesis using Super Script VILO Master Mix kit (Thermo Fisher Scientific). Reactions were primed with random hexamers. For each sample, a negative control (RT−) was made lacking Super Script VILO Master Mix. Quantitative real-time polymerase chain reaction (qPCR) was performed using PowerUp SYBR Green Master Mix (final volume 10 μL) and was run with the default cycle parameters of ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). The expression levels of target genes were normalized to 18S ribosomal RNA. Primers used for qPCR are listed in supplemental Table 3.
PCR screening of CRISPR-Cas9 modified clones
PCR and Sanger sequencing were used to select clonal cell populations with desired mutations following CRISPR-Cas9 genome editing. Genomic DNA (gDNA) was extracted from cells using QuickExtract DNA Extraction Solution (Gene Target Solutions, Sydney, Australia) following the manufacturer’s protocol. PCR was performed using Q5 High-Fidelity DNA Polymerase (New England BioLabs, Melbourne, Australia) according to the manufacturer’s instructions. Primers used to screen clones can be found in supplemental Table 4. Sanger Sequencing of PCR products was used to confirm modifications (Australian Genome Research Facility).
Copy number analysis
Copy number analysis was performed on gDNA extracts prepared using the Pure Link Genomic DNA mini kit. gDNA was made up to a concentration of 1 ng/μl, and qPCR was performed for the target genes using a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). qPCR was performed against the region of interest (the HBB promoter or HBB gene) and reference gene hypersensitive site-2 of the locus control region (HS-2). Copy number was performed by looking at the relative abundance of the targeted region against the unedited reference regions (HS-2). This was then compared with the wild-type (WT) clonal cells. A list of primers used for copy number analysis is shown in supplemental Table 5.
Hemoglobin HPLC
5 × 106 cells were harvested, washed with PBS, and lysed in 50 µL of 0.1% sodium dodecyl sulfate. The cell lysate was centrifuged, and the clear cell lysate was filtered using Ultrafree-MC devices Hydrophilic Polyvinylidene Fluoride (PVDF, 0.45 µM). Twenty microliters of filtered cell lysate was mixed with 80 µL of buffer A (20 mM Bis-Tri, pH 6.8) with 2 mM NaCN and incubated for 30 minutes on ice. HUDEP-2 samples were injected into PolyCat A column (3.54CT0315, PolyLC) fitted to a Bio inert high-performance liquid chromatography (HPLC) system (1260 Infinity Agilent Technologies HPLC) (University of New South Wales Recombinant Products Facility, Sydney, Australia). Hemoglobins were eluted from the PolyCat column at a flow rate of 1.2 mL/m during a gradient of 2% to 25% buffer B (20 mM Bis-Tris, 200 mM NaCl, pH 6.9) in buffer A for 10 minutes followed by a gradient of 25% to 100% buffer B in buffer A for 10 to 22 minutes. Absorbance measurements for hemoglobin proteins were detected at 415 nm. Quantification of hemoglobin was assessed by determining the area under the HbF, HbA, HbA2, and free α-globin peaks. For each sample of HUDEP-2, WT sample lysates were used as a reference for the retention times of HbF and HbA.
For the CD34+ HSPCs HPLC was performed using ion-exchange and reverse-phase columns on a Prominence HPLC System (Shimadzu Corporation, Columbia, MD). Proteins eluted from the column were identified at 220 and 415 nm with a diode array detector. The relative amounts of hemoglobins were calculated from the area under the 415-nm peak and normalized based on the dimethyl sulfoxide control. Percent HbF = [HbF/(HbA + HbF)] × 100; % γ-globin = [(Gγ-chain + Aγ-chain)/β-like chains (β + Gγ + Aγ)] × 100.
Western blots
CD34+ HSPC cells were lysed on day 15 of differentiation. Western blot was performed as described with FBXO11 protein.23 Antibodies used can be found in supplemental Table 6.
Flow cytometry
CD34+ HSPC erythroblast maturation was monitored by immuno-flow cytometry for the cell surface markers CD235a, CD49d, CD71, and Band3. 5 × 105 cells were collected on culture day 15, spun down at 500 g/m for 3 minutes, and then resuspended in 0.1% bovine serum albumin PBS containing antibody CD235a (1:100), CD49d (1:20), and Band3 (1:100) (supplemental Table 7). The cells were kept in the dark for 20 minutes, washed once with 0.1% bovine serum albumin PBS, and then analyzed by flow cytometry (BD Biosciences Fortessa instruments, Franklin Lakes, NJ). CD235a-FITC was included as a negative control for CD49d and Band3. Gating strategy can be found in supplemental Figure 5C. Data were analyzed using FlowJo software, version 10.7.1 (FlowJo).
Capture-C
HUDEP-2 cells were differentiated for 5 days in Iscove's modified Dulbecco's medium supplemented with 330 mg/mL holo-transferrin (Sigma-Aldrich), 10 mg/mL insulin (Sigma-Aldrich), 2 units/mL heparin (Sigma-Aldrich), 5% fetal bovine serum, EPO (3 IU/mL), 1 mg/mL doxycycline, and 1% PSG. Cell. Capture-C was performed as described using NlaIII enzyme for gDNA digestion.24 Biotin-labeled DNA probes are listed in supplemental Table 8.
Statistical analysis
The mean and standard error of the mean (SEM) were calculated and 2-tailed Mann-Whitney U tests performed to assess the significance of differences in expression between the various mutants and WT clones for all datasets using GraphPad Prism, version 8, for Windows (GraphPad Software).
Results
The core common region found in deletions associated with elevated HbF contains the adult HBB gene promoter
We tabulated known deletions associated with elevated (>2%) HbF, including heterozygous individuals formally identified as exhibiting either HPFH, δβ-thalassemia, or β0-thalassemia with increased fetal globin. HPFH-associated deletions included HPFH6 (Thai),25 HPFH7 (Kenyan),26 HPFH10 (Indian),27 Algerian HPFH,28 HPFH3 (Indian),29 HPFH9 (Indian),27 HPFH2 (African),30 HPFH4 (Italian),31 HPFH1 (African),30 HPFH5 (Italian),32 French HPFH,28 Caucasian HPFH,28 and South East Asian HPFH.29 δβ-thalassemia- or β0-thalassemia–associated deletions included Turkish (Aγδβ)0,33 German (Aγδβ)0,34 Malaysian-2 (Aγδβ)0,35 Italian (Aγδβ)0,36 Chinese (Aγδβ)0,37 Black (Aγδβ)0,33 Indian (δβ)0,27 Japanese 2 (δβ)0,38 Japanese (δβ)0,39 Thai 11.3 (δβ)0,40 Spanish (δβ)0,41 Black (δβ)0,42 Thai (δβ)0,43 Sicilian (δβ)0,44 Laotian (δβ)0,45 Sri Lankan (δβ)0,9 East European (δβ)0,46 AS Indian (β)0,47 Dutch (β)0,48 Czech (β)0,49 Croatian (β)0,50 British Black (β)0,51 and 1393 deletion (β)0.7 Although a few other deletions in the β-globin locus have been reported in patients,8,52 we did not include these or the common −619 bp (As Indian) β-thalassemia deletion53,54 that removes the 3′ end of the HBB gene because it is not associated with HbF elevation.55 We also did not include the Corfu deletion, which is associated with upregulated HbF in patients but does not appear to be directly causative in cell models.13 The alignments identified a ∼1100 bp core region, which we have defined as the region between the 5′ breakpoint of the 1393 deletion and the 3′ breakpoint of the Croatian deletion (Figure 1). This region spanned the proximal promoter and the first exon of the HBB gene and harbored transcription factor binding sites for Krüppel-like factor 1 (KLF1), nuclear transcription factor Y (NFY), and TATA box binding protein (TBP) along with the beginning of the HBB coding region.56
Targeted deletion of the adult HBB promoter, but not of the gene itself, significantly increases fetal HBG gene expression
The finding that deletions of the adult HBB promoter correlated with impaired fetal HBG silencing prompted us to test whether deleting the HBB promoter in a cellular model was sufficient to boost fetal HBG production. We used CRISPR-Cas9–mediated mutagenesis in HUDEP-2 erythroid cells to delete the HBB promoter and also generated a control deletion that removed the HBB gene exons but left its promoter intact. Despite including 2 sgRNAs and a ssOND spanning the 2 end points, in all cases the cells did not use the ssOND for repair and instead used non-homologous end joining to fuse together the cut ends. We therefore selected clonal populations harboring identical deletions of the promoter or transcribed region for further analysis (Figure 2A). To confirm that deletions had occurred on both alleles and that the clones were homozygous for the deletions, copy number analysis was performed (supplemental Figure 1).
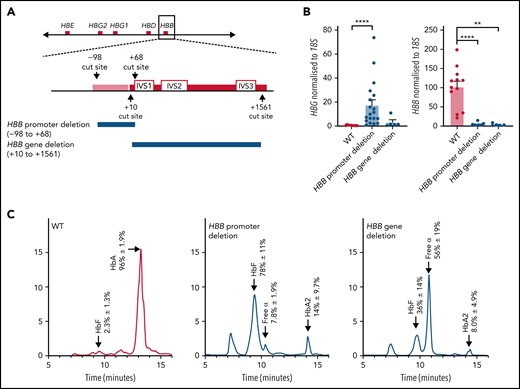
Deletion of the HBB promoter leads to increased expression of HBG and increased HbF in HUDEP-2 cells, whereas deletion of the HBB coding region does not significantly affect HBG expression. (A) The CRISPR-Cas9 sgRNA cut sites for the introduction of the HBB promoter deletion (above) and the HBB gene deletion (below). The promoter (pink bar) and gene (red bar) are indicated with the intron (IVS) locations provided. (B) HBG and HBB mRNA levels in WT HUDEP-2 clonally sorted (n = 12), HBB promoter deletion (n = 19), and HBB gene deletion clonal populations (n = 5) (mean plus or minus SEM). Significance was determined by a Mann-Whitney U test (**P < .01, ****P < .0001). (C) Hemoglobin protein levels as determined by HPLC for clonally sorted WT HUDEP-2 (n = 3), HBB promoter deletion (n = 3), and HBB gene deletion (n = 2), with peaks for HbF, HbA, HbA2, and free α-globin chains indicated by the arrows above the peaks. A mean trace is shown for each with mean percent value plus or minus SEM for each hemoglobin peak.
Deletion of the HBB promoter leads to increased expression of HBG and increased HbF in HUDEP-2 cells, whereas deletion of the HBB coding region does not significantly affect HBG expression. (A) The CRISPR-Cas9 sgRNA cut sites for the introduction of the HBB promoter deletion (above) and the HBB gene deletion (below). The promoter (pink bar) and gene (red bar) are indicated with the intron (IVS) locations provided. (B) HBG and HBB mRNA levels in WT HUDEP-2 clonally sorted (n = 12), HBB promoter deletion (n = 19), and HBB gene deletion clonal populations (n = 5) (mean plus or minus SEM). Significance was determined by a Mann-Whitney U test (**P < .01, ****P < .0001). (C) Hemoglobin protein levels as determined by HPLC for clonally sorted WT HUDEP-2 (n = 3), HBB promoter deletion (n = 3), and HBB gene deletion (n = 2), with peaks for HbF, HbA, HbA2, and free α-globin chains indicated by the arrows above the peaks. A mean trace is shown for each with mean percent value plus or minus SEM for each hemoglobin peak.
HUDEP-2 clones containing the adult HBB promoter deletion produced more fetal HBG messenger (mRNA) and HbF protein than WT HUDEP-2 clones and showed significantly reduced HBB transcript and protein levels (Figure 2B-C). In contrast, deletion of the HBB transcribed region resulted in only a minor (nonsignificant) upregulation of HBG mRNA and a modest increase in percent HbF protein (Figure 2B-C). HBB mRNA and protein were essentially undetectable, as expected (Figure 2B-C). Adult hemoglobin HbA2 was also elevated with both the HBB promoter and gene deletion, as has previously been observed with reduced HBB expression in β-thalassemia patients.7,48-50 (Figure 2C). Additionally, clones carrying the adult HBB gene deletion showed an increase in free α-globin chains (Figure 2C). Our results suggest that deleting the coding region of the HBB gene itself has limited effects on increasing HbF levels, but removing the promoter is sufficient to impair fetal globin silencing and generate robust fetal HBG expression and protein production.
We also used CRISPR-Cas9–mediated mutagenesis to delete the HBB promoter in HUDEP-2Δɛγδβ/GγAγ cells, which lack the entire β-like globin gene cluster on one chromosome (Δɛγδβ) and contain an in-frame HBG2-HBG1 (GγAγ) fusion gene on the other chromosome.21 Thus, these cells contain a single HBG gene linked to HBB in the native chromosomal context. HUDEP- 2Δɛγδβ/GγAγ clones containing the HBB promoter deletion had significantly higher levels of HBG mRNA and significantly lower levels of HBB mRNA than HUDEP- 2Δɛγδβ/GγAγ control clones with an intact HBB gene (supplemental Figure 2), suggesting that part, if not all, of the effect of the HBB promoter deletion on γ-globin gene expression occurs in cis.
Specifically disrupting known transcription factor binding sites in the adult HBB promoter is sufficient to elevate fetal HBG expression
To fine map the adult HBB promoter regions responsible for inhibiting fetal HBG expression, we sought to test the effect of disabling the promoter via very small deletions, targeted at specific transcription factor binding sites. We designed sgRNAs to cut at these sites (Figure 3A)56 and selected clones in which non-homologous end joining had led to on-target deletions of >1 base pair. The 5 most common deletions recovered from editing with each sgRNA are listed in supplemental Figure 3. Small deletions generated using each of the 4 guides individually led to elevation of HBG mRNA and reduction of HBB mRNA (Figure 3B). More detailed analysis of individual clones was carried out to assess which specific transcription factor binding site or combination of sites had been disrupted. Interestingly, all mutations that disrupted the HBB promoter led to an upregulation of fetal HBG expression except for deletion of the NFY binding site alone (Figure 3C). All mutations that disrupted the HBB promoter led to a decrease in HBB expression, except for deletion of the KLF1 binding sites (Figure 3C). Deletion of the TATA box also caused elevated HbF protein levels (Figure 3D). These data suggest that a functional adult HBB promoter, rather than any specific transcription factor binding site, is essential for proper fetal HBG silencing, consistent with our hypothesis that disrupting HBB promoter activity is sufficient to significantly activate HBG expression.
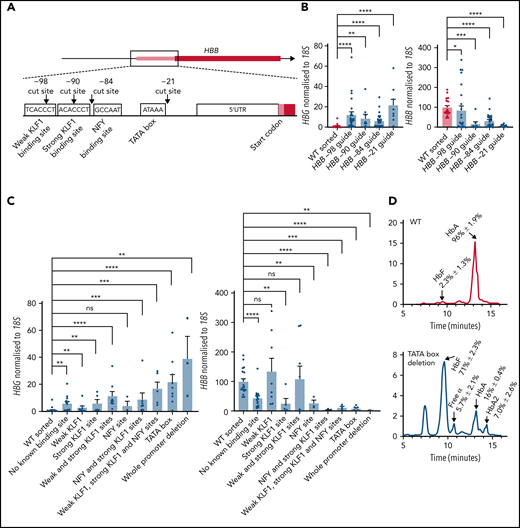
Disruption of known transcription factor binding sites or intervening sequence in the adult HBB promoter is sufficient to elevate fetal HBG expression. (A) CRISPR-Cas9 sgRNA cut sites (black arrows) and transcription factor binding sites (white boxes). (B) HBG and HBB mRNA levels in WT HUDEP-2 (n = 18) and cells cut with −21 (n = 9), −84 (n = 26), −90 (n = 7), and −98 (n = 27) guides that resulted in >1bp deletions. (C) HBG and HBB mRNA levels in WT HUDEP-2 (n = 18) and cells cut with the −21, −84, −90, and −98 guides: no known binding site (n = 15), weak KLF1 site (n = 8), strong KLF1 site (n = 5), 2 KLF1 sites (n = 8), NFY site (n = 3), NFY and strong KLF1 sites (n = 7), 2 KLF1 sites and NFY site (n = 5), TATA box (n = 9), and whole HBB promoter (n = 3). For panels B-C, values are relative to a mean WT level of 1 and 100 for HBG and HBB, respectively (mean plus or minus SEM). Significance determined by a Mann-Whitney U test (ns P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001). (D) Hemoglobin protein levels (as determined by HPLC) for WT HUDEP-2 (n = 3) and TATA box mutation (n = 3), with peaks for HbF, HbA and HbA2 indicated. A mean trace is shown with mean percent value plus or minus SEM for each peak.
Disruption of known transcription factor binding sites or intervening sequence in the adult HBB promoter is sufficient to elevate fetal HBG expression. (A) CRISPR-Cas9 sgRNA cut sites (black arrows) and transcription factor binding sites (white boxes). (B) HBG and HBB mRNA levels in WT HUDEP-2 (n = 18) and cells cut with −21 (n = 9), −84 (n = 26), −90 (n = 7), and −98 (n = 27) guides that resulted in >1bp deletions. (C) HBG and HBB mRNA levels in WT HUDEP-2 (n = 18) and cells cut with the −21, −84, −90, and −98 guides: no known binding site (n = 15), weak KLF1 site (n = 8), strong KLF1 site (n = 5), 2 KLF1 sites (n = 8), NFY site (n = 3), NFY and strong KLF1 sites (n = 7), 2 KLF1 sites and NFY site (n = 5), TATA box (n = 9), and whole HBB promoter (n = 3). For panels B-C, values are relative to a mean WT level of 1 and 100 for HBG and HBB, respectively (mean plus or minus SEM). Significance determined by a Mann-Whitney U test (ns P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001). (D) Hemoglobin protein levels (as determined by HPLC) for WT HUDEP-2 (n = 3) and TATA box mutation (n = 3), with peaks for HbF, HbA and HbA2 indicated. A mean trace is shown with mean percent value plus or minus SEM for each peak.
In CD34+ primary cells, adult HBB promoter deletions also cause increased fetal HBG expression
Experiments were also carried out in primary human erythroblasts derived from in vitro culture of peripheral blood CD34+ HSPCs. We electroporated normal donor CD34+ cells with RNP complexes containing Cas9 complexed with sgRNAs targeting the −98 or +68 region of the adult HBB gene (Figure 2A), either individually or in pairs, followed by in vitro culture to support erythroid differentiation. As a positive control for HBG gene induction, we used an RNP containing sgRNA targeting the BCL11A repressor binding site at positions −118 to −114 of the fetal HBG promoter. Indels in this region recapitulate a naturally occurring form of HPFH associated with ∼20% to 30% HbF.57,58 The 5 most common indels and their frequencies in each population are shown in supplemental Figure 4. In general, a single sgRNA-Cas9 RNP generated small deletions at the target site, whereas combined RNPs targeting the −98 or +68 sites in HBB resulted in deletion of the intervening region consisting of the entire promoter.
HBG mRNA (Figure 4A), γ-globin protein (Figure 4B), individual HbF-expressing cells (F-cells) (Figure 4C), and HbF levels (Figure 4D) increased significantly in all 3 populations, with the highest increase occurring with the entire adult HBB promoter deletion. As expected, the level of HBB expression also decreased significantly in all 3 populations, with both guides used together generating the largest reduction in expression (Figure 4A). Some residual HBB expression was still observed in these pools because the efficiency of editing never achieved 100%. Disruption of the adult HBB promoter did not alter the morphology of late-stage erythroblasts, indicating that differentiation was unaffected (supplemental Figure 5A-B). Taken together, these results confirm that disruption of the adult HBB promoter is associated with an impairment of fetal HBG silencing and upregulation of HBG expression in both HUDEP-2 cells and primary CD34+ cell–derived erythroblasts.
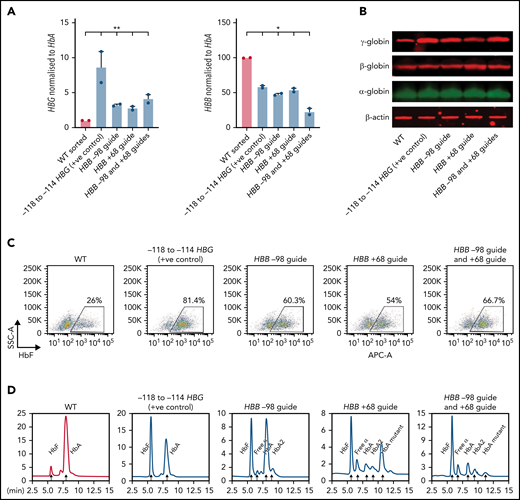
Deletion of the HBB promoter leads to increased expression of HBG and increased HbF in CD34+ HSPCs. (A) HBB and HBG mRNA levels measured by qPCR in indicated sgRNA/s combination edited erythroid cells derived from CD34+ cells (n = 2) (mean plus or minus SEM). (B) Western blot analysis of γ-globin, β-globin, and α-globin and β-actin at day 15 differentiated erythroid cells derived from indicated sgRNA/s combination edited CD34+ cells. (C) Representative flow cytometric analysis of day 15 erythroid cells derived from indicated sgRNA/s combination edited CD34 cells stained with anti-HbF antibody. (D) Hemoglobin protein levels as determined by HPLC for day 15 erythroid cells derived from indicated sgRNA/s combination edited CD34+ cells. Peaks for each hemoglobin types or variants are indicated by the arrows above the peaks. SSC-A, flow cytometry side scatter.
Deletion of the HBB promoter leads to increased expression of HBG and increased HbF in CD34+ HSPCs. (A) HBB and HBG mRNA levels measured by qPCR in indicated sgRNA/s combination edited erythroid cells derived from CD34+ cells (n = 2) (mean plus or minus SEM). (B) Western blot analysis of γ-globin, β-globin, and α-globin and β-actin at day 15 differentiated erythroid cells derived from indicated sgRNA/s combination edited CD34+ cells. (C) Representative flow cytometric analysis of day 15 erythroid cells derived from indicated sgRNA/s combination edited CD34 cells stained with anti-HbF antibody. (D) Hemoglobin protein levels as determined by HPLC for day 15 erythroid cells derived from indicated sgRNA/s combination edited CD34+ cells. Peaks for each hemoglobin types or variants are indicated by the arrows above the peaks. SSC-A, flow cytometry side scatter.
Deletion of the adult HBB promoter or disruption of the TATA box switches the association of globin genes with the locus control region
The observation that a functional adult HBB promoter is required for proper fetal HBG silencing fits best with a model where the adult HBB promoter competes with the fetal HBG promoters for the upstream globin super enhancer or LCR. To determine whether contact with the LCR is affected by adult HBB promoter deletions, we carried out Capture-C experiments on WT HUDEP-2 cells and 2 of the mutant lines from Figures 2 and 3, including those containing the full proximal HBB promoter deletion and those in which the TATA box had been disabled. Capture-C was used to assess the interaction of DNase hypersensitive site 3 (HS3) of the LCR with the fetal HBG or adult HBB genes. In WT HUDEP-2 cells, a strong association between the LCR and the adult HBB gene was evident, as seen in the strong peaks (Figure 5A). In contrast, when the adult HBB promoter was disabled by deletion or by disruption of the TATA box, the pattern changed dramatically, and the fetal HBG genes then associated more strongly with the LCR, whereas the adult HBB gene associated less strongly with the LCR (Figure 5A; supplemental Figure 6). BGLT3 and HBBP1 pseudogenes, which both lie just downstream of HBG1 within the β-globin locus, also associated more strongly with the LCR in HUDEP-2 cell clones with the HBB promoter deletion or TATA box disruption, whereas the interaction of HBD with the LCR was unaltered by these mutations (supplemental Figure 6). Interestingly, the effect is somewhat more pronounced in the lines where only the TATA box is disrupted (supplemental Figure 6), suggesting that minor changes that disrupt promoter function, rather than larger deletions that may alter spacing in the locus, are sufficient to reverse promoter competition and allow HBG to contact the LCR and be more strongly expressed.
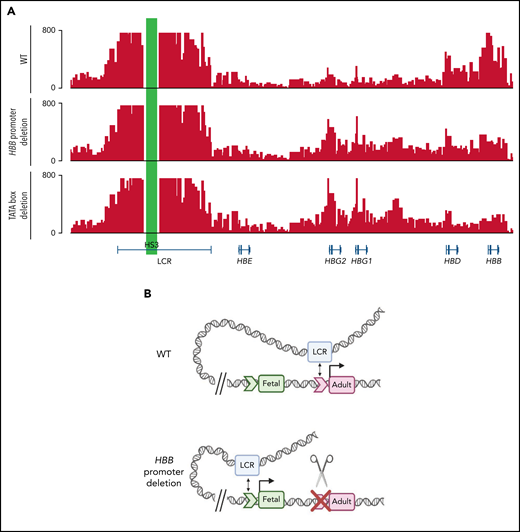
Deletion of the HBB promoter or disruption of the TATA box alters association of globin genes with the LCR. (A) Capture-C profiles of HS3 in clonal populations of WT HUDEP-2 cells (n = 2), HBB promoter deletion cells (n = 2), and TATA box mutation cells (n = 2). The positions of the β-globin locus genes are indicated below the profiles. (B) Proposed model of the molecular mechanism underlying the reversal of globin switching following the removal or disruption of the HBB promoter. In WT adult erythroid cells, the LCR loops to the HBB gene and promotes the expression of the HBB gene. When the promoter is unavailable to the LCR, the LCR loops to the promoter of another β-like globin gene, most commonly the HBG gene, and promotes expression. Created with BioRender.com.
Deletion of the HBB promoter or disruption of the TATA box alters association of globin genes with the LCR. (A) Capture-C profiles of HS3 in clonal populations of WT HUDEP-2 cells (n = 2), HBB promoter deletion cells (n = 2), and TATA box mutation cells (n = 2). The positions of the β-globin locus genes are indicated below the profiles. (B) Proposed model of the molecular mechanism underlying the reversal of globin switching following the removal or disruption of the HBB promoter. In WT adult erythroid cells, the LCR loops to the HBB gene and promotes the expression of the HBB gene. When the promoter is unavailable to the LCR, the LCR loops to the promoter of another β-like globin gene, most commonly the HBG gene, and promotes expression. Created with BioRender.com.
Discussion
HPFH has been studied for several decades and is known to be associated either with specific mutations in the fetal HBG promoters or with large deletions downstream of the fetal gene HBG2. Interestingly, other deletions in the locus that are associated with different but well-defined clinical conditions, such as δβ-thalassemia and β-thalassemia, also exhibit elevated HbF levels of varying magnitude.7,8 Here, rather than focusing on only the HPFH individuals with a set clinical definition and high HbF, we have considered as many patients as possible where HbF is detectably elevated. Our analysis of deletions associated with increased HbF clearly identifies a common deleted region that contains the adult HBB proximal promoter. Our conclusion that loss of the HBB promoter stimulates HBG upregulation is supported by the 619 bp (As Indian) β-thalassemia deletion,53,54 which removes the 3′ end of the HBB gene but leaves its promoter intact and is not associated with elevated HbF. Our data indicate that disruption of the adult HBB promoter leads to upregulation of the fetal HBG genes through the promoter competition model that has been previously postulated8 and is supported by observations in patients with homozygous deletions of the HBB promoter50 but has not been demonstrated experimentally until now. Multiple different small deletions in the adult HBB promoter were generated to determine whether any key binding site was particularly important, but it appears that any mutation that caused reduced HBB promoter activity led to an increase in HBG promoter activity, with the exception of deleting the NFY binding site. Interestingly, some patients with point mutations within the HBB promoter have been reported to have elevated HbF levels,8,59-63 correlating with our findings in cell models. Although in most cases we saw a reciprocal increase in HBG and decrease in HBB with these promoter mutations in HUDEP-2 cells, no significant decreases in HBB expression were seen with the weak KLF1 or weak and strong KLF1 site mutations. This was unexpected given that the strong KLF1 site alone both increased HBG and decreased HBB expression. It is also perhaps unexpected that mutations in the binding site for the basal transcription factor TBP were also effective in derepressing HBG because basal factors are not usually viewed as having fundamental roles in higher order chromatin architecture and looping. However, it is possible TBP does have such roles or that the overall configuration of the active HBB promoter is required for LCR competition.
The reciprocal effects of HBB and HBG promoter activity fit well with the model where both promoters compete for the LCR19,64,65 (Figure 5B). Capture-C analysis confirmed that association of the WT HBB gene with the LCR is markedly impaired if the HBB promoter is deleted or even if just the TATA box is disrupted. In these cases, the HBG gene outcompetes the HBB gene for binding to the LCR and is more highly expressed, providing a clear example that promoter competition plays a key role in normal globin switching in humans, as outlined in a model that was first proposed by Choi and Engel more than thirty years ago under the term “dueling lollipops”.18 Their original studies on the chicken β-globin locus suggested that early in development, the enhancer might align with the embryonic promoter, creating a loop, or “lollipop,” and then later in development a mutually exclusive loop would form as the adult promoter bound the enhancer. More recent studies in various experimental systems with marked reporter genes has provided additional evidence for promoter-enhancer competition models.66-69
Our results provide a unifying explanation for deletional HPFH and related deletions associated with other clinical conditions associated with elevated HbF levels. However, availability of the adult HBB promoter for LCR association is not the only determinant of HbF levels. Detailed analysis of specific HPFH deletions has implicated other regions within the extended β-globin locus, such as the HBBP1 and BGLT3 genes, and alterations in the spacing of various putative enhancers that may be brought into the proximity of the fetal HBG genes.11,12,24,70 The deletion of silencer elements, deletion of regions encoding RNAs, and the juxtaposition of enhancer elements are likely to contribute to the variations in HbF levels associated with particular deletions.9-12 However, we present experimental data showing that disruption of the adult HBB promoter is a major unifying feature that likely contributes to HbF induction in most or all forms deletional HPFH.
Our work illustrates an important general principle in the field of gene regulation. Namely, that disrupting the expression of one gene may influence the expression of adjacent genes. Here, it is remarkable that disabling the adult HBB promoter, irrespective of the precise mutation or which transcription factor binding site is affected, causes the upregulation of the nearby fetal HBG genes, a clear demonstration of action at a distance and further physiologically compelling evidence for promoter competition in the enhancer-promoter looping model.
Acknowledgments
PX458 plasmid was a gift from Feng Zhang, Broad Institute (Addgene #48138). HUDEP-2 cells were kindly provided by Ryo Kurita and Yukio Nakamura, RIKEN BioResource Center, Japan. The authors acknowledge Helene Lebhar and the University of New South Wales (UNSW) Sydney Recombinant Products Facility for HPLC assistance, and Christopher Brownlee and Emma Johansson Beves and the Biological Resources Imaging Laboratory (Mark Wainwright Analytical Centre, UNSW Sydney) for assistance with flow cytometry.
This work was supported by funding from the Australian National Health and Medical Research Council to M.C. (APP1164920). S.K.T., L.C.L., and G.E.M. were supported by Australian Government Research Training Program postgraduate scholarships. G.A.B. is supported by National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI) grant HL119479. M.J.W. is supported by NIH NHLBI grant P01HL053749 and NIH National Institute of Diabetes and Digestive and Kidney Diseases grant R24 DK106766. K.G.R.Q. is a UNSW Sydney Scientia Associate Professor.
Authorship
Contribution: S.K.T., G.E.M., G.A.B., M.J.W., K.G.R.Q., and M.C. designed the study and experiments; S.K.T. performed the genome editing and analysis of the HUDEP-2 cells; R.F. performed the genome editing and analysis of the CD34+ cells; P.H. performed the Capture-C experiments and analysis; L.C.L. provided essential experimental reagents; S.K.T., G.A.B., M.J.W., K.G.R.Q., and M.C. wrote the manuscript; G.A.B., M.J.W., K.G.R.Q., and M.C. supervised the study; and all authors have read and approved the final version of the manuscript, and have agreed both to be personally accountable for the author's own contributions and to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved and the resolution documented in the literature.
Conflict-of-interest disclosure: M.J.W. is on advisory boards for Novartis, Forma Therapeutics and Cellarity Inc. The remaining authors declare no competing financial interests.
Correspondence: Merlin Crossley, School of Biotechnology and Biomolecular Sciences, University of New South Wales, High Street, Kensington, NSW 2052, Australia; e-mail: m.crossley@unsw.edu.au.
The Capture-C data have been deposited to the Gene Expression Omnibus (GEO) database (GSE194064).
Requests for data sharing may be submitted to Merlin Crossley (m.crossley@unsw.edu.au).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal