

Key Points
APC contribution to the downregulation of thrombin generation depends on the vascular context.
PC activation during vascular injury is regulated by thrombin spatial distribution in conjunction with local physical conditions.

Abstract
Rebalancing the hemostatic system by targeting endogenous anticoagulant pathways, like the protein C (PC) system, is being tested as a means of improving hemostasis in patients with hemophilia. Recent intravital studies of hemostasis demonstrated that, in some vascular contexts, thrombin activity is sequestered in the extravascular compartment. These findings raise important questions about the context-dependent contribution of activated PC (APC) to the hemostatic response, because PC activation occurs on the surface of endothelial cells. We used a combination of pharmacologic, genetic, imaging, and computational approaches to examine the relationships among thrombin spatial distribution, PC activation, and APC anticoagulant function. We found that inhibition of APC activity, in mice either harboring the factor V Leiden mutation or infused with an APC-blocking antibody, significantly enhanced fibrin formation and platelet activation in a microvascular injury model, consistent with the role of APC as an anticoagulant. In contrast, inhibition of APC activity had no effect on hemostasis after penetrating injury of the mouse jugular vein. Computational studies showed that differences in blood velocity, injury size, and vessel geometry determine the localization of thrombin generation and, consequently, the extent of PC activation. Computational predictions were tested in vivo and showed that when thrombin generation occurred intravascularly, without penetration of the vessel wall, inhibition of APC significantly increased fibrin formation in the jugular vein. Together, these studies show the importance of thrombin spatial distribution in determining PC activation during hemostasis and thrombosis.
Introduction
The coagulation system is tightly regulated by multiple endogenous anticoagulant mechanisms, such as antithrombin, tissue factor pathway inhibitor, and the protein C (PC) system. PC is unique among the major endogenous anticoagulants in that it requires an activation step. Thrombin generated as a product of the coagulation cascade binds to thrombomodulin on the endothelial cell surface, where it can activate circulating PC to become activated PC (APC).1 PC activation is enhanced by its localization on the endothelial cell surface via binding to the endothelial cell PC receptor (EPCR).2 Once activated, APC exerts its anticoagulant activity in association with its cofactor, protein S (PS), by proteolytically inactivating the coagulation cofactors factor Va (FVa) and FVIIIa.1,3,4 Consequently, APC provides negative feedback to downregulate thrombin generation.
The importance of APC as an endogenous anticoagulant is demonstrated clinically by the association of mutations or deficiencies in the PC system with prothrombotic conditions. FV Leiden, characterized by a mutation in FV that renders it resistant to APC-mediated cleavage, is the most commonly inherited thrombophilia.5,6 Severe homozygous PC or PS deficiency is rare and results in thrombosis manifesting as purpura fulminans in neonates.7-9 Heterozygous deficiency is associated with a prothrombotic state, including susceptibility to warfarin-induced skin necrosis.10
These clinical manifestations of PC or PS deficiency highlight the importance of this endogenous anticoagulant as a critical antithrombotic mechanism, preventing unwarranted or deleterious intravascular coagulation. However, the contribution of APC to the downregulation of thrombin generation in settings of normal hemostasis is less well defined. Clinical studies examining the effect of recombinant APC administration in settings of sepsis resulted in major bleeding as an adverse effect, consistent with the powerful anticoagulant properties of APC.11,12 Similarly, a thrombomodulin mutation resulting in elevated soluble thrombomodulin and systemic PC activation resulted in excessive bleeding.13 In these cases, however, systemic APC administration or generation bypassed the physiologic PC activation step that normally regulates APC activity. In mouse models, homozygous PC or PS deficiency recapitulates human phenotypes, resulting in perinatal or embryonic lethality resulting from thrombosis, coagulopathy, and vascular defects.14-16 Neither heterozygous PC nor PS deficiency has been reported to affect bleeding, although a mouse strain expressing ∼3% normal levels of PC exhibited enhanced hemostasis.17 Furthermore, inhibition of PC or PS prevented excessive hemorrhage in the setting of hemophilia, which suggests that this anticoagulant pathway contributes to the regulation of thrombin generation during hemostasis, at least in some settings.18,19
A previously unexplored facet of the PC system is the relationship among physical factors affecting thrombin spatial distribution at a site of injury and PC activation on the endothelial cell surface. Thrombin spatial distribution is limited by both biochemical and physical constraints. Physical factors include localization of procoagulant surfaces, convection of thrombin away from the injury site by flowing blood, and hindered molecular transport within the platelet plug. The combined effect of these factors on thrombin localization can vary in different vascular contexts as physical conditions change throughout the vasculature. For example, intravital imaging studies have shown differences in thrombin spatial distribution between the intravascular and extravascular compartments after vascular injury in the microcirculation vs macrocirculation.20,21 Because PC activation occurs on the endothelial cell surface, we hypothesized that the variability of thrombin localization in different contexts may determine the extent to which APC contributes to the regulation of thrombin generation during hemostasis. We used a combination of in vivo models, where the spatial distribution of thrombin could be inferred, and in silico approaches to test this hypothesis, examining the relationships among local physical factors, thrombin spatial distribution, and PC anticoagulant function.
Methods
Mice and reagents
The Institutional Animal Care and Use Committee of the University of Pennsylvania approved all animal procedures. C57Bl/6 mice (#000664) and mice harboring the FV Leiden mutation (B6.129S2-F5tm2Dgi/J; #004080) were both from The Jackson Laboratory (Bar Harbor, ME). FV Leiden heterozygote mice were bred to generate the homozygous wild-type (WT) and FV Leiden mice used. Homozygous FV Leiden mice are referred to throughout as FVQ/Q. Anti-PC blocking antibody (clone 1609; referred to throughout as Ab1609) was previously described.22,23 Vehicle (normal saline) was administered for control experiments. Fluorescent labeling reagents are described in the data supplement.
Response to vascular injury in mouse cremaster arterioles
Laser-induced injury in mouse cremaster muscle arterioles was performed as described.20 Male C57Bl/6 or FVQ/Q mice were infused with fluorescently labeled imaging reagents via the jugular vein. For PC-blocking antibody studies, Ab1609 (10 mg/kg) or vehicle was infused along with the labeling antibodies. Full details regarding microscopy and image analysis are included in the data supplement.
Mouse jugular vein puncture injury
The jugular vein puncture injury model was performed as previously described.21 Male C57Bl/6 or FVQ/Q mice were infused with fluorescently labeled imaging reagents 5 minutes before puncture injury. For PC-blocking antibody studies, Ab1609 (10 mg/kg) or vehicle was infused along with the labeling reagents. Additional details are provided in the data supplement.
Two-photon fluorescence and scanning electron microscopy
Microscopy was performed as previously described.21 Details are provided in the data supplement.
Mouse jugular vein thrombosis
Mice were prepared as described for the jugular vein puncture injury model. After exposure of the right jugular vein, a 0.5-mm2 piece of Whatman #1 filter paper soaked with 5% ferric chloride (FeCl3; anhydrous; Sigma-Aldrich, St Louis, MO) was applied to the extravascular surface for 1 minute. The filter paper was then removed, the vein was quickly rinsed in saline, and real-time fluorescence imaging was initiated. Imaging was performed using an Olympus MVX10 fluorescence macroscope with a 0.63× objective and a 5× zoom factor. Excitation illumination was provided by a DG4 high-speed wavelength changer (Sutter), and images were captured using an OrcaFlash 4.0 sCMOS digital camera (Hamamatsu). Samples were fixed at predetermined time points and excised for high-resolution imaging.
Statistics
Statistical analysis and graphs were produced using Prism 6.0 software (GraphPad). For cremaster arteriole injury studies, statistics were performed using the number of injuries as n, as is typical for this assay. Data sets were compared using either the Mann-Whitney test or a 2-way analysis of variance with Bonferroni post hoc test for multiple comparisons, as noted in figure legends.
Computational approach
A 3-dimensional (3D) steady-state computational model was developed combining fluid dynamics, chemical species transport, and a chemical reaction network within 3D vessel geometries characterized by an inlet, a vessel outlet, and an injury outlet. Vessel geometries were informed by experimental evidence from cremaster20 and jugular21 models (supplemental Figure 4). Computational fluid dynamics and chemical engineering packages in COMSOL 5.4 were used to simulate fluid flow, chemical reactions, and molecular species transport. Maximum velocity was set to 0.2 cm/s in the cremaster24 and 10 cm/s in the jugular.25 Thrombin and APC generation were simulated using a simplified surface reaction scheme (supplemental Figure 5). Thrombin is generated from activation of bound prothrombin on procoagulant surfaces lining the extravascular compartment and the injury channel wall. PC activation is simulated by thrombin-mediated cleavage when thrombin binds to intravascular endothelial cell surfaces where PC is also bound (supplemental Figure 6). We did not consider reactions occurring on platelet surfaces. Our choice is supported by experimental studies showing that fibrin deposition, an indicator of thrombin activity, was not affected by platelet deposition in the mouse cremaster microcirculation.26 Parameter values were taken from previous studies when possible or otherwise arbitrarily set so that the maximum concentrations of thrombin and APC at steady state were in the low nanomolar range (supplemental Table 1). This choice was motivated by the following: (1) the uncertainty in the amount of thrombin and APC being generated during an in vivo hemostatic response, and 2) some single reactions in our model represent a set of reactions, and thus rates measured from single reactions cannot be used. Following our previous studies,24 hemostatic plugs were represented using a homogeneous porous medium. Full model details can be found in the data supplement.
Results
APC regulates the response to vascular injury in the microcirculation
We first examined the contribution of APC anticoagulant activity to the regulation of thrombin generation after laser-induced vascular injury in mouse cremaster arterioles. Prior studies have shown that laser-induced injury as performed here recapitulates mechanical puncture injury with a glass micropipette.20 The injury results in thrombin generation and fibrin formation, and platelet activation as measured by α-granule secretion (P-selectin expression) is largely thrombin dependent.20,27 Thrombin activity seems localized to the injury site, extending into the extravascular space and, to a limited degree, within the platelet mass that forms intravascularly.20,28 We employed 2 distinct approaches to inhibit APC activity, either a genetically modified mouse mimicking the FV Leiden mutation in humans (annotated here as FVQ/Q)29 or a pharmacologic approach using an anti–PC-blocking antibody.22
Consistent with prior reports,20,30 laser-induced injury of mouse cremaster arterioles resulted in rapid platelet accumulation over the injury site (Figure 1A top panels; Figure 1B,E). Platelet P-selectin expression as well as fibrin formation appeared more gradually over the first 3 minutes postinjury (Figure 1A,C-D,F-G). In FVQ/Q mice, the rate of initial platelet accumulation as well as total platelet mass size was significantly enhanced as compared with WT mice (Figure 1B,E; P < .05 vs WT). The increase in platelet accumulation was accompanied by significantly increased P-selectin expression and fibrin formation, both indicators of enhanced thrombin activity (Figure 1C-D,F-G; P < .0001 vs WT). Confocal imaging showed that P-selectin expression and fibrin formation were substantially increased within the intravascular platelet mass (Figure 1A bottom panels; supplemental Video 1).
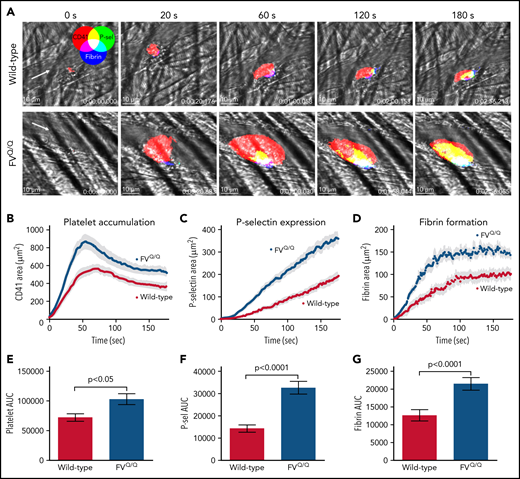
Laser-induced injury in WT vs FV Leiden mice. (A) Time lapse images from representative WT (top row) and FVQ/Q mice (bottom row) after laser injury in cremaster arterioles. Images are binary representations of 2D confocal fluorescence images overlaid on the brightfield. Platelets are shown in red, P-selectin+ area in green, and fibrin area in blue (overlay colors are shown in the color wheel). White arrow indicates the direction of blood flow. Scale bar in each image, 10 μm. (B-D) Graphs show CD41+, P-selectin+, and fibrin+ areas over time after laser injury of cremaster arterioles in WT (red) and FVQ/Q (blue) mice. Values are mean ± standard error of the mean (SEM). (E-G) Graphs show area under the curve (AUC; mean ± SEM) for platelets and P-selectin and fibrin areas over time. Statistics were calculated using the Mann-Whitney test; n = 49 injuries from 4 WT mice and 46 injuries from 4 FVQ/Q mice.
Laser-induced injury in WT vs FV Leiden mice. (A) Time lapse images from representative WT (top row) and FVQ/Q mice (bottom row) after laser injury in cremaster arterioles. Images are binary representations of 2D confocal fluorescence images overlaid on the brightfield. Platelets are shown in red, P-selectin+ area in green, and fibrin area in blue (overlay colors are shown in the color wheel). White arrow indicates the direction of blood flow. Scale bar in each image, 10 μm. (B-D) Graphs show CD41+, P-selectin+, and fibrin+ areas over time after laser injury of cremaster arterioles in WT (red) and FVQ/Q (blue) mice. Values are mean ± standard error of the mean (SEM). (E-G) Graphs show area under the curve (AUC; mean ± SEM) for platelets and P-selectin and fibrin areas over time. Statistics were calculated using the Mann-Whitney test; n = 49 injuries from 4 WT mice and 46 injuries from 4 FVQ/Q mice.
In separate studies, APC was inhibited using a blocking antibody that prevents PC from binding to membranes (Ab1609).22 We confirmed that Ab1609 (10 mg/kg IV) significantly inhibits APC anticoagulant activity using a thrombin generation assay in which control or antibody-treated mouse plasma was spiked with recombinant mouse APC (supplemental Figure 1). We found that Ab1609-treated mice had significantly increased platelet accumulation, P-selectin expression, and fibrin formation after laser-induced injury in cremaster arterioles as compared with control mice that received vehicle alone (Figure 2; supplemental Video 2). The effect of the antibody was even more pronounced than the enhanced response observed in FV Leiden mice. Platelet accumulation was so robust within the first minute postinjury that nearly all the injuries resulted in at least transient occlusion of blood flow within the vessel, and 25% of injuries resulted in stable vessel occlusion that did not reestablish flow within the 3-minute observation period (supplemental Figure 2). These occlusive events are in stark contrast to vehicle-treated mice, in which none of the vessels occluded, even transiently, as a result of platelet accumulation after laser injury. Confocal imaging revealed enhanced intravascular fibrin formation within the platelet mass and extending downstream from the injury site in Ab1609-treated mice (Figure 2A). Taken together, the results from the FVQ/Q mice and APC-blocking antibody–treated mice show that APC anticoagulant activity is an important regulator of thrombin generation after vascular injury in the mouse microcirculation.
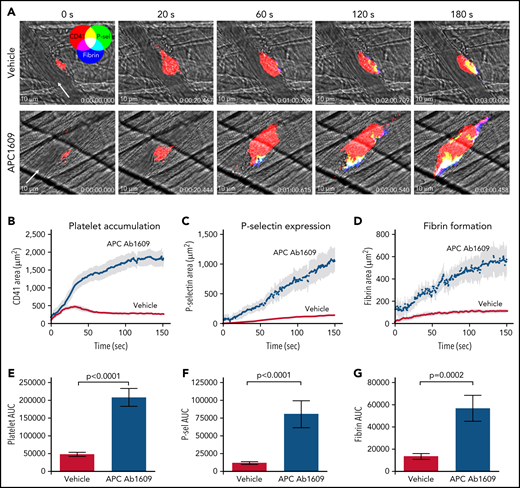
Inhibition of APC enhances the response to laser-induced injury in mouse cremaster arterioles. (A) Time lapse images from representative vehicle-treated (top row) and APC monoclonal Ab1609–treated mice (bottom row) after laser injury in cremaster arterioles. Images are binary representations of 2D confocal fluorescence images overlaid on the brightfield. Platelets are shown in red, P-selectin+ area in green, and fibrin area in blue (overlay colors are shown in the color wheel). White arrow indicates the direction of blood flow. Scale bar in each image, 10 μm. (B-D) Graphs show CD41+, P-selectin+, and fibrin+ areas over time after laser injury of cremaster arterioles in vehicle-treated (red) and Ab1609-treated mice (blue). Values are mean ± standard error of the mean (SEM). (E-G) Graphs show area under the curve (AUC; mean ± SEM) for platelets and P-selectin and fibrin areas over time. Statistics were calculated using the Mann-Whitney test; n = 13 injuries from 3 vehicle-treated mice and 10 injuries from 3 Ab1609-treated mice.
Inhibition of APC enhances the response to laser-induced injury in mouse cremaster arterioles. (A) Time lapse images from representative vehicle-treated (top row) and APC monoclonal Ab1609–treated mice (bottom row) after laser injury in cremaster arterioles. Images are binary representations of 2D confocal fluorescence images overlaid on the brightfield. Platelets are shown in red, P-selectin+ area in green, and fibrin area in blue (overlay colors are shown in the color wheel). White arrow indicates the direction of blood flow. Scale bar in each image, 10 μm. (B-D) Graphs show CD41+, P-selectin+, and fibrin+ areas over time after laser injury of cremaster arterioles in vehicle-treated (red) and Ab1609-treated mice (blue). Values are mean ± standard error of the mean (SEM). (E-G) Graphs show area under the curve (AUC; mean ± SEM) for platelets and P-selectin and fibrin areas over time. Statistics were calculated using the Mann-Whitney test; n = 13 injuries from 3 vehicle-treated mice and 10 injuries from 3 Ab1609-treated mice.
Regulation of the hemostatic response by APC in the mouse jugular vein
We next asked whether APC contributes to the regulation of thrombin generation in a large-vessel hemostasis model. In contrast to our observations in the microcirculation, we found no significant difference in total platelet accumulation between WT and FVQ/Q mice, either on the intraluminal side of the injury site or on the extraluminal component of the hemostatic plug (Figure 3A). Similarly, we did not observe any difference in P-selectin expression or fibrin formation in FVQ/Q mice as compared with WT (Figure 3B-C). There was no evidence of intravascular fibrin formation on the intact endothelium surrounding and downstream from the injury site. Accordingly, there was no difference in bleeding time in WT vs FVQ/Q mice in this injury model (Figure 3D).
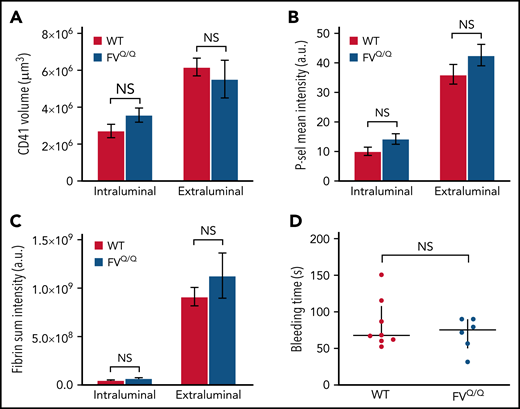
No effect of FV Leiden mutation on hemostatic plug formation after jugular vein puncture injury in mice. (A-C) Graphs show platelet accumulation (A), P-selectin expression (B), and fibrin accumulation (C) on the intra- and extravascular sides of the mouse jugular vein after puncture injury. Red columns are WT and blue FVQ/Q. Values are mean ± standard error of the mean. Statistics were performed using a 2-way analysis of variance with Bonferroni post hoc test for multiple comparisons; n = 8 WT and 6 FVQ/Q mice. (D) Bleeding time after jugular vein puncture injury in WT and FVQ/Q mice. Horizontal line is median and error bars interquartile range. Statistics were performed using the Mann-Whitney test. a.u., arbitrary unit; NS, not significant.
No effect of FV Leiden mutation on hemostatic plug formation after jugular vein puncture injury in mice. (A-C) Graphs show platelet accumulation (A), P-selectin expression (B), and fibrin accumulation (C) on the intra- and extravascular sides of the mouse jugular vein after puncture injury. Red columns are WT and blue FVQ/Q. Values are mean ± standard error of the mean. Statistics were performed using a 2-way analysis of variance with Bonferroni post hoc test for multiple comparisons; n = 8 WT and 6 FVQ/Q mice. (D) Bleeding time after jugular vein puncture injury in WT and FVQ/Q mice. Horizontal line is median and error bars interquartile range. Statistics were performed using the Mann-Whitney test. a.u., arbitrary unit; NS, not significant.
Similarly, we found that the hemostatic response in mice receiving the same concentration of Ab1609 as used in the microvascular injury model was not significantly different than that observed in vehicle-treated control mice (Figure 4). There was no difference in the extent of platelet accumulation, P-selectin expression, or fibrin formation (Figure 4A-C). As in the FVQ/Q mice, there was no evidence of fibrin formation on the intact endothelium adjacent to or downstream from the injury site in either vehicle- or APC-blocking antibody–treated mice. There was no significant difference in bleeding time after jugular vein puncture injury in vehicle- vs antibody-treated mice (Figure 4D). Taken together, the results from the FVQ/Q mice and Ab1609-treated mice suggest that APC does not participate in the regulation of thrombin activity after puncture injury of the mouse jugular vein.
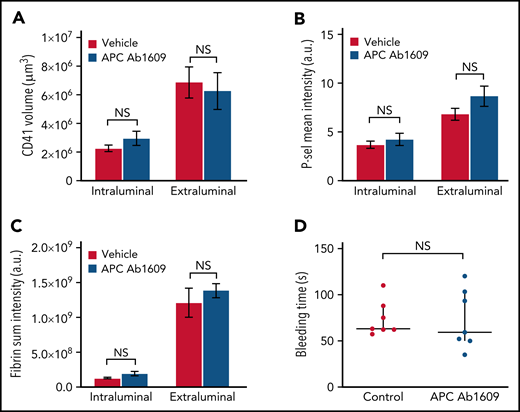
Inhibition of APC does not affect hemostatic plug formation after jugular vein puncture injury in mice. (A-C) Graphs show platelet accumulation (A), P-selectin expression (B), and fibrin accumulation (C) on the intra- and extravascular sides of the mouse jugular vein after puncture injury. Red columns are vehicle treated and blue Ab1609 treated. Values are mean ± standard error of the mean. Statistics were performed using a 2-way analysis of variance with Bonferroni post hoc test for multiple comparisons; n = 7 vehicle- and 7 Ab1609-treated mice. (D) Bleeding time after jugular vein puncture injury in vehicle- and Ab1609-treated mice. Horizontal line is median and error bars interquartile range. Statistics were performed using the Mann-Whitney test. a.u., arbitrary unit; NS, not significant.
Inhibition of APC does not affect hemostatic plug formation after jugular vein puncture injury in mice. (A-C) Graphs show platelet accumulation (A), P-selectin expression (B), and fibrin accumulation (C) on the intra- and extravascular sides of the mouse jugular vein after puncture injury. Red columns are vehicle treated and blue Ab1609 treated. Values are mean ± standard error of the mean. Statistics were performed using a 2-way analysis of variance with Bonferroni post hoc test for multiple comparisons; n = 7 vehicle- and 7 Ab1609-treated mice. (D) Bleeding time after jugular vein puncture injury in vehicle- and Ab1609-treated mice. Horizontal line is median and error bars interquartile range. Statistics were performed using the Mann-Whitney test. a.u., arbitrary unit; NS, not significant.
We confirmed that both thrombomodulin and EPCR are expressed on the endothelium of the mouse jugular vein (supplemental Figure 3) and cremaster microcirculation (data not shown). These findings rule out absence of thrombomodulin and/or EPCR expression as an explanation for the lack of involvement of PC anticoagulant activity during jugular vein hemostasis.
Computational studies on thrombin spatial distribution during hemostasis in different physiologic contexts
An alternative explanation for the finding that PC does not regulate thrombin activity in the setting of jugular vein hemostasis is that the thrombin generated remains primarily extravascular, with little intravascular thrombin available to activate PC on the endothelial cell surface. To investigate this possibility, we constructed computational simulations based on the geometry and physics of either the mouse jugular vein or cremaster arterioles to help visualize the spatial localization of thrombin and APC (supplemental Figure 4). Our model combined blood flow, molecular transport, and a simplified surface reaction network within 3D geometries of the vessel, injury site, and hemostatic plug. Hemostatic plugs were simulated as homogeneous porous media, as previously done by our group,24 with geometries informed by experimental studies. Prothrombin and PC were perfused at constant concentrations of 1000 and 60 nM, respectively. Thrombin was generated from the activation of bound prothrombin on procoagulant surfaces. APC was generated by thrombin-mediated cleavage of PC on intraluminal surfaces (supplemental Figures 5 and 6). The cremaster model was characterized by slow blood flow (0.2 cm/s; Figure 5Bi), small injury size (3-4 μm in diameter), and a thin vessel wall (5 μm). In the jugular injury model, blood flow was faster (10 cm/s; Figure 5Ci), injury size larger (300 μm in diameter), and vessel wall thicker (50 μm; Figure 5).
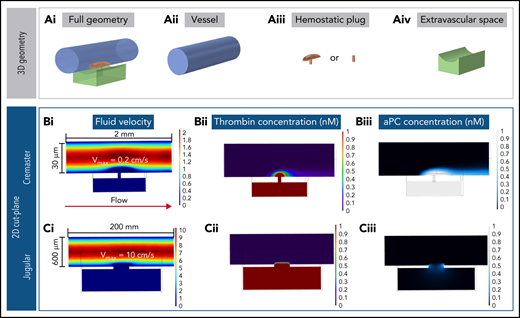
Computational modeling simulations recapitulate experimental results. (A) Geometries used for the computational models, shown in full (i) and deconstructed into the individual components: –vessel (ii), hemostatic plug (iii), and extravascular space (iv). Supplemental Figure 5 and supplemental Table 1 provide more detailed illustrations and dimensions. (B) Fluid velocity field (i), thrombin (ii), and APC (iii) concentration fields in the cremaster geometry model. The sheltering action of platelets allows the thrombin generated both intra- and extravascularly to diffuse through the hemostatic mass; more intravascular thrombin results in APC being generated, and the APC has increased residence time within the platelet mass. (C) Fluid velocity field (i), thrombin (ii), and APC (iii) concentration fields in the jugular geometry model. Thrombin backpropagates via diffusion through the hemostatic mass, but as soon as it diffuses out of the platelet mass, it is quickly washed away. The thrombin that diffuses through the sides of the plug produces only low amounts of APC. Vmax, maximum velocity.
Computational modeling simulations recapitulate experimental results. (A) Geometries used for the computational models, shown in full (i) and deconstructed into the individual components: –vessel (ii), hemostatic plug (iii), and extravascular space (iv). Supplemental Figure 5 and supplemental Table 1 provide more detailed illustrations and dimensions. (B) Fluid velocity field (i), thrombin (ii), and APC (iii) concentration fields in the cremaster geometry model. The sheltering action of platelets allows the thrombin generated both intra- and extravascularly to diffuse through the hemostatic mass; more intravascular thrombin results in APC being generated, and the APC has increased residence time within the platelet mass. (C) Fluid velocity field (i), thrombin (ii), and APC (iii) concentration fields in the jugular geometry model. Thrombin backpropagates via diffusion through the hemostatic mass, but as soon as it diffuses out of the platelet mass, it is quickly washed away. The thrombin that diffuses through the sides of the plug produces only low amounts of APC. Vmax, maximum velocity.
In both the cremaster and jugular vein simulations, when blood flowed freely through the injury site (ie, simulating conditions before platelet plug formation), prothrombin encountered procoagulant surfaces in the extravascular compartment and was converted to thrombin. The thrombin generated was unable to backpropagate across the injury site because of the convective effects of the extravasating blood (supplemental Figure 7). Next, platelet plugs were added to seal the injury site (Figure 5A). In the cremaster simulations, thrombin formed a spatial gradient that extended throughout the platelet mass and diffused to adjacent endothelial surfaces in the intravascular compartment (Figure 5Bii) to generate APC (Figure 5Biii). By contrast, in the jugular vein model, the combination of increased flow, thicker vessel wall, and larger injury site was found to prevent thrombin from backpropagating into the vessel lumen (Figure 5Cii) and generating APC (Figure 5Ciii).
To gain further insight into how blood flow and vessel injury geometry interact to produce the observed differences in PC activation, we ran additional simulations using the jugular model. We created 4 model geometries with increasing injury size diameter (20, 50, 100, and 300 μm) and ran simulations under 3 flow velocity profiles (0.2, 2, and 10 cm/s). We found that increasing injury size led to reduced average thrombin concentration (supplemental Figure 8A) because larger injuries present smaller surface area/volume ratios. Furthermore, although flow velocity had little effect on thrombin generation, it had profound effects on APC generation because of the rapid convection of thrombin and/or APC away from endothelial cell surfaces exposed to flow (supplemental Figure 8A-B). Notably, a decrease in thrombin concentration was not necessarily followed by a decrease in APC generation (supplemental Figure 8A-B).
We also created 2 additional cremaster models: a model with thrombin generation limited to extraluminal surfaces, and a model with extraluminal thrombin generation and a much smaller hemostatic plug (Figure 6A). Interestingly, the localization of thrombin generation did not significantly alter APC concentration, because thrombin was able to diffuse throughout the platelet mass efficiently at the small length scales involved (Figure 6Bi,Ci vs 6Bii,Cii). However, platelet plug geometry had a dramatic effect, with significant APC generation only in the case where the platelet mass covered endothelial cell surfaces adjacent to the injury site (Figure 6Bii,Cii vs 6Biii,Ciii). These observations suggest that the existence of a sheltered region for thrombin activity is important for thrombin to find suitable endothelial cell surfaces for APC generation.
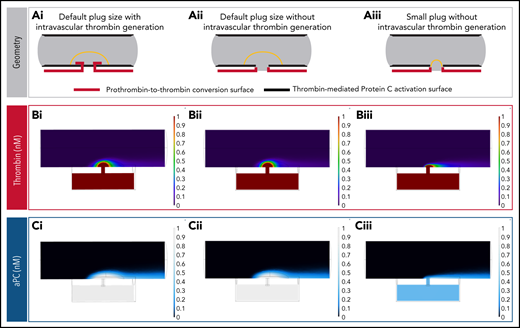
Impact of hemostatic plug size on thrombin and APC concentration. (A) Reaction surface distribution. PC is converted to APC on all surfaces lining the vessel lumen (bold black lines). Thrombin generation occurs on surfaces of the extravascular compartment and the injury site walls, with or without intravascular surfaces that are protected by the hemostatic plug (bold red lines): default plug size with intravascular thrombin generation (i), default plug size without intravascular thrombin generation (ii), and small plug size without intravascular thrombin generation (iii). (B) Thrombin concentration fields for each of the geometries described in panel A. When the intravascular thrombin generation surfaces are removed without changing the shape or size of the hemostatic plug, the resulting thrombin concentration field remains virtually unchanged (i vs ii). This occurs because the thrombin generated on the injury walls and extravascular space can backpropagate in the lumen sheltered by the hemostatic plug via diffusion. (C) APC concentration fields for each of the geometries described in panel A. Note the reduction in APC concentration when reaction surfaces capable of generating APC become unsheltered from flow (iii).
Impact of hemostatic plug size on thrombin and APC concentration. (A) Reaction surface distribution. PC is converted to APC on all surfaces lining the vessel lumen (bold black lines). Thrombin generation occurs on surfaces of the extravascular compartment and the injury site walls, with or without intravascular surfaces that are protected by the hemostatic plug (bold red lines): default plug size with intravascular thrombin generation (i), default plug size without intravascular thrombin generation (ii), and small plug size without intravascular thrombin generation (iii). (B) Thrombin concentration fields for each of the geometries described in panel A. When the intravascular thrombin generation surfaces are removed without changing the shape or size of the hemostatic plug, the resulting thrombin concentration field remains virtually unchanged (i vs ii). This occurs because the thrombin generated on the injury walls and extravascular space can backpropagate in the lumen sheltered by the hemostatic plug via diffusion. (C) APC concentration fields for each of the geometries described in panel A. Note the reduction in APC concentration when reaction surfaces capable of generating APC become unsheltered from flow (iii).
Overall, the results of the computational studies show that the combination of convective forces from extravasating blood, the shape of the hemostatic plug, and the ability of thrombin generated extravascularly to diffuse across the platelet mass determine the extent of PC activation in different vascular contexts.
Contribution of PC anticoagulant activity after intravascular thrombin generation in the mouse jugular vein
To further examine the importance of thrombin localization for APC anticoagulant activity, we examined the effect of APC inhibition after FeCl3-induced vascular injury, because prior studies have demonstrated that this model results in intravascular thrombin activity.31,32 We performed the model in the mouse jugular vein so that direct comparisons could be made with our jugular vein hemostasis studies. Application of 5% FeCl3 to the adventitial surface of the mouse jugular vein resulted in a subocclusive thrombus composed of platelets, fibrin, and red blood cells in vehicle-treated mice without causing a penetrating injury. Real-time imaging showed platelet and fibrin formation occurred with similar kinetics after an initial lag of ∼2 minutes, followed by a growth phase that began to plateau by 7 minutes post-FeCl3 application (Figure 7Ai,Bi). Interestingly, platelet accumulation and fibrin formation occurred with faster kinetics in Ab1609-treated mice (Figure 7A-B), as demonstrated by a statistically significant increase in platelet and fibrin accumulation 3 minutes post-FeCl3 application (Figure 7Aii,Bii). The difference in the early stages of thrombus formation was dramatic when imaged using scanning electron microscopy (Figure 7C-F; supplemental Figure 9). Initial platelet and red blood cell accumulation with minimal fibrin formation on the surface of the endothelium was observed 3 minutes post-FeCl3 application in vehicle-treated mice (Figure 7C-D). In contrast, there was a thick fibrin mat embedded with red blood cells and platelet aggregates at the same time point in mice treated with the APC-blocking antibody (Figure 7E-F). These results demonstrate the critical role that APC anticoagulant activity plays in the regulation of thrombin generation in the early stages of intravascular thrombus formation.
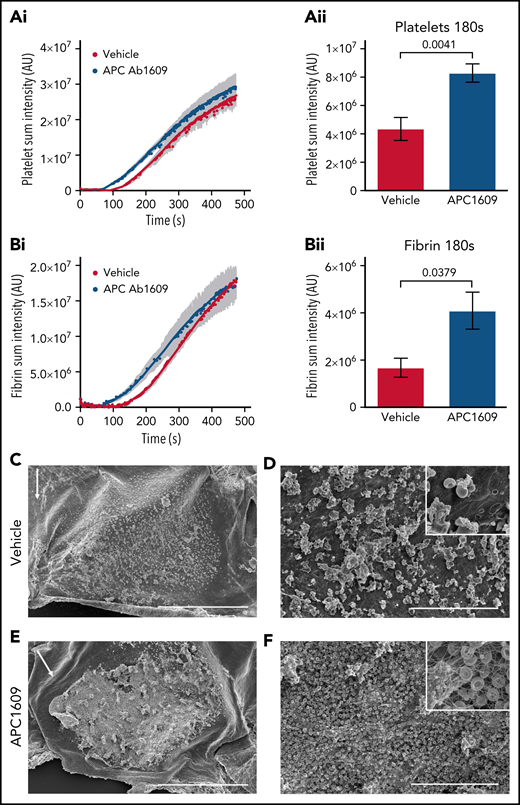
Inhibition of APC enhances thrombus formation after FeCl3 injury of the mouse jugular vein. (A) Graphs show platelet accumulation in vehicle- (red) and Ab1609-treated (blue) mice after FeCl3 induced injury of the mouse jugular vein: platelet accumulation over the entire time course of the experiment (i), and platelet accumulation 180 seconds post-FeCl3 removal (ii). (B) Corresponding graphs showing fibrin accumulation after FeCl3 induced injury. All values are mean ± standard error of the mean. Statistics were performed using the Mann-Whitney test; n = 7 vehicle- and 7 Ab1609-treated mice. (C-F) Scanning electron microscopy images of the intravascular side of the jugular vein after FeCl3 injury. (C-D) Representative thrombus formed in a vehicle-treated mouse 3 minutes after removal of FeCl3. Scale bars, 500 and 30 μm, respectively. (E-F) Corresponding images of a representative thrombus from an Ab1609-treated mouse. Insets in panels D and F show a higher-magnification view of the vessel surface; note the exposed endothelium and lack of fibrin formation in the vehicle-treated mouse (panel D inset) vs a fibrin-, red blood cell–, and platelet-rich thrombus in the presence of Ab1609. White arrows indicate the direction of blood flow (C,E). Images are representative of n = 4 mice in each group (images from additional mice are included in supplemental Figure 9).
Inhibition of APC enhances thrombus formation after FeCl3 injury of the mouse jugular vein. (A) Graphs show platelet accumulation in vehicle- (red) and Ab1609-treated (blue) mice after FeCl3 induced injury of the mouse jugular vein: platelet accumulation over the entire time course of the experiment (i), and platelet accumulation 180 seconds post-FeCl3 removal (ii). (B) Corresponding graphs showing fibrin accumulation after FeCl3 induced injury. All values are mean ± standard error of the mean. Statistics were performed using the Mann-Whitney test; n = 7 vehicle- and 7 Ab1609-treated mice. (C-F) Scanning electron microscopy images of the intravascular side of the jugular vein after FeCl3 injury. (C-D) Representative thrombus formed in a vehicle-treated mouse 3 minutes after removal of FeCl3. Scale bars, 500 and 30 μm, respectively. (E-F) Corresponding images of a representative thrombus from an Ab1609-treated mouse. Insets in panels D and F show a higher-magnification view of the vessel surface; note the exposed endothelium and lack of fibrin formation in the vehicle-treated mouse (panel D inset) vs a fibrin-, red blood cell–, and platelet-rich thrombus in the presence of Ab1609. White arrows indicate the direction of blood flow (C,E). Images are representative of n = 4 mice in each group (images from additional mice are included in supplemental Figure 9).
Discussion
The studies presented here explicitly examined the relationship between thrombin spatial distribution at sites of vascular injury in vivo, PC activation, and APC anticoagulant function. They show that activation of PC and its anticoagulant activity are highly dependent on thrombin spatial distribution. Furthermore, the results show that differences in thrombin spatial distribution result in disparate contributions of the APC system to the regulation of thrombin generation in different vascular contexts. Specifically, using a combination of in vivo and in silico approaches, we found that thrombin diffusion across a platelet plug results in intravascular thrombin activity and PC activation during hemostasis in the microcirculation. In contrast, localization of thrombin activity to the extravascular compartment in the context of large-vessel hemostasis results in minimal PC activation and no appreciable impact of APC anticoagulant activity on the regulation of thrombin generation. Finally, we found that intravascular thrombin activity is regulated by APC anticoagulant activity in the early stages of large-vessel thrombosis, consistent with the known antithrombotic function of the PC system. These findings provide a mechanistic basis for context-dependent contribution of APC to the regulation of hemostasis and thrombosis in vivo.
Prior studies have suggested that the contribution of APC to thrombin regulation may vary in different vascular beds. It was proposed that APC may contribute a more substantial role in the microcirculation than in large vessels, consistent with the clinical manifestation of microvascular thrombosis in the setting of PC or PS deficiency.7 A proposed mechanistic explanation for the apparent context-dependent contribution of APC is the difference in vessel wall surface area/blood volume ratio in the microcirculation vs macrocirculation and its consequences for thrombin binding to thrombomodulin.33 The results of the current study show that thrombin spatial distribution at a site of vascular injury, influenced by vessel geometry, injury size, and local flow conditions, is also an important determinant of APC anticoagulant function.
The in vivo studies reported show that the spatial distribution of thrombin activity at a site of vascular injury is context dependent. In a microvasculature injury model, fibrin formation and platelet surface P-selectin expression, both markers of thrombin activity, were observed extending from the injury site into the platelet plug within the intravascular compartment. In a macrovasculature injury model, fibrin formation and P-selectin expression were found primarily at the injury site and extending into the extravascular compartment, with no evidence of intravascular thrombin activity extending beyond the injury site. The mechanisms responsible for these distinct spatial distributions are multifactorial, as explored by computational simulations in the present study. In both cases, immediately after injury, the convective forces associated with blood flowing out through the injury site prevent any thrombin initially generated in the extravascular compartment from backpropagating into the vessel lumen. Once platelets fill the injury site and stop extravasating flow, thrombin transport occurs through the platelet mass and is dominated by hindered diffusion.24,34,35 Although not incorporated into the computational simulations, thrombin inhibition by antithrombin would further restrict its spatial distribution as it diffuses through the platelet mass. Finally, any thrombin that escapes the platelet mass in the vessel lumen is rapidly convected away by flowing blood, which highlights the important role of the platelet plug as a sheltered environment for thrombin and other biochemical species during the hemostatic process.
The combination of in vivo and in silico results presented here further shows that thrombin spatial distribution influences the extent to which APC is generated and contributes to the regulation of thrombin formation. The large impact of APC inhibition in the microvascular injury model is associated with the localization of thrombin activity in the vascular lumen. These findings are consistent with recent studies showing the potential of APC system inhibition to restore the hemostatic balance in the setting of hemophilia.18,19,36 In contrast, APC did not seem to play a role in the regulation of thrombin activity in the large-vessel hemostasis model used here. This finding is unlikely to be explained by a diminished potential for PC activation on the endothelium of the mouse jugular vein. Both thrombomodulin and EPCR are expressed and an APC anticoagulant function is observed during thrombosis when thrombin activity is localized intravascularly. Instead, the lack of a role for APC in the jugular vein hemostasis model may be explained by the primarily extravascular localization of thrombin activity in this setting, such that minimal APC is generated. This explanation is supported by results of computation simulations that showed very little APC formation, thus recapitulating experimental results obtained in vivo.
The computational studies reported demonstrate a role for convective flow in regulating APC generation, both by limiting thrombin binding to the endothelium and by rapidly removing any APC that may be generated immediately adjacent to the injury site. Thus, the APC anticoagulant system is particularly adapted to settings of intravascular thrombin generation with low flow. Indeed, recent studies have shown upregulation of anticoagulant proteins, including thrombomodulin and EPCR, on the surface of endothelial cells in regions of the vasculature exposed to abnormal flow regimes, such as in the pocket formed behind venous valves.37-39 The contribution of APC anticoagulant activity is critical in such regions, as demonstrated by the enhanced risk of deep vein thrombosis in people with the FV Leiden mutation.
In summary, the studies reported here show that APC anticoagulant function is context dependent in settings of hemostasis and thrombosis. The extent of PC activation at sites of vascular injury is determined in large part by spatial distribution of thrombin activity, in conjunction with local physical conditions. These findings provide a mechanistic basis for context-dependent contributions of APC anticoagulant activity and its role in the regulation of thrombin generation.
Acknowledgments
The authors thank Andrea Stout and Yuri Veklich of the CDB Microscopy Core at the University of Pennsylvania for assistance with 2-photon and scanning electron microscopy imaging. The visual abstract was created with BioRender.com.
This study was supported by research funding from the National Heart, Lung and Blood Institute, National Institutes of Health (P01-HL139420 [T.J.S.], P01-HL146373 [LF.B. and T.J.S.], and R21-HL153946 [M.T. and T.S.]), the American Heart Association (19TPA34880016 [T.J.S.]), and the Basic Research Award from the Bayer Hemophilia Awards Program (M.T.).
Authorship
Contribution: T.T.M., C.N.M., and J.W. performed experiments and data analysis; C.T.E. provided vital reagents and data interpretation; T.J.S. contributed to computational studies and data analysis; L.F.B. provided data interpretation and analysis; T.J.S. and M.T. designed studies, performed experiments, and wrote the manuscript; and all authors edited and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Timothy J. Stalker, Cardeza Foundation for Hematologic Research, Thomas Jefferson University, 1020 Locust St, JAH 394B, Philadelphia, PA 19107; e-mail: timothy.stalker@jefferson.edu; and Maurizio Tomaiuolo, Vickie and Jack Farber Vision Research Center at Wills Eye, Wills Eye Hospital, 840 Walnut St, Philadelphia, PA 19107; e-mail: mtomaiuolo@willseye.org.
Requests for data sharing may be made directly to the corresponding authors via e-mail.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal