


Abstract
Reactive aldehydes are potent genotoxins that threaten the integrity of hematopoietic stem cells and blood production. To protect against aldehydes, mammals have evolved a family of enzymes to detoxify aldehydes, and the Fanconi anemia DNA repair pathway to process aldehyde-induced DNA damage. Loss of either protection mechanisms in humans results in defective hematopoiesis and predisposition to leukemia. This review will focus on the impact of genotoxic aldehydes on hematopoiesis, the sources of endogenous aldehydes, and potential novel protective pathways.
Introduction
The human body turns over approximately 280 billion blood cells every day.1 This voracious demand is met by as few as 50 000 hematopoietic stem cells actively making mature blood cells at any one time.2 To maintain blood production, the protection of the hematopoietic stem cell (HSC) genome from DNA damage is an essential requirement to ensure HSC integrity. This is highlighted by diseases caused by inherited defects of DNA repair such as Fanconi anemia (FA), where afflicted children have catastrophic bone marrow failure early in life because of the loss of HSCs and are at significantly increased risk of leukemia.3 Furthermore, murine models deficient in several different DNA repair pathways all result in progressive HSC defects with age.4-6 This not only indicates that DNA damage is a constant physiologic threat to HSCs throughout life but also suggests the HSC genome must be protected against different sources of DNA damage.
Endogenous aldehydes are hematopoietic genotoxins
Our DNA is under constant assault by a broad range of chemical threats.7 The chemical reactivity of DNA makes it vulnerable to spontaneous hydrolytic depurination and deamination. Purposeful or aberrant enzymatic methylation and deamination of DNA also contribute to the DNA damage burden. Another source of chemical modification comes from consequences of transcription and replication (polymerase error, replication fork collapse, topoisomerase trapping). Last, DNA can be attacked by large number of reactive metabolites produced either within the cell (endogenous sources) or from environmental exposure (exogenous sources). These include reactive oxygen and nitrogen species, peroxides, alkylating agents such as S-adenosyl methionine, and reactive aldehydes. These can lead to a range of lesions that interfere with replication and transcription. The identity and origin of reactive genotoxins in hematopoietic cells is a fundamental question. One approach is to study congenital deficiencies of DNA repair to identify the putative genotoxins that cause DNA damage. In FA, the deficiency of a DNA cross-link repair pathway results in profound loss of HSCs and failure of hematopoiesis early in life, suggesting a ubiquitous endogenous genotoxin underpins the disease.
Genetic interaction between aldehyde catabolism and DNA repair
Studies over the last 11 years have pointed to endogenous aldehydes as the genotoxins that cause HSC attrition in FA. Aldehydes are reactive chemicals found in the environment and produced endogenously in our body that are able to covalently modify DNA to produce a range of adducts and cross-links.8-11 The genotoxic potential of aldehydes is well characterized; for example, environmental formaldehyde has been extensively investigated as a potential carcinogen.12 Exposure of FA-deficient eukaryotic cells to aldehydes results in accumulation of DNA breaks and chromosomal aberrations, supporting a role for the FA pathway in protecting against aldehyde genotoxicity.13-15 The major breakthrough demonstrating endogenous aldehydes are potent genotoxins stems from the discovery that aldehyde detoxification enzymes and the FA pathway act in concert to maintain hematopoiesis. This is demonstrated in 2 key mouse models, the Aldh2−/−Fancd2−/− and Adh5−/−Fancd2−/− double knockout mice,16-18 where genetic deletion of the FA pathway was combined with deficiency in either ALDH2 (acetaldehyde dehydrogenase 2), a NAD+ (nicotinamide adenine dinucleotide)-dependent mitochondrial enzyme that oxidizes acetaldehyde to acetate,19 or the cytosolic ADH5 (alcohol dehydrogenase 5) that metabolizes formaldehyde as a reversible glutathione conjugate.20 Both Aldh2−/−Fancd2−/− and Adh5−/−Fancd2−/− mice exhibited striking phenotypes characterized by significantly shortened survival (median survival, 145 and 33 days, respectively, compared with >600 days in controls), with mice succumbing to bone marrow failure and/or acute leukemia.16-18 The HSCs were severely compromised, with a 600- to 900-fold reduction in HSC number, as well as in their ability to reconstitute bone marrow when transplanted into recipient mice. Furthermore, the hematopoietic defect can be exacerbated in Aldh2−/−Fancd2−/− and Adh5−/−Fancd2−/− mice by treatment with ethanol (precursor of acetaldehyde) and methanol (precursor of formaldehyde), respectively, suggesting that aldehydes are indeed the causative toxin.
Endogenous aldehydes lead to genomic instability in blood stem cells
Bone marrow cells from Aldh2−/−Fancd2−/− and Adh5−/−Fancd2−/− mice exhibit increased markers of DNA damage, including y-H2A.X (marker of DNA double-strand breaks), chromosomal translocations, red cell micronucleation, and sister chromatid exchanges indicative of active homologous recombination to repair DNA double-strand breaks.17,21,22 To interrogate DNA damage originating from rare HSCs, single HSCs from Aldh2−/−Fancd2−/− mice were transplanted into recipients to amplify the stem cell genome through clonal hematopoiesis. Whole genome sequencing of the clonal progeny revealed that Aldh2−/−Fancd2−/− HSCs harbor significant increases in single base substitutions, deletions, and chromosomal rearrangements.21 The deletion of p53 in Aldh2−/−Fancd2−/− mice was able to rescue both number and function of HSCs, indicating the role of the DNA damage response in removing HSCs with damaged genomes. Unexpectedly, Aldh2−/−Fancd2−/−Trp53−/− HSCs did not exhibit a higher mutation burden than Aldh2−/−Fancd2−/− HSCs. This suggests that, although the immediate induction of p53 in response to endogenous DNA damage is a potent trigger of apoptosis, it appears not to suppress the accumulation of DNA mutations. This parallels a previous study showing the acute p53 response to genotoxic stress from chemotherapy and irradiation in mice induces widespread apoptosis but contributes negligibly to tumor suppression.23 It also raises the possibility that the relationship between p53, DNA damage, and repair is different in stem cells compared with mature cells.
Beyond hematopoiesis defects, Aldh2−/−Fancd2−/− mice more closely phenocopy the clinical features found in patients with FA compared with Fancd2−/− mice. This includes increased frequency of developmental abnormalities, although these defects do not entirely parallel the developmental defects observed in human FA for reasons currently unknown and beyond the scope of this review. In addition, ALDH2-deficient mothers are unable to bear Aldh2−/−Fancd2−/− or Aldh2−/−Fanca−/− pups, suggesting a critical role for maternal aldehyde catabolism to protect against the toxic effect of aldehydes in utero.16,24
Formaldehyde detoxification is essential for hematopoiesis
The severe genetic interaction between aldehyde clearance enzymes ALDH2 and ADH5 and the FA DNA repair pathway strongly supports endogenous aldehydes as potent metabolic genotoxins. ALDH2 and ADH5 clearly play important roles in the detoxification of aldehydes that threaten HSC viability. However, are these detoxification enzymes dispensable if the FA DNA repair pathway remains intact? Mice deficient in ALDH2 or ADH5 exhibit a preserved HSC compartment and intact hematopoiesis, whereas in contrast, mice deficient in both ALDH2 and ADH5 show a severe phenotype. Aldh2−/−Adh5−/− mice are born very small, fail to gain weight, and most die within 30 days of birth.22,25,26 Therefore, an intact FA pathway in these mice was insufficient to protect hematopoiesis, resulting in anemia, loss of immature B and T lymphocyte populations, and deficiency in common lymphoid progenitors and HSCs. A simple prediction in Aldh2−/−Adh5−/− mice would be that accumulation of both acetaldehyde and formaldehyde drives the severe phenotype. However, the level of acetaldehyde DNA adducts (a biomarker of DNA exposure to the aldehyde) was unchanged in these mice compared with wildtype and Aldh2−/− mice, suggesting endogenous acetaldehyde cannot be the offending aldehyde in these mice. Strikingly, both the level of formaldehyde DNA adducts and the serum formaldehyde concentration were found to be significantly elevated in Aldh2−/−Adh5−/− mice beyond that of single mutants. Therefore, the key discoveries from the study of Aldh2−/−Adh5−/− mice are that ALDH2 in fact acts on endogenous formaldehyde and that under basal conditions mammals produce lethal levels of formaldehyde that can overwhelm DNA repair pathways, thus necessitating detoxification by ALDH2 and ADH5. Intriguingly, the mutational pattern of Aldh2−/−Adh5−/− hematopoietic stem and progenitor cell (HSPC) genomes resembles two signatures (SBS5 and SBS40) found commonly in human cancer and aged somatic tissue.27-29 This raises the possibility that formaldehyde is ubiquitously produced throughout life and a major mutational influence on the mammalian genome.
Impact of aldehydes in human hematopoiesis
Several human studies now support endogenous aldehydes, and, in particular, formaldehyde, as a major genotoxin in human HPCs. A dominant-negative destabilizing polymorphism in ALDH2 (rs671) is highly prevalent in East Asian populations, affecting approximately 500 million people worldwide.30 This most often manifests as an acute flushing reaction after ingestion of alcohol, known as the Asian flushing syndrome.31 A study of a group of Japanese patients with FA revealed that coinheritance with the human ALDH2 polymorphism greatly accelerates bone marrow failure to within the first 7 months of life.32ALDH2 has also been shown to be epigenetically silenced in a subset of human acute myeloid leukemia, leading to a dependence on the FA pathway to maintain viability.33 In addition, recent studies have characterized several families with children carrying autosomal recessive mutations in ADH5 in combination with ALDH2 polymorphism.22,25 Analogous to Aldh2−/−Adh5−/− mice, these children are born with low birth weight, short stature, and developmental abnormalities and exhibit intellectual disability. All affected individuals develop myelodysplastic syndrome very early in life, with many progressing to acute leukemia. Termed aldehyde degradation deficiency syndrome (ADDS; also referred to as AMeDS for anemia, mental retardation, and dwarfism syndrome), the disease shares clinical features reminiscent of FA and necessitates the same treatment strategy with bone marrow transplantation.25,34 However, patients with ADDS do not harbor mutations in FA genes, and their cells are not hypersensitive to DNA cross-linkers. Instead, they are hypersensitive to formaldehyde and demonstrate spontaneously elevated y-H2A.X foci and sister chromatid exchanges in patient-derived lymphocytes and induced pluripotent stem cells.35 The similarity of this formaldehyde accumulation syndrome with FA suggests that formaldehyde genotoxicity likely underpins the pathogenesis of both diseases. Future treatment options for both conditions could involve lowering endogenous formaldehyde. Indeed, a proof-of-principle experiment by Takata et al35 showed partial rescue of the hematopoiesis defect in induced pluripotent stem cells derived from patients with ADDS when treated with a drug restoring ALDH2 enzyme activity.
Difference between human and murine FA
The significance of endogenous formaldehyde as a physiologic genotoxin in both mice and mammals raises an interesting question regarding why FA in human is much more severe than murine models of FA deficiency? In mice, loss of the FA pathway results in mild loss of bone marrow stem cells with little-to-no functional impact. In this case, murine tier 1 enzymes ALDH2 and ADH5 appear to provide near-complete protection against formaldehyde in the absence of the FA pathway. This is not the case in humans where loss of the FA pathway leads to a devastating bone marrow failure and leukemia syndrome despite intact ALDH2 and ADH5. This might be because of differences in life span and stem cell number, with FA mice harboring decreased but sufficient HSCs necessary to support blood production to last its short lifespan. In addition, the more diverse and stressful environmental influences experienced by human HSCs, including inflammation and infection, could accelerate stem cell attrition in FA.36 Another simple reason could be that humans harbor higher endogenous levels of formaldehyde compared with mice. This could arise because of increased production, decreased catabolism of formaldehyde, or both. It will be interesting to compare both the formaldehyde level and efficiency of formaldehyde clearance in both species using the same assay method. If true, then this raises the possibility toward therapies that reduce the human formaldehyde burden to murine levels as a strategy to treat FA in humans.35,37
Protection against formaldehyde genotoxicity beyond the FA pathway
Intriguingly, some clinical features in people affected by loss of ALDH2 and ADH5 are distinct from FA. This includes certain skeletal abnormalities affecting the lower extremities in ADDS rather than the hand/arm in FA. Other distinctive features such as intellectual disability and small stature closely resemble another genetic disease: Cockayne syndrome (CS). This genetic disorder arises because of mutations in CSA (also known as ERCC8) or CSB (also known as ERCC6). Both CSA and CSB proteins play an important role in triggering transcription-coupled nucleotide excision repair pathway (TC-NER) to excise DNA lesions such as DNA cross-links that can inhibit the progression of RNA polymerase. Interestingly, we have shown that mice deficient in ADH5 and CSB, a critical component of TC-NER, recapitulate many clinical features of human CS, including neurologic defects, small stature, and renal failure.38 This strongly suggests that formaldehyde is also the endogenous genotoxin that drives CS. Thus, whereas FA and CS might reflect failure of specific cellular protection pathways against formaldehyde, ADDS more likely represents a clinical amalgamation of several clinical syndromes because of excess formaldehyde overwhelming multiple distinct cellular protection pathways.
In summary, the profound hematopoiesis defect in mice and humans deficient in aldehyde clearance and DNA cross-link repair reveals that endogenous aldehydes are potent sources of DNA damage in hematopoietic cells. Ten years ago, we originally proposed a 2-tier model for the protection against aldehyde genotoxicity. We can now update this model to highlight formaldehyde as a major endogenous aldehyde genotoxin produced at abundant levels in mammals (Figure 1). Tier 1 protection against formaldehyde requires detoxification by the enzymes ALDH2 and ADH5 that exhibit functional redundancy. Tier 2 DNA repair is provided by the FA pathway. Both tiers are essential to protect hematopoiesis against formaldehyde genotoxicity and explain why combined loss of ALDH2 and ADH5 or the FA pathway in humans results in diseases such as ADDS or FA that are characterized by blood defects and leukemia predisposition.

Two-tier protection against endogenous aldehydes and consequences of its failure. (A) Hematopoietic stem and progenitor cells are protected against endogenous aldehydes through high expression of ALDH2 (detoxifies formaldehyde and acetaldehyde) and ADH5 (detoxifies formaldehyde) (tier 1 protection). Any aldehyde-induced DNA damage is repaired by the FA DNA repair pathway (tier 2 protection). (B) In humans, the loss of tier 1 protection through the combined genetic deficiency of ALDH2 and ADH5 leads to ADDS, whereas loss of tier 2 protection through deficiency of the FA pathway (or combined loss of tier 1 and tier 2) results in the eponymous FA syndrome. Both conditions share common clinical features that stem from overwhelming formaldehyde genotoxicity, in particular, aplastic anemia and predisposition to acute leukemia. Beyond the hematopoietic defect, both conditions also exhibit microcephaly, short stature, skin pigmentation such as café-au-lait spots, and skeletal malformations.
Two-tier protection against endogenous aldehydes and consequences of its failure. (A) Hematopoietic stem and progenitor cells are protected against endogenous aldehydes through high expression of ALDH2 (detoxifies formaldehyde and acetaldehyde) and ADH5 (detoxifies formaldehyde) (tier 1 protection). Any aldehyde-induced DNA damage is repaired by the FA DNA repair pathway (tier 2 protection). (B) In humans, the loss of tier 1 protection through the combined genetic deficiency of ALDH2 and ADH5 leads to ADDS, whereas loss of tier 2 protection through deficiency of the FA pathway (or combined loss of tier 1 and tier 2) results in the eponymous FA syndrome. Both conditions share common clinical features that stem from overwhelming formaldehyde genotoxicity, in particular, aplastic anemia and predisposition to acute leukemia. Beyond the hematopoietic defect, both conditions also exhibit microcephaly, short stature, skin pigmentation such as café-au-lait spots, and skeletal malformations.
Genotoxic lesion formed by aldehydes in vivo
Aldehydes can chemically react with DNA to form base adducts, and covalent cross-links between adjacent bases on the same DNA strand (intrastrand cross-link) or bases on opposite DNA strands (interstrand cross-link; Figure 2).8-11 These DNA modifications are not equally genotoxic or mutagenic, and thus it is important to understand which aldehyde-DNA modifications underpin aldehyde genotoxicity. The interrogation of genomic DNA exposed to aldehydes in vivo using mass spectrometry has permitted quantification of aldehyde-DNA adducts. For example, the level of the formaldehyde-DNA adduct N2-hydroxymethyl-guanine in genomic DNA correlates with both exogenous and endogenous formaldehyde burden in mice.17,22 Endogenous formaldehyde-DNA cross-links between guanine bases have also been detected in vivo but at 10- to 30-fold lower levels compared with the formaldehyde-DNA adducts N2-hydroxymethyl-guanine and N6-hydroxymethyl-adenine.39
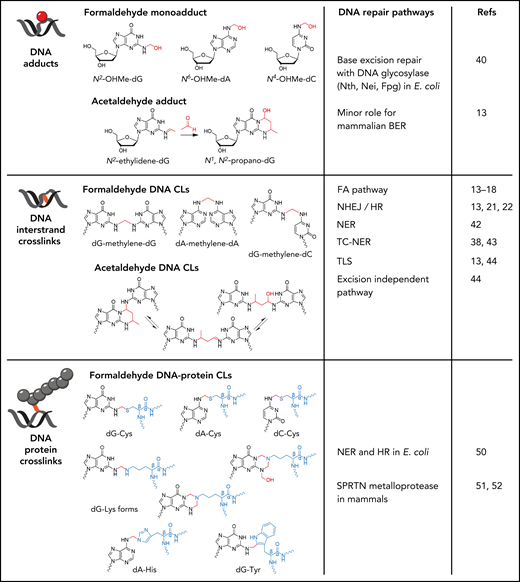
Genotoxic lesions formed by formaldehyde and acetaldehyde. Formaldehyde reacts with the amine group on guanine, adenine, and cytosine to form N2-hydroxymethyl-guanine (N2-OHMe-dG), N6-hydroxymethyl-adenine (N6-OHMe-dA), and N4-hydroxymethyl-cytosine (N4-OHMe-dC), respectively. Acetaldehyde initially forms an unstable N2-ethylidene-guanine adduct, which spontaneously reacts with an additional acetaldehyde molecule to form the cyclic adduct N1,N2-propano-guanine. Formaldehyde can form DNA interstrand cross-links through the formation of a methylene bridge that covalently links guanine, adenine, and cytosine on opposing DNA strands. Although only dG-dG, dA-dA, and dG-dC are shown, any combinations of dG, dA, and dC can be cross-linked by formaldehyde. Acetaldehyde reacts with opposing guanines to form cross-links that exist in equilibrium between 3 chemical states. Formaldehyde can also form methylene cross-links between guanine and the amino acids cysteine, histidine, tryptophan, and lysine to give rise to DNA-protein cross-links. The DNA repair pathways with activity toward the respective aldehyde-DNA lesions are shown.
Genotoxic lesions formed by formaldehyde and acetaldehyde. Formaldehyde reacts with the amine group on guanine, adenine, and cytosine to form N2-hydroxymethyl-guanine (N2-OHMe-dG), N6-hydroxymethyl-adenine (N6-OHMe-dA), and N4-hydroxymethyl-cytosine (N4-OHMe-dC), respectively. Acetaldehyde initially forms an unstable N2-ethylidene-guanine adduct, which spontaneously reacts with an additional acetaldehyde molecule to form the cyclic adduct N1,N2-propano-guanine. Formaldehyde can form DNA interstrand cross-links through the formation of a methylene bridge that covalently links guanine, adenine, and cytosine on opposing DNA strands. Although only dG-dG, dA-dA, and dG-dC are shown, any combinations of dG, dA, and dC can be cross-linked by formaldehyde. Acetaldehyde reacts with opposing guanines to form cross-links that exist in equilibrium between 3 chemical states. Formaldehyde can also form methylene cross-links between guanine and the amino acids cysteine, histidine, tryptophan, and lysine to give rise to DNA-protein cross-links. The DNA repair pathways with activity toward the respective aldehyde-DNA lesions are shown.
Genetic approaches to identify genotoxic lesions
Another approach to deduce the genotoxic lesion of aldehydes is to extrapolate from the substrate specificity of DNA repair pathways required to protect against aldehyde-DNA damage. This will not reveal the identity of the DNA lesion but gives insight into which type of aldehyde-DNA damage necessitates repair to mitigate toxicity. In Escherichia coli, deficiency of base excision repair DNA glycosylases (nth, nei, and fpg), recombination repair (eg, recA), and nucleotide excision repair (NER) confers sensitivity to formaldehyde, suggesting both base adducts and cross-links must be repaired to mitigate genotoxicity.40 In mammals, the genetic requirement for the FA pathway to protect against formaldehyde and acetaldehyde is clearly established. In addition, homologous recombination (HR) and nonhomologous end joining also play a role in repair of formaldehyde- and acetaldehyde-induced DNA damage.13,21,41 In particular, aldehyde-DNA damage in primary cells from humans and mice triggers robust activation of HR as observed by elevation of sister chromatid exchanges.21,22,35 This all supports the notion that aldehyde cross-links are repaired by the concerted action of the FA, HR, and nonhomologous end joining pathways to avoid DNA double-strand breaks. Interestingly, although both NER and TC-NER DNA repair pathways protect against formaldehyde toxicity,38,42,43 these pathways appear to be dispensable in protection against acetaldehyde.38,42 This suggest that the reactivity of formaldehyde and acetaldehyde toward DNA and the types of DNA lesions that are formed are not identical.
DNA repair biochemistry of aldehyde-induced DNA cross-links
The biochemistry of how such an aldehyde-cross-link is repaired has been further dissected using DNA replication and repair proteins from Xenopus egg extracts,44 a model used extensively to study the mechanism of vertebrate DNA cross-link repair.45 The repair of a plasmid containing an acetaldehyde-DNA interstrand cross-link in Xenopus egg extracts is coupled to DNA replication; it requires the FA pathway to excise and unhook the cross-link, followed by translesion synthesis (TLS) to bypass the unhooked cross-link on one DNA strand and homologous recombination to resolve the double-strand break. Interestingly, a second repair mechanism operates independently of the FA pathway, where the cross-link is not excised but directly cleaved apart by an unidentified factor. The Nei-like DNA glycosylase 3 has exhibited direct cleavage activity against cross-links derived from abasic sites (which chemically are aldehydes themselves) and psoralen46 but is not the enzyme responsible for cleavage of the acetaldehyde-DNA cross-link.44 This highlights that chemically distinct DNA cross-links are processed by different repair pathways, and because an in vitro synthesized aldehyde cross-link may differ from aldehyde DNA cross-links encountered in vivo, there may be differences in repair depending on chemical similarity. Indeed, despite the critical requirement for TLS in the repair of interstrand cross-links formed from chemotherapy drugs mitomycin C and cisplatin,47 surprisingly, cells deficient in the TLS polymerase REV1 are only mildly sensitive to acetaldehyde and formaldehyde.13,14,16,24 Furthermore, Aldh2−/−Rev1−/− double knockout mice do not exhibit the developmental defects or loss of hematopoietic stem and progenitor cells seen in Aldh2−/−Fancd2−/− mice.24 The dispensability of TLS in repair of aldehyde-DNA cross-links in vivo is intriguing, one possible explanation being that the unhooked aldehyde cross-link is chemically small and does not impede the canonical DNA polymerases, thus not requiring bypass by a TLS DNA polymerase.
Aldehyde-induced DNA-protein cross-links
Another class of aldehyde-induced DNA lesions that may be responsible for aldehyde genotoxicity are DNA-protein cross-links (DPCs), bulky conjugates between DNA and neighboring proteins such as histones that can impede DNA replication and transcription. Reactive aldehydes such as formaldehyde can catalyze the formation of such DNA-protein cross-links and have been detected in animal tissues as a result of both endogenous and exogenous formaldehyde exposure (Figure 2).48,49 In E coli, DNA repair pathways such as NER and HR exhibit repair activity against DPCs.50 In yeast, the dedicated metalloprotease WSS1 can directly bind to DNA and cleave bound proteins to dissolve the DPC.51 Mammals have also evolved a metalloprotease called SPRTN that exhibits proteolytic activity against DPCs.52 Cells deficient in SPRTN are more sensitive to formaldehyde treatment with reduced survival and increased levels of the DNA damage marker y-H2A.X. Genetic deficiency of SPRTN in humans is responsible for the rare Ruijs-Aalfs syndrome; clinical features include low weight, lipodystrophy, and developmental abnormalities, and all afflicted individuals developed hepatocellular carcinoma.53,54 Several of these clinical features overlap with ADDS from deficiency of ALDH2 and ADH5, and ADH5-deficient mice are more susceptible to hepatocellular carcinomas.55 This raises the possibility that endogenous formaldehyde could be the main catalyst for genotoxic DPCs that underlie Ruijs-Aalfs syndrome. Although no hematopoiesis defect has been described in Ruijs-Aalfs syndrome or in mice carrying hypomorphic mutations in Sprtn,56 lymphocytes from patients deficient in SPRTN do harbor increased chromosomal aberrations, indicating blood cells are susceptible to genotoxic DPCs.54 It will be interesting to assess whether other novel DPC proteases similar to SPRTN operate in the hematopoietic compartment to protect against DPC-derived genotoxicity.
Origins of endogenous formaldehyde and acetaldehyde
Aldehydes are found abundantly in the environment but can also be produced endogenously in our own bodies at significant levels (Figure 3). What are the major metabolic sources of endogenous formaldehyde and acetaldehyde?
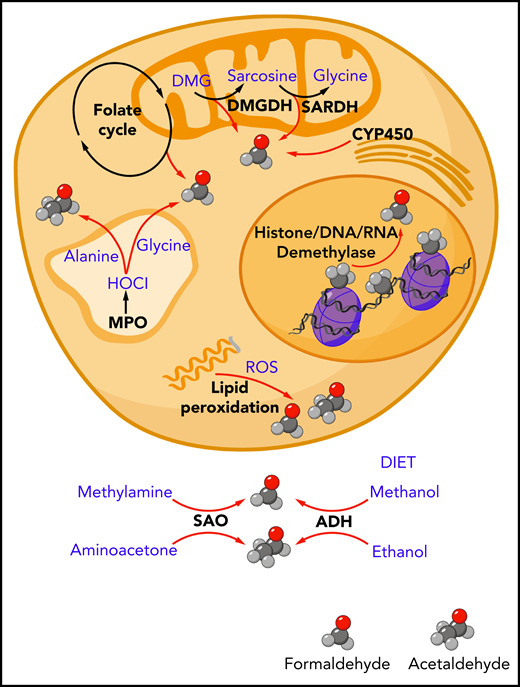
Endogenous sources of formaldehyde and acetaldehyde. The spontaneous chemical decomposition of tetrahydrofolate and dihydrofolate releases formaldehyde. Mitochondrial enzymes dimethylglycine dehydrogenase (DMGDH) and sarcosine dehydrogenase (SARDH) release formaldehyde as catalytic byproducts. Cytochrome P450 (CYP450) enzymes located in the mitochondria and endoplasmic reticulum can generate formaldehyde through the oxidation of glycerol. Oxidative demethylases release formaldehyde through cleavage of methyl groups found on histone proteins, DNA, and RNA. Polyunsaturated lipid species can be oxidized by reactive oxygen species (ROS) in a process called lipid peroxidation to release a range of aldehydes including formaldehyde and acetaldehyde. In myeloid cells such as neutrophils and monocytes, myeloperoxidase enzyme found in the lysosomes and secreted into the surrounding extracellular environment can generate hypochlorous acid (HOCl), which can oxidize the amino acids glycine and alanine to release formaldehyde and acetaldehyde, respectively. Outside of the cell, the oxidative deamination of methylamine and aminoacetone by serum amine oxidase can release formaldehyde and acetaldehyde, respectively. Ingested methanol and ethanol can be enzymatically converted to formaldehyde and acetaldehyde by alcohol dehydrogenase enzymes expressed mainly in the liver (ADH).
Endogenous sources of formaldehyde and acetaldehyde. The spontaneous chemical decomposition of tetrahydrofolate and dihydrofolate releases formaldehyde. Mitochondrial enzymes dimethylglycine dehydrogenase (DMGDH) and sarcosine dehydrogenase (SARDH) release formaldehyde as catalytic byproducts. Cytochrome P450 (CYP450) enzymes located in the mitochondria and endoplasmic reticulum can generate formaldehyde through the oxidation of glycerol. Oxidative demethylases release formaldehyde through cleavage of methyl groups found on histone proteins, DNA, and RNA. Polyunsaturated lipid species can be oxidized by reactive oxygen species (ROS) in a process called lipid peroxidation to release a range of aldehydes including formaldehyde and acetaldehyde. In myeloid cells such as neutrophils and monocytes, myeloperoxidase enzyme found in the lysosomes and secreted into the surrounding extracellular environment can generate hypochlorous acid (HOCl), which can oxidize the amino acids glycine and alanine to release formaldehyde and acetaldehyde, respectively. Outside of the cell, the oxidative deamination of methylamine and aminoacetone by serum amine oxidase can release formaldehyde and acetaldehyde, respectively. Ingested methanol and ethanol can be enzymatically converted to formaldehyde and acetaldehyde by alcohol dehydrogenase enzymes expressed mainly in the liver (ADH).
Sources of acetaldehyde
The major source of acetaldehyde exposure in humans likely comes from alcohol consumption, whereby ethanol is subsequently converted to acetaldehyde by alcohol dehydrogenase enzymes. In ALDH2-deficient mice, a single dose of ethanol was sufficient to significantly elevate DNA damage as measured by sister chromatid exchanges.21 The scale of alcohol consumption worldwide would suggest this to be a widespread genotoxin in humans, especially in populations that harbor the inactive ALDH2 polymorphism. Endogenous acetaldehyde can also arise spontaneously from lipid peroxidation, enzymatic byproducts of amine oxidases, and myeloperoxidase.57 Surprisingly, we found the level of endogenous acetaldehyde-DNA adducts in Aldh2−/− mice to be unchanged compared with wildtype mice, suggesting either that endogenous sources do not supply a major acetaldehyde burden under basal metabolism or that other enzymes redundant to ALDH2 such as ALDH1A1 can provide sufficient functional compensation.58 In contrast to acetaldehyde-DNA adducts, Adh5−/− mice that are deficient in formaldehyde clearance exhibit significant elevation of formaldehyde-DNA adducts, which is further elevated in Aldh2−/−Adh5−/− mice. This indicates that there is a very major physiologic burden of formaldehyde arising from endogenous metabolic sources rather than environmental exposure.22 This finding is supported by other studies showing that endogenous formaldehyde-DNA adducts far exceed those obtained through nasal inhalation of environmental formaldehyde in animal models.59,60
Formaldehyde-generating reactions in cell metabolism
One potential endogenous source of formaldehyde is folic acid, an essential vitamin required as carrier of 1-carbon units.41 In ex vivo cell culture, formaldehyde is released spontaneously from dihydrofolate, tetrahydrofolate, and 5,10-methylene-tetrahydrofolate at sufficient quantities to exert toxicity to ADH5-deficient cells. However, the contribution to endogenous formaldehyde from physiologic levels of folate in vivo is unknown. Oxidative demethylation reactions also release formaldehyde from methyl groups removed from RNA,61 DNA,62-64 or proteins.65,66 In hematopoiesis, histone demethylases such as LSD1 (also known as KDM1A), and JMJC play essential roles in definitive hematopoiesis and terminal differentiation of blood cells, and these demethylases could be responsible for a shower of nuclear formaldehyde during intense epigenetic reprogramming as a stem cell differentiates to a mature blood cell. In support of this, a recent study correlated the increased transcriptional activity from differentiating HSPCs with elevated formaldehyde. Inactivation of the FA DNA repair pathway resulted in increased DNA damage and defective differentiation of the HSPCs.67 Mitochondrial oxidative demethylases such as dimethylglycine dehydrogenase and sarcosine dehydrogenase can also give rise to formaldehyde,68 and the mitochondrial localization of ALDH2 suggests that mitochondria could be a significant source of aldehydes. Other potential sources of formaldehyde could come from oxidation of the amino acid glycine by hypochloric acid produced by neutrophil myeloperoxidase,69 deamination of methylamine by monoamine oxidases,70,71 and glycerol oxidation by cytochrome P450 enzymes.72 Lipids and fatty acids under certain oxidizing conditions have also been shown to release formaldehyde among other aldehydes such as hydroxynonenal and malondialdehyde.73 The accumulation of redox active iron can trigger lipid peroxidation of polyunsaturated fatty acids in phospholipids to release aldehydes. This process triggers ferroptosis, a nonapoptotic form of cell death that results in the disruption of the mitochondrial membrane. It is tempting to speculate that cells particularly reliant on tier 1 and tier 2 protection against aldehydes might be most susceptible to ferroptosis. Indeed, overexpression of mitochondrially localized ALDH2 has been associated with protection against ferroptosis in a β-amyloid transgenic mouse model,74 although it remains unclear whether ALDH2 is directly detoxifying lipid peroxidation-derived aldehydes. It will be interesting to assess whether ferroptosis could play a role in some of the pathologies observed in humans and mice deficient in ALDH2 and ADH5, and whether antioxidant scavengers of lipid peroxidation products, such as Trolox, could alleviate these pathologies.
Novel tools to identify sources of endogenous aldehyde
Although several biochemical pathways are plausible sources of aldehydes, it remains to be determined how much these processes contribute to the systemic burden, under which (patho)physiologic situations, and whether treatment (eg, with antioxidants) might suppress these. Another interesting question is whether the cellular aldehyde burden comes primarily from cell-intrinsic metabolism, or whether aldehydes derive from a few tissues responsible for the bulk of production in the body. For formaldehyde, tissue-specific analysis of DNA adducts reveals the highest formaldehyde adduct level to be found in the liver, followed by the kidney, brain, and hematopoietic tissues.17,22 However, DNA adduct levels across tissues as a biomarker for formaldehyde exposure may be influenced by cell turnover, thus not directly reflecting the level of formaldehyde exposure when comparing tissues of vastly different cell division rates. New methods of aldehyde detection in vivo will be critical to facilitate the identification of the molecular origins of formaldehyde. Over the last few years, significant progress has been made toward development of such tools. Several reactivity-based chemical fluorescent probes sensitive to formaldehyde have been developed to visualize formaldehyde in living cells, such as 2-aza-Cope reactive probes75-77 and a 1,8-naphthalimide-based two-photon fluorescence probe.78 These chemical probes have been used to detect endogenous formaldehyde production in the nucleus67 and in mitochondria.79 However, these chemical probes are restricted by their ability to be introduced into living biological systems, are currently mainly used in cell lines, and are less amenable to interrogating whole organisms. Another promising tool is a genetically encoded fluorescent protein with reactivity to formaldehyde. This sensor links the formaldehyde-sensing transcription factor HxIR from bacteria Bacillus subtilis to a fluorescent protein and is sensitive to elevated endogenous formaldehyde in HeLa cells treated with ALDH2 and ADH5 inhibitors.80
Two-tier protection against other aldehydes
Altogether in humans, there are 19 aldehyde dehydrogenase genes (22 in mice) and 7 alcohol dehydrogenase genes (5 in mice),81 suggesting we must defend against a diverse range of aldehydes. The expression profile of Aldh and Adh family members in murine HSCs and progenitor cells shows that Aldh2 and Adh5 are the highest expressed genes in this family.22 Other less-expressed members include Aldh1a1, Aldh1a7, Aldh3a2, Aldh7a1, Aldh9a1, Aldh16a1, and Aldh18a1.22,82 These other ALDH enzymes may play a role in protecting hematopoiesis against aldehydes, but functional redundancy between these enzymes limits the inference of relevant genotoxic aldehydes purely based on enzymatic activity. Indeed, hematopoiesis remains intact in mice singly deficient in Aldh1a1, possibly because of functional compensation by other family homologs.82
α-Oxoaldehydes (dicarbonyls)
A number of aldehydes other than formaldehyde are produced at abundant levels in our body, including dicarbonyl compounds containing a further carbonyl group in addition to an aldehydic group. These include 3-deoxyglucosone, glyoxal, and methylglyoxal (all of which can derive from sugar),83 as well as lipid peroxidation products including (again) glyoxal and malondialdehyde. The endogenous level of methylglyoxal in human blood has been reported to range between 100 and 200 nM.84,85 In patients with diabetes with elevated blood glucose, methylglyoxal can increase up to 900 nM,84 along with a two- to threefold elevation of glyoxal and 3-deoxyglucosone.86,87 α-Oxoaldehydes covalently modify proteins and nucleic acids (particularly on the guanine base) in multistep reactions ultimately yielding advanced glycation endproducts.88 Analogous to the 2-tier protection against formaldehyde and acetaldehyde, a similar 2-tier system could exist to defend against dicarbonyls. For methylglyoxal, tier 1 detoxification consists of enzymes such as AKR1A1 and ALDH1B1 (nicotinamide adenine dinucleotide (phosphate)-dependent) and GLO1 and 2 (glutathione-dependent) that catalyze conversion of methylglyoxal to inert lactate.89 Tier 2 DNA repair pathways could involve NER and HR based on observations of E coli and eukaryotic cells deficient in these respective DNA repair pathways to exhibit increased mutagenesis and cytotoxicity to exogenous methylglyoxal challenge.90-92 However, avian DT40 cells deficient in the FA pathway were not hypersensitive to methylglyoxal,13 and the physiologic roles of NER and HR in protecting against endogenous dicarbonyl DNA damage remain poorly characterized. Beyond the canonical DNA repair pathways, a deglycase enzyme known as DJ-1 (also named PARK7) has been shown to detoxify methylglyoxal by conversion to lactate93 but also exhibit repair activity through cleavage of methylglyoxal adducts from proteins and guanine in both RNA and DNA.94,95 Indeed, human cells deficient in DJ-1 exhibit increased markers of DNA double-strand breaks, p53 activation, and apoptosis.95 Thus, it would appear that DJ-1 evolved potential roles as a combined tier 1 and tier 2 enzyme against methylglyoxal. Genetic deficiency in GLO1 and DJ-1 has been associated with neurologic disorders such as schizophrenia and Parkinson disease, respectively,96,97 and mice deficient in DJ-1 show an age-dependent hematopoietic stem cell defect.98 It will be interesting to dissect potential functional redundancy between the 2 tiers of defense against α-oxoaldehydes that analogous to formaldehyde might mask their full toxicity.
Fatty aldehydes
Another group of endogenous aldehydes are fatty aldehydes, arising mainly from intermediate byproducts of lipid metabolism.99 These aldehydes are structurally diverse, ranging in length from 6 to more than 26 carbons arranged in straight or branching chains, and can be saturated or contain varying numbers of unsaturated carbons. To prevent the toxic accumulation of these fatty aldehydes, cells rely on alcohol dehydrogenase and aldehyde dehydrogenase enzymes to convert the fatty aldehydes to their corresponding fatty alcohol or fatty acid, respectively. The enzyme best characterized for detoxification of fatty aldehydes is ALDH3A2 (also known as fatty aldehyde dehydrogenase), which catalyses the NAD+-dependent oxidation of fatty aldehydes (with preference for C14-C18 chain length substrates) to fatty acids.100 Genetic deficiency in ALDH3A2 is responsible for the neurocutaneous disease Sjögren-Larsson syndrome, characterized by widespread thickened and dry skin (ichthyosis), intellectual disability, and spasticity.101 In acute myeloid leukemia, ALDH3A2 has been implicated to protect against fatty aldehydes generated by ferroptosis.102 In nonhematologic malignancies, reduced expression of ALDH3A2 along with several other ALDH family members has also been observed in human esophageal adenocarcinomas; this correlated with accumulation of medium-chain aldehydes including nonanal (C9) and decanal (C10), increased markers of DNA damage, and worse overall survival.103
Both glucose and fatty acids have been suggested to provide energy to ensure HSPC viability and normal hematopoiesis.104-106 If so, HSPCs are likely to produce significant levels of dicarbonyls and fatty aldehydes as metabolic byproducts. It is clear that multiple cellular pathways are deployed to defend against aldehydes, and beyond Aldh and Adh family members, the question remains whether glyoxalases, DJ-1, or other enzyme families such as aldoketoreductases107 may operate to protect against the toxic accumulation of these endogenous aldehydes. In addition, HSCs may be protected in part by virtue of their quiescent state with reduced uptake of glucose,108 which could restrict their production of sugar-derived dicarbonyls.
Future directions
Over the last decade, endogenous aldehydes have been firmly established as potent genotoxins that threaten hematopoiesis. Their role as likely drivers of the disease FA, along with the discovery of a novel bone marrow failure syndrome ADDS because of deficiency of aldehyde clearance enzymes, highlights the severe pathology that can arise from aldehyde genotoxicity. Although the ultimate metabolic source of these aldehydes remains to be determined, both conditions can be considered aldehydeopathies because of either failure of aldehyde clearance or DNA repair of aldehyde-DNA damage. Although these syndromes are rare, there are approximately 500 million people that are ALDH2 deficient. It is important to establish whether chronically elevated endogenous aldehydes such as formaldehyde in these people play a role in shaping the clonal landscape in the bone marrow and play a role in the pathogenesis of preleukemic disorders and leukemia, because ALDH2 deficiency has been observed to affect the risk of other cancers.109-111 Another important area to address is whether endogenous aldehydes other than formaldehyde are a source of genotoxicity, and if so, which cellular pathways defend against these aldehydes? Along with the development of better tools for tracking and quantifying aldehydes in vivo and interventions to modulate aldehyde levels, these research questions should provide a focus for the field of DNA damage and hematopoiesis in the coming years.
Acknowledgments
The authors thank the K.J.P. laboratory members, Sarah L. Caddy, and Michael R. Hodskinson for critically reading the manuscript.
This work was supported by funding to M.W. by Cancer Research UK (C60150/A23919), to F.A.D. and K.J.P. by the Medical Research Council, and the Wellcome Trust and Cancer Research UK to K.J.P.
Authorship
Contribution: M.W., F.A.D., and K.J.P. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Meng Wang, Wellcome-MRC Cambridge Stem Cell Institute, Jeffrey Cheah Biomedical Centre, University of Cambridge, Cambridge, CB2 0AW, United Kingdom; email: mw323@cam.ac.uk; and K. J. Patel, MRC Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, OX3 9DS, United Kingdom; email: ketan.patel@imm.ox.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal