


Abstract
Primary cutaneous T-cell lymphomas (CTCLs) constitute a heterogeneous group of non-Hodgkin T-cell lymphomas that present in the skin. In recent years, significant progress has been made in the understanding of the pathogenesis of CTCLs. Progress in CTCL classifications combined with technical advances, in particular next-generation sequencing, enabled a more detailed analysis of the genetic and epigenetic landscape and transcriptional changes in clearly defined diagnostic entities. These studies not only demonstrated extensive heterogeneity between different CTCL subtypes but also identified recurrent alterations that are highly characteristic for diagnostic subgroups of CTCLs. The identified alterations, in particular, involve epigenetic remodeling, cell cycle regulation, and the constitutive activation of targetable oncogenic pathways. In this respect, aberrant JAK-STAT signaling is a recurrent theme; however, it is not universal for all CTCLs and has seemingly different underlaying causes in different entities. A number of the mutated genes identified are potentially actionable targets for the development of novel therapeutic strategies. Moreover, these studies have produced an enormous amount of information that will be critically important for the further development of improved diagnostic and prognostic biomarkers that can assist in the clinical management of patients with CTCL. In the present review, the main findings of these studies in relation to their functional impact on the malignant transformation process are discussed for different subtypes of CTCLs.
Introduction
Primary cutaneous T-cell lymphomas (CTCLs) constitute a heterogeneous group of non-Hodgkin T-cell lymphomas that present in the skin. Clinicopathologic studies combined with long-term follow-up were critically important in defining different types of CTCLs with highly characteristic clinical and histologic features. These studies also demonstrated that CTCL differ in clinical behavior and prognosis compared with morphologically similar nodal lymphomas that may involve the skin secondarily.
As a result of these studies, primary CTCLs were included as distinct entities in current lymphoma classifications. The first classification dedicated to cutaneous lymphomas was the World Health Organization–European Organization for Research and Treatment of Cancer (WHO-EORTC) consensus classification published in 2005 that was subsequently incorporated in the 2008 WHO classification and its 2018 revision.1 These advances in lymphoma classification allow clinicians to correctly diagnose and select appropriate treatment in CTCL (see Table 1 for updated classification of CTCLs).
Relative frequency and prognosis of primary cutaneous T-cell lymphomas included in the 2018 update of the WHO-EORTC classification
| WHO-EORTC Classification 2018 | Frequency, % | 5-y DSS, % |
|---|---|---|
| CTCL | ||
| MF | 39 | 88 |
| MF variants | ||
| Folliculotropic MF | 5 | 75 |
| Pagetoid reticulosis | <1 | 100 |
| Granulomatous slack skin | <1 | 100 |
| Sz | 2 | 36 |
| Adult T-cell leukemia/lymphoma | <1 | NDA |
| Primary cutaneous CD30+ LPDs | ||
| C-ALCL | 8 | 95 |
| LyP | 12 | 99 |
| Subcutaneous panniculitis-like T-cell lymphoma | 1 | 87 |
| Extranodal NK/T-cell lymphoma, nasal type | <1 | 16 |
| Chronic active Epstein-Barr virus infection | <1 | NDA |
| Primary cutaneous peripheral T-cell lymphoma, rare subtypes | ||
| Primary cutaneous γ/δ T-cell lymphoma | <1 | 11 |
| CD8+ AECyTCL (provisional) | <1 | 31 |
| Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder (provisional) | 6 | 100 |
| Primary cutaneous acral CD8+ T-cell lymphoma (provisional) | <1 | 100 |
| Primary cutaneous peripheral T-cell lymphoma, NOS | 2 | 15 |
| WHO-EORTC Classification 2018 | Frequency, % | 5-y DSS, % |
|---|---|---|
| CTCL | ||
| MF | 39 | 88 |
| MF variants | ||
| Folliculotropic MF | 5 | 75 |
| Pagetoid reticulosis | <1 | 100 |
| Granulomatous slack skin | <1 | 100 |
| Sz | 2 | 36 |
| Adult T-cell leukemia/lymphoma | <1 | NDA |
| Primary cutaneous CD30+ LPDs | ||
| C-ALCL | 8 | 95 |
| LyP | 12 | 99 |
| Subcutaneous panniculitis-like T-cell lymphoma | 1 | 87 |
| Extranodal NK/T-cell lymphoma, nasal type | <1 | 16 |
| Chronic active Epstein-Barr virus infection | <1 | NDA |
| Primary cutaneous peripheral T-cell lymphoma, rare subtypes | ||
| Primary cutaneous γ/δ T-cell lymphoma | <1 | 11 |
| CD8+ AECyTCL (provisional) | <1 | 31 |
| Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder (provisional) | 6 | 100 |
| Primary cutaneous acral CD8+ T-cell lymphoma (provisional) | <1 | 100 |
| Primary cutaneous peripheral T-cell lymphoma, NOS | 2 | 15 |
Modified from reference 1.
MF, mycosis fungoides; Sz, Sézary syndrome; CD30+ LPDs, CD30+ lymphoproliferative diseases, C-ALCL, cutaneous anaplastic large cell lymphoma; LyP, Lymphomatoid papulosis; CD8+ AECyTCL, primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma; DSS, disease-specific survival; NDA, no data available; NOS, not otherwise specified.
However, until recently, the molecular alterations underlying initiation and progression of different CTCL subtypes remained largely unknown. Early cytogenetic studies demonstrated widespread genomic instability and complex karyotypes, but recurrent translocations or other recurrent genetic alterations were not found. The heterogeneity in the results of these studies combined with the lack of appropriate mouse models and limited number of informative cell lines have hampered progress in this field.
Recent technical advances including array-based comparative genome hybridization (aCGH) and, in particular, next-generation sequencing (NGS) enabled a more detailed analysis of genetic, epigenetic, and transcriptional changes in CTCL tumor cells. These studies confirmed the extensive heterogeneity between and within CTCL subtypes but also identified recurrent molecular alterations affecting oncogenic pathways in different types of CTCLs. In the present review, the main findings of these studies for different subtypes of CTCL are presented.
Genetic alterations in Sézary syndrome: copy number alterations
Early studies on genetic alterations in Sézary syndrome (Sz; mainly using arrays and cytogenetics) already identified extensive genetic instability with complex karyotypes.2-4 A series of genomic studies using aCGH or single nucleotide polymorphism arrays identified broad chromosomal regions affected by recurrent copy number alterations (CNAs). Significant CNA gains were detected in regions with known and bona fide oncogenes, like MYC,2TOX,5 and losses in regions harboring tumor suppressor genes like TP53,2,6PTEN, RB1, and ZEB1,5,7,8CDKN2A-CDKN2B,9E2A,10DNMT3A, USP28, CAAP, NCOR1,11 and TNFAIP3.12 In addition to the genes above, deletions of RPA1, HIC1, and DUSP5 and gain of STAT3/STAT5 and IL-2 (receptor) genes were reported.2 Caprini et al13 noted that more than 3 of these recurrent chromosomal alterations (gain or loss) are significantly correlated with Sz prognosis.
This characteristic pattern of gains and losses was confirmed, with a higher resolution, by massive parallel sequencing (NGS) of large cohorts of patients with Sz. Choi et al14 not only identified identical regions as being recurrently deleted and gained in Sz but, strikingly, also with a similar prevalence as described earlier.2,13 This hallmark CNA signature, also in agreement with results from Wang et al,15 is depicted in Figure 1 and, on the one hand, underscores the heterogeneity of the genetic alterations in this disease, but on the other hand, illustrates the similarity (in terms of tumor genetics) of seemingly heterogenous patients in cohorts analyzed in different centers using different platforms. Based on the NGS data, an 11-probe fluorescence in situ hybridization panel was developed that largely confirmed the findings for Sz and thus may offer the capacity to facilitate diagnosis.16 Cyrenne et al17 linked the common gene alterations in Sz (STAT3/5B amplifications, P53 deletions) to inhibition of apoptosis by increased BCL2 activity and showed clinical efficacy of the BCL2 inhibitor venetoclax in the treatment of advanced CTCL with blood involvement.
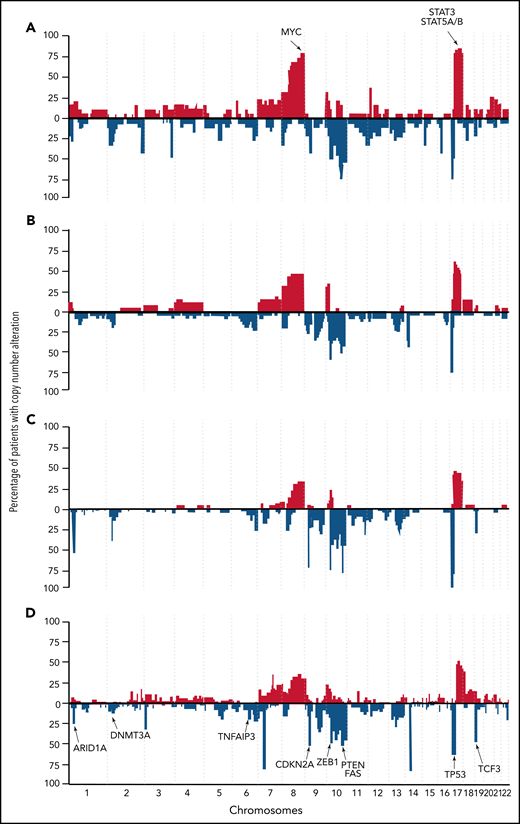
Schematic representation of CNAs in DNA of tumor cells of patients with Sz determined for independent cohorts using different platforms. Gains/amplifications are in red (on top of the x-axis) and losses/deletions in blue (below the x-axis). (A) Twenty patients using aCGH (Vermeer et al2). (B) Twenty-eight patients using single nucleotide polymorphism arrays (Caprini et al13). (C) Forty patients using NGS (Choi et al14). (D) Thirty-seven patients using NGS (Wang et al15). Location of genes recurrently affected by CNA in Sz are indicated. Irrespective of the technique/laboratory/protocol used, not only comparable aberrant chromosomal areas are identified but also with a similar frequency.
Schematic representation of CNAs in DNA of tumor cells of patients with Sz determined for independent cohorts using different platforms. Gains/amplifications are in red (on top of the x-axis) and losses/deletions in blue (below the x-axis). (A) Twenty patients using aCGH (Vermeer et al2). (B) Twenty-eight patients using single nucleotide polymorphism arrays (Caprini et al13). (C) Forty patients using NGS (Choi et al14). (D) Thirty-seven patients using NGS (Wang et al15). Location of genes recurrently affected by CNA in Sz are indicated. Irrespective of the technique/laboratory/protocol used, not only comparable aberrant chromosomal areas are identified but also with a similar frequency.
Genetic alterations in Sz: DNA rearrangements and fusion transcripts
To date, the only recurrent DNA rearrangement giving rise to a recurring fusion transcript in Sz is a CTLA4/CD28 transcript identified in 3 patients by independent groups.15,18,19 CD28 is expressed in normal T cells and involved in a cascade of activation signals including cell proliferation but also transcription of CTLA4. CTLA4, in turns, counteracts the effects of CD2820 and inhibits proliferation. In the fusion gene, the activating tail of CD28 replaces the inhibitory cytoplasmic tail of CTLA4. Therefore, this fusion could be considered a novel aberrant stimulatory mechanism enhancing oncogenic proliferation and survival.20
Many more in-frame fusion transcripts are detected by RNA-seq analysis (Table 2). Wang et al15 identified 41 in 37 patients (29 of them were successfully validated), whereas Prasad et al21 identified 86 potential fusion transcripts (7 validated) in 10 Sezary samples. Iżykowska et al11 identified 9 new fusion events in frame in 9 Sz patients; 5 fusion transcripts were described as ectopically expression of fragments of genes not expressed in normal T lymphocytes. These observations collectively underscore the genetic instability of Sz genome but have no clinal value (yet).
Fusion transcripts in Sézary syndrome
| Sample | Fusion transcript | Breakpoints (DNA) | Breakpoint type | Event class | WGS confirmed |
|---|---|---|---|---|---|
| Izykowska et al11 | |||||
| P1 | EHD1-CAPN12 | chr11: 64 628,862-chr19: 39 235,710 | Genic-genic | CTX | Yes |
| P4 | CEP57-NAT10 | ch11:95 529,671-chr16:8 872 293 | Genic-genic | CTX | Yes |
| P4 | TMEM66-BAIAP2 | chr8:29 928,614-chr17:79 602,847 | Genic-genic | CTX | Yes |
| P6 | MBD4-PTPRC | chr3:129 151,514-chr1:198 647,866 | Genic-genic | CTX | Yes |
| P6 | PTPRC-CPN2 | chr1: 198 644,193-chr2:39 030,368 | Genic-genic | CTX | Yes |
| P6 | MYB-MBNL1 | chr6:135 518,448-chr3:152 111,859 | Genic-genic | CTX | Yes |
| P6 | TFG-GPR128 | chr3: 100 334,867-chr3:100 446,145 | Genic-genic | iDel | Yes |
| P8 | ARMC6-DHX57-CNRIP | chr2:68 543,463-chr2:39 049,173–39 050, 512-chr2: 69 247,504 | Genic-genic-Genic | ITX | Yes |
| P8 | MAP4K3-FIGLA | chr2:71 014,668-chr2:39 563,447 | Genic-genic | ITX | Yes |
| P8 | DCP1-CCl27 | chr3: 53 370,567-chr15: 44 079,110 | Genic-genic | CTX | Yes |
| Wang et al15 | |||||
| SPZ-002 | CARD11-PIKR3 | chr7:2968232-chr1:46512297 | Genic-genic | CTX | Yes |
| SPZ-002 | USP32-RAD51B | chr17:58422842-chr14:68878141 | Genic-genic | CTX | Yes |
| SPZ-010 | CTSC-RAB38 | chr11:88033698-chr11:87883123 | Genic-genic | ITX | Yes |
| SPZ-011 | ICOS-CD28 | chr 2:204801595-chr2:204591356 | Genic-nongenic | iDel | Yes |
| SPZ-012 | C15orf57-CBX3 | chr15:40854180-chr7:26241365 | Nongenic genic | CTX | Yes |
| SPZ-013 | PSMG4-WNK2 | chr6:3259430-chr9:95991978 | Genic-genic | CTX | Yes |
| SPZ-014 | MYCBP-SLC25A33 | chr1:39338690-chr1:9613684 | Genic-genic | ITX | Yes |
| SPZ-015 | MED22-SURF6 | chr9:136210989-chr9:136202883 | Genic-genic | iDel | Yes |
| SPZ-015 | FAM186A-DIP2B | chr12:50750031-chr12:50899023 | Genic-genic | iDel | Yes |
| SPZ-016 | RBMS1-GLI2 | chr2:161223727-chr2:121727969 | Genic-genic | ITX | Yes |
| SPZ-016 | C15orf57-CBX3 | chr15:40854974-chr7:26241386 | Genic-genic | CTX | Yes |
| SPZ-017 | SLC35A3-HIAT1 | chr1:100480976-chr1:100515465 | Genic-genic | iDel | Yes |
| SPZ-017 | RPL11-TCEB3 | chr1:24021281-chr1:24075511 | Genic-genic | iDel | Yes |
| SPZ-020 | SLC35A3-HIAT1 | chr1:100480976-chr1:100515465 | Genic-genic | iDel | Yes |
| SPZ-021 | ZFP64-WBP1L | chr20:50776662-chr10:104569650 | Genic-genic | CTX | Yes |
| SPZ-022 | ELMO1-TNRC18 | chr7:37136224-chr7:5434226 | Genic-genic | ITX | Yes |
| SPZ-028 | GPR155-CIR1 | chr2:175304628-chr2:175260365 | Genic-genic | iDel | Yes |
| SPZ-028 | CAB39-C%orf15 | chr2:231682612-chr5:133292681 | Genic-genic | CTX | Yes |
| SPZ-028 | GOLGB1-IQCG | chr3:121445760-chr3:197665651 | Genic-genic | ITX | Yes |
| SPZ-029 | IKZF2-GLI2 | chr2:213921635-chr2:121684937 | Genic-genic | iDel | Yes |
| SPZ-030 | MORF4L1-B2M | chr15:79170555-chr15:45003811 | Genic-genic | ITX | Yes |
| SPZ-030 | TTC39C-MEP1B | chr18:21663045-chr18:29771785 | Genic-genic | ITX | Yes |
| SPZ-031 | CIC-EXOSC5 | chr19:42790923-chr19:41903086 | Genic-genic | iDel | Yes |
| SPZ-033 | PPFIA1-NFKB2 | chr11:70218351-chr10:104160534 | Genic-genic | CTX | Yes |
| SPZ-033 | KDM4C-UNC13B | chr9:6990524-chr9:35228012 | Genic-genic | ITX | Yes |
| SPZ-033 | USP32-TTC18 | chr17:58365884-chr10:75108279 | Genic-genic | CTX | Yes |
| SPZ-036 | DR1-HIPK1 | chr1:93819628-chr1:114500688 | Genic-genic | ITX | Yes |
| SPZ-037 | IRF2BP2-IRF2BP2 | chr1:234738670-chr1:234742706 | Nongenic-genic | iDel | Yes |
| SPZ-038 | RINL-SYK | chr19:39359887-chr9:93636486 | Genic-genic | CTX | Yes |
| Prasad et al21 | |||||
| SS01 | SPATA21-RASA2 | Not reported | Not reported | Not reported | Yes |
| SS04 | FASN-SGMS1 | Not reported | Not reported | Not reported | Yes |
| SS05 | PITRM1-HK1 | Not reported | Not reported | Not reported | Yes |
| SS13 | COL25A1-NFKB2 | Not reported | Not reported | Not reported | Yes |
| SS13 | TYK2-UPF1 | Not reported | Not reported | Not reported | Yes |
| SS15 | NDUFAF6-BCR | Not reported | Not reported | Not reported | Yes |
| Sample | Fusion transcript | Breakpoints (DNA) | Breakpoint type | Event class | WGS confirmed |
|---|---|---|---|---|---|
| Izykowska et al11 | |||||
| P1 | EHD1-CAPN12 | chr11: 64 628,862-chr19: 39 235,710 | Genic-genic | CTX | Yes |
| P4 | CEP57-NAT10 | ch11:95 529,671-chr16:8 872 293 | Genic-genic | CTX | Yes |
| P4 | TMEM66-BAIAP2 | chr8:29 928,614-chr17:79 602,847 | Genic-genic | CTX | Yes |
| P6 | MBD4-PTPRC | chr3:129 151,514-chr1:198 647,866 | Genic-genic | CTX | Yes |
| P6 | PTPRC-CPN2 | chr1: 198 644,193-chr2:39 030,368 | Genic-genic | CTX | Yes |
| P6 | MYB-MBNL1 | chr6:135 518,448-chr3:152 111,859 | Genic-genic | CTX | Yes |
| P6 | TFG-GPR128 | chr3: 100 334,867-chr3:100 446,145 | Genic-genic | iDel | Yes |
| P8 | ARMC6-DHX57-CNRIP | chr2:68 543,463-chr2:39 049,173–39 050, 512-chr2: 69 247,504 | Genic-genic-Genic | ITX | Yes |
| P8 | MAP4K3-FIGLA | chr2:71 014,668-chr2:39 563,447 | Genic-genic | ITX | Yes |
| P8 | DCP1-CCl27 | chr3: 53 370,567-chr15: 44 079,110 | Genic-genic | CTX | Yes |
| Wang et al15 | |||||
| SPZ-002 | CARD11-PIKR3 | chr7:2968232-chr1:46512297 | Genic-genic | CTX | Yes |
| SPZ-002 | USP32-RAD51B | chr17:58422842-chr14:68878141 | Genic-genic | CTX | Yes |
| SPZ-010 | CTSC-RAB38 | chr11:88033698-chr11:87883123 | Genic-genic | ITX | Yes |
| SPZ-011 | ICOS-CD28 | chr 2:204801595-chr2:204591356 | Genic-nongenic | iDel | Yes |
| SPZ-012 | C15orf57-CBX3 | chr15:40854180-chr7:26241365 | Nongenic genic | CTX | Yes |
| SPZ-013 | PSMG4-WNK2 | chr6:3259430-chr9:95991978 | Genic-genic | CTX | Yes |
| SPZ-014 | MYCBP-SLC25A33 | chr1:39338690-chr1:9613684 | Genic-genic | ITX | Yes |
| SPZ-015 | MED22-SURF6 | chr9:136210989-chr9:136202883 | Genic-genic | iDel | Yes |
| SPZ-015 | FAM186A-DIP2B | chr12:50750031-chr12:50899023 | Genic-genic | iDel | Yes |
| SPZ-016 | RBMS1-GLI2 | chr2:161223727-chr2:121727969 | Genic-genic | ITX | Yes |
| SPZ-016 | C15orf57-CBX3 | chr15:40854974-chr7:26241386 | Genic-genic | CTX | Yes |
| SPZ-017 | SLC35A3-HIAT1 | chr1:100480976-chr1:100515465 | Genic-genic | iDel | Yes |
| SPZ-017 | RPL11-TCEB3 | chr1:24021281-chr1:24075511 | Genic-genic | iDel | Yes |
| SPZ-020 | SLC35A3-HIAT1 | chr1:100480976-chr1:100515465 | Genic-genic | iDel | Yes |
| SPZ-021 | ZFP64-WBP1L | chr20:50776662-chr10:104569650 | Genic-genic | CTX | Yes |
| SPZ-022 | ELMO1-TNRC18 | chr7:37136224-chr7:5434226 | Genic-genic | ITX | Yes |
| SPZ-028 | GPR155-CIR1 | chr2:175304628-chr2:175260365 | Genic-genic | iDel | Yes |
| SPZ-028 | CAB39-C%orf15 | chr2:231682612-chr5:133292681 | Genic-genic | CTX | Yes |
| SPZ-028 | GOLGB1-IQCG | chr3:121445760-chr3:197665651 | Genic-genic | ITX | Yes |
| SPZ-029 | IKZF2-GLI2 | chr2:213921635-chr2:121684937 | Genic-genic | iDel | Yes |
| SPZ-030 | MORF4L1-B2M | chr15:79170555-chr15:45003811 | Genic-genic | ITX | Yes |
| SPZ-030 | TTC39C-MEP1B | chr18:21663045-chr18:29771785 | Genic-genic | ITX | Yes |
| SPZ-031 | CIC-EXOSC5 | chr19:42790923-chr19:41903086 | Genic-genic | iDel | Yes |
| SPZ-033 | PPFIA1-NFKB2 | chr11:70218351-chr10:104160534 | Genic-genic | CTX | Yes |
| SPZ-033 | KDM4C-UNC13B | chr9:6990524-chr9:35228012 | Genic-genic | ITX | Yes |
| SPZ-033 | USP32-TTC18 | chr17:58365884-chr10:75108279 | Genic-genic | CTX | Yes |
| SPZ-036 | DR1-HIPK1 | chr1:93819628-chr1:114500688 | Genic-genic | ITX | Yes |
| SPZ-037 | IRF2BP2-IRF2BP2 | chr1:234738670-chr1:234742706 | Nongenic-genic | iDel | Yes |
| SPZ-038 | RINL-SYK | chr19:39359887-chr9:93636486 | Genic-genic | CTX | Yes |
| Prasad et al21 | |||||
| SS01 | SPATA21-RASA2 | Not reported | Not reported | Not reported | Yes |
| SS04 | FASN-SGMS1 | Not reported | Not reported | Not reported | Yes |
| SS05 | PITRM1-HK1 | Not reported | Not reported | Not reported | Yes |
| SS13 | COL25A1-NFKB2 | Not reported | Not reported | Not reported | Yes |
| SS13 | TYK2-UPF1 | Not reported | Not reported | Not reported | Yes |
| SS15 | NDUFAF6-BCR | Not reported | Not reported | Not reported | Yes |
CTX, interchromosomal rearaangement; iDel, interstitial deletion; ITX, intrachromosomal rearrangement.
Genetic alterations in Sz: single nucleotide variations
Many studies have focused on somatic mutations in Sz, initially directing on individual genes, followed by targeting genes involved in signaling pathways, and most recently by whole exome and whole genome analysis using NGS.
NGS (supported by proper bio-informatical tools) offers, in addition to the identification of single nucleotide variations and CNA determination, the possibility to resolve the mutational rate and characteristic mutational signatures (so-called genomic scars).22 Several groups used these approaches and found that the somatic mutation rate in Sz is on average approximately 4 mutations/Mb,15,23 being similar to solid tumors.22 Most somatic single nucleotide substitutions observed in Sz were C>T transitions (40%-75%),14,15,23 which is much less common in other hematologic cancers.22 Closer inspection (using the data from refs. 14 and 23 and algorithms from ref. 22) shows resemblance with so-called single-base substitution signatures SBS5 (aging) and SBS7a/b (UV radiation), although whether exposure to UV light plays a causative role in the onset of the disease15,24 is still under debate.
From analyses of individual mutations (often supported by CNAs), the picture emerges that genes affected in Sz cluster mainly into 5 cellular processes: cell cycle regulation, MAP-kinase signaling, T-cell receptor (TCR)/NF-κB signaling, JAK-STAT signaling, and chromatin modification.15,24,25
Two studies reanalyzed published datasets of NGS studies on CTCLs (mainly Sz) to generate high-quality homogeneous data in combination with uniform metrics and methods to create a more representative overview of point mutation frequencies.25,26 Park et al25 identified 55 putative driver genes in a genomic study conducted in 220 CTCLs (Figure 2). In addition to previously identified genes, the authors discovered novel mutations all in agreement with the above-described themes, MAPK kinase signaling (MAP2K1 and NF1), NF-κB signaling (PRKCB and CSNK1A1), TCR signaling (PTPRN2 and RLTPR), chromatin remodeling (BCOR, KDM6A, SMARCB1, and TRRAP), and T-cell differentiation (RARA). In addition, mutations in phosphatidylinositol 3-kinase signaling (PIK3R1and VAV1) and immune surveillance (CD58 and RFXAP) were found. Point mutations either predicted or confirmed to be gain-of-function were reported in JAK1 (0.9% of cases), JAK3 (2.7%), STAT3 (0.9%), and STAT5B (3.6%).25 Chang et al26 analyzed genomic data of 139 CTCL cases in a uniform matter and discovered that mutations within P53 and NF-κB pathway genes (PLCG1, CARD11, and TNFRSF1B) were mutually exclusive in Sz.
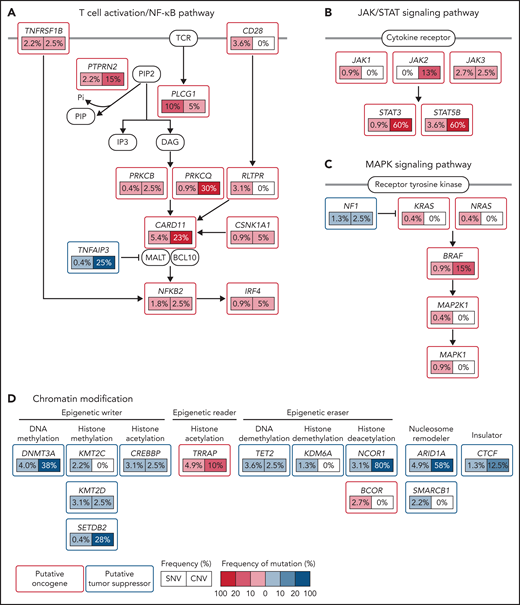
Schematic view of mutations in recurrently mutated signaling pathways in CTCLs. CTCL harbors recurrent mutations that are predicted to affect (A) T-cell activation/NF-κB signaling, (B) JAK/STAT signaling, (C) MAPK signaling, and (D) chromatin modifications. SNV, single nucleotide variation. Adapted from Park et al 2017. Used with permission from the American Society of Hematology.
Schematic view of mutations in recurrently mutated signaling pathways in CTCLs. CTCL harbors recurrent mutations that are predicted to affect (A) T-cell activation/NF-κB signaling, (B) JAK/STAT signaling, (C) MAPK signaling, and (D) chromatin modifications. SNV, single nucleotide variation. Adapted from Park et al 2017. Used with permission from the American Society of Hematology.
Genetic alterations in Sz: epigenetic alterations
Given the (initially reported) success of epigenetic therapies for CTCLs, epigenetic changes in CTCLs (tumor genomes) have been studied extensively and continue to be a focus of refined drug development. The epigenetic regulation of gene expression is accomplished primarily through histone/chromatine modifications, DNA (promoter) methylation, and gene silencing via the action of microRNAs. Recent technological innovations greatly facilitated detection of alterations in the epigenome and epigenetic machinery.
Histone/chromatin modifications
Histone/chromatin modifications in Sz have been studied indirectly (by determining alterations in genes encoding enzymes that modulate nucleosome structure and chromatin activity) and directly using the assay for transposase-accessible chromatin with sequencing (ATAC-seq).
Kiel et al8 identified recurrent loss-of-function aberrations targeting members of the chromatin remodeling/histone modification and trithorax families in 40% of Sz genomes (n = 66) analyzed. Similar results (although with less prevalence) were found in other NGS studies.14,19,23,27 The actual consequences of these alterations for treatment with epigenetic medication are not clear yet. Qu et al28 demonstrated (using ATACseq) that histone deacetylase inhibitor (HDACi) treatment emphasizes the differences between normal host CD4+ cells and leukemic Sz cells. Highly expressed signature elements unique to Sz cells (or normal T cells) residing in open chromatin regions were found to be even more highly expressed after treatment with HDACi, without recovery of silenced genes in either group. Using single-cell RNA sequencing and flow cytometry, Buus et al29 demonstrated that patients with Sz display a high degree of single-cell heterogeneity within the malignant T-cell population and that distinct subpopulations of malignant T cells carry HDACi resistance.29 A combination of sequencing of the TCR-encoding genes with ATAC-seq at the single-cell level, termed transcript-indexed ATAC-seq (T-ATAC-seq), was used to start unraveling epigenomic signatures in immortalized leukemic T cells, primary human T cells from healthy volunteers, and primary leukemic T cells from patient samples including Sz.30 The technique enabled identification of leukemic and nonleukemic regulatory pathways in T cells from the same individual by comparing the information that arose from the malignant Sz clone from the background T-cell signal.
MicroRNAs
In house–developed miRNA microarrays were initially used to conduct a genome wide analysis of miRNA expression of peripheral blood–derived malignant T cells from patients with Sz.31 More than 100 differentially expressed miRNAs (compared with normal peripheral blood–derived CD4+ T cells) were identified. miR-145, miR-574-5p, miR200c, miR-199a*, miR-143, and miR-214 were among the most prominently upregulated miRs, whereas miR-342, miR-223, and miR-150 were found to be downregulated in Sz compared with normal CD4+ T cells. Of note, these findings were in line with earlier observations that microRNAs exhibit high-frequency genomic alterations in human cancers because most observed changes in miR expression levels correlated with reported genomic copy number abnormalities.32–34 miR-214 and miR-199a* upregulation in Sz was also identified using a commercial array-based miR expression detection platform.35 In this study, furthermore upregulation of miR-7 and decreased expression of miR-342, miR-223, miR-92, miR-181a, and miR-191 was confirmed in Sz. Downregulation of miR-181a and upregulation of miR-214 and miR-199a* and miR-486 in Sz cells was confirmed by an NGS-based approach.36 This latter study also showed (using a quantitative polymerase chain reaction [Q-PCR]–based analysis of miR precursors) that the miR-199a2/214 cluster within the DNM3os transcript embodies most of the aberrantly expressed microRNAs in Sz.36 Given the recurrent upregulation of miR-214 in CTCLs, a transgenic mouse model (spontaneously overexpressing miR-214 in malignant T cells) was used to study its functional role. AntagomiR-214 treatment led to significant decrease in disease severity. It was also shown that aberrantly expressed TWIST1 and BET protein BRD4 cooperate to drive miR-214 expression in CTCL cell lines and in samples from patients.37
DNA methylation
The most elaborate study on the DNA methylome in Sz was carried out by van Doorn et al,38 showing that Sz is characterized by widespread yet distinct DNA methylation alterations, which can be used clinically as epigenetic diagnostic markers. Several studies focused on individual genes, revealing distinctive promoter (hypo)methylation of genes encoding PLS3,39,40 SAMHD1,41 RUNX3,42 TWIST1, and GATA640 in Sz.
JAK-STAT signaling in Sz
Activation of the JAK-STAT signaling cascade starts after binding of cognate cytokines to cytokine receptors at the cell surface. This results in STAT protein dimerization followed by nuclear translocation and subsequent transcriptional activation of genes regulating (T)-cell proliferation, differentiation, and apoptosis. Aberrant JAK-STAT signaling is a hallmark of T-cell lymphoma43 and appears to be of critical importance in Sz. It was shown that Staphylococcus aureus (isolated from the skin of patients with Sz) can directly activate oncogenic STAT3 signaling in malignant cells.44 Using various coculture models, it was demonstrated that Staphylococcal enterotoxin A type has an indirect effect on malignant T cells, by activating infiltrating bystander nonmalignant T cells, which in response to this stimulus produce IL-2 and other regulatory cytokines. In combination with (epi) genetic changes in genes being part of or controlling this signaling pathway can further derail and fuel the oncogenic process resulting in activation of STAT3. In Sz, copy number gains of STAT3 and STAT5B are found in >60% of the patients (Figure 1), a change that can be oncogenic in itself.45 Although with a lower prevalence, gain-of-function mutations are described for JAK and STAT8,23 in Sz cells. Combined, these alterations are potentially targetable with JAK-STAT inhibitors, with a preference for STAT inhibitors because they have the potency to target STAT mutations and overexpression of STAT effectively. It is shown that cucurbitacins can induce Sz cell death by an increase in apoptosis and a decrease in proliferative signaling through inactivation of STAT3,46,47 and hence are attractive candidates to treat Sezary. In analogy, small molecule inhibitors of JAK signaling might find their way to the clinic. Ruxolitinib (JakaviTM), a JAK1/2 inhibitor, and AZD1480 (a JAK2 inhibitor) are effective on Sz cell lines.48
Genetic alterations in mycosis fungoides
The tumor (epi) genome of mycosis fungoides (MF) is far less studied, but, seemingly paradoxical, much better understood than Sz. Nearly all NGS studies on CTCLs focused primarily on Sz and included only a handful of MF biopsy specimens as an additional group. Exceptions are the whole genome study by McGirt et al,49 whole exome sequencing by Iyer et al,50 and combined whole transcriptome and whole genome study by Bastidas Torres et al,51 all focusing on MF only.
Early studies, using aCGH, revealed a pattern distinct from Sz52 and emphasized that Sz and MF are distinct diseases. On the level of individual genes, differences are found; for example, the most recurrent finding in MF is inactivation/deletion of the CDKN2A-B locus encoding the tumor suppressor protein P16,51,53-55 a finding hardly ever encountered in Sz. In Sz, the classical tumor suppressor TP53 is recurrently deleted; this loss of the chromosomal region encoding TP53 is less prevalent in MF. Using fluorescence in situ hybridization analysis, Garaico et al56 demonstrated CDKN2A disbalances are a particular frequent finding in folliculotropic MF and transformed MF vs tumor stage MF.
Genetic alterations in MF: DNA rearrangements, CNA, and fusion transcripts
McGirt et al49 were the first to resolve genomic alterations in MF by NGS. In this small cohort study, only recurrent focal loss of a region containing ZEB1 (2 of 5 patients) was identified. An integrated NGS analysis (DNA/RNA) of genomic rearrangements in MF at base‐level resolution revealed a complex and heterogeneous landscape of inter‐ and intrachromosomal rearrangements.51 This study showed, similar to what was seen in Sz,11,14 a large variety of novel gene rearrangements resulting in recurrent deletion of tumor suppressors involved in pathways that are commonly deregulated in patients with MF (eg, ARID1A, CDKN2A/B, PTPRC, SOCS1, and STK11). This analysis also identified 24 fusion transcripts, including 6 containing bona fide cancer genes, which although not recurrent, could contribute to MF development in individual cases (Table 3).
Fusion transcripts in MF51
| Sample | Fusion transcript | Breakpoints (DNA) | Breakpoint type | Event class | WGS confirmed |
|---|---|---|---|---|---|
| MF1 | KDM6A–IL1RAPL1 | chrX:44746566–chrX:29451290 | Genic–genic | ITX | Yes |
| MF1 | CHIC1–RP2 | chrX:72844450–chrX:46680435 | Genic–nongenic | ITX | Yes |
| MF3 | ANKRD13A–CUL9 | chr12:110448655–chr6:43160142 | Genic–genic | CTX | Yes |
| MF3 | CLEC16A–SCARB1 | chr16:11067010–chr12:125350896 | Genic–nongenic | CTX | Yes |
| MF3 | SSH2–GRAP2 | chr17:28059210–chr22:40314573 | Genic–genic | CTX | Yes |
| MF3 | LMF1–TAF15 | chr16:986148–chr17:34145925 | Genic–genic | CTX | Yes |
| MF3 | ATXN1–TP63 | chr6:16307814–chr3:189470345 | Genic–genic | CTX | Yes |
| MF4 | CCR7–DOT1L | chr17:38718403–chr19:2181252 | Genic–genic | CTX | Yes |
| MF5 | PHACTR4–EPB41 | chr1:28755797–chr1:29246304 | Genic–genic | iDel | Yes |
| MF5 | ADAM12–MMRN2 | chr10:127935628–chr10:88698606 | Genic–genic | iDel | Yes |
| MF5 | TRAPPC10–TRPM2 | chr21:45487448–chr21:45795335 | Genic–genic | iDel | Yes |
| MF5 | ARHGAP26–TENM2 | chr5:142272242–chr5:167448836 | Genic–genic | ITX | Yes |
| MF6 | ANK3–RNLS | chr10:62168031–chr10:90101461 | Genic–genic | ITX | Yes |
| MF6 | ELF1–SATB2 | chr13:41540637–chr2:200369768 | Genic–nongenic | CTX | Yes |
| MF7 | TP53–GPR3 | chr17:7579754–chr1:27718138 | Genic–nongenic | CTX | Yes |
| MF7 | CLPP–NR3C1 | chr19:6365544–chr5:142800539 | Genic–genic | CTX | Yes |
| MF7 | SARNP–WRAP53 | chr12:56161974–chr17:7593927 | Genic–genic | CTX | Yes |
| MF8 | SETD5–RNF19A | chr3:9497374–chr8:101391443 | Genic–nongenic | CTX | Yes |
| MF8 | SUDS3–TMEM132B | chr12:118847216–chr12:125987360 | Genic–genic | ITX | Yes |
| MF8 | AACS–STAB2 | chr12:125625503–chr12:104094623 | Genic–genic | ITX | Yes |
| MF8 | RPUSD3–RNF19A | chr3:9882804–chr8:101303862 | Genic–genic | CTX | Yes |
| MF8 | YTHDF3–LIFR | chr8:64081882–chr5:38586949 | Genic–genic | CTX | Yes |
| MF9 | DPM1–UBE2V1 | chr20:49574368–chr20:48703893 | Genic–genic | iDel | Yes |
| MF9 | KCNAB2–ESPN | chr1:6071941–chr1:6493075 | Genic–genic | iDel | Yes |
| Sample | Fusion transcript | Breakpoints (DNA) | Breakpoint type | Event class | WGS confirmed |
|---|---|---|---|---|---|
| MF1 | KDM6A–IL1RAPL1 | chrX:44746566–chrX:29451290 | Genic–genic | ITX | Yes |
| MF1 | CHIC1–RP2 | chrX:72844450–chrX:46680435 | Genic–nongenic | ITX | Yes |
| MF3 | ANKRD13A–CUL9 | chr12:110448655–chr6:43160142 | Genic–genic | CTX | Yes |
| MF3 | CLEC16A–SCARB1 | chr16:11067010–chr12:125350896 | Genic–nongenic | CTX | Yes |
| MF3 | SSH2–GRAP2 | chr17:28059210–chr22:40314573 | Genic–genic | CTX | Yes |
| MF3 | LMF1–TAF15 | chr16:986148–chr17:34145925 | Genic–genic | CTX | Yes |
| MF3 | ATXN1–TP63 | chr6:16307814–chr3:189470345 | Genic–genic | CTX | Yes |
| MF4 | CCR7–DOT1L | chr17:38718403–chr19:2181252 | Genic–genic | CTX | Yes |
| MF5 | PHACTR4–EPB41 | chr1:28755797–chr1:29246304 | Genic–genic | iDel | Yes |
| MF5 | ADAM12–MMRN2 | chr10:127935628–chr10:88698606 | Genic–genic | iDel | Yes |
| MF5 | TRAPPC10–TRPM2 | chr21:45487448–chr21:45795335 | Genic–genic | iDel | Yes |
| MF5 | ARHGAP26–TENM2 | chr5:142272242–chr5:167448836 | Genic–genic | ITX | Yes |
| MF6 | ANK3–RNLS | chr10:62168031–chr10:90101461 | Genic–genic | ITX | Yes |
| MF6 | ELF1–SATB2 | chr13:41540637–chr2:200369768 | Genic–nongenic | CTX | Yes |
| MF7 | TP53–GPR3 | chr17:7579754–chr1:27718138 | Genic–nongenic | CTX | Yes |
| MF7 | CLPP–NR3C1 | chr19:6365544–chr5:142800539 | Genic–genic | CTX | Yes |
| MF7 | SARNP–WRAP53 | chr12:56161974–chr17:7593927 | Genic–genic | CTX | Yes |
| MF8 | SETD5–RNF19A | chr3:9497374–chr8:101391443 | Genic–nongenic | CTX | Yes |
| MF8 | SUDS3–TMEM132B | chr12:118847216–chr12:125987360 | Genic–genic | ITX | Yes |
| MF8 | AACS–STAB2 | chr12:125625503–chr12:104094623 | Genic–genic | ITX | Yes |
| MF8 | RPUSD3–RNF19A | chr3:9882804–chr8:101303862 | Genic–genic | CTX | Yes |
| MF8 | YTHDF3–LIFR | chr8:64081882–chr5:38586949 | Genic–genic | CTX | Yes |
| MF9 | DPM1–UBE2V1 | chr20:49574368–chr20:48703893 | Genic–genic | iDel | Yes |
| MF9 | KCNAB2–ESPN | chr1:6071941–chr1:6493075 | Genic–genic | iDel | Yes |
Known cancer genes (NCG 5.0) are in bold.
CTX, interchromosomal rearrangement; iDel, interstitial deletion; ITX, intrachromosomal rearrangement.
Genetic alterations in MF: genes and mutations
Mutations in phospholipase C γ1 (PLCG1) were studied intensively since initial reports described a prevalence of 20% of a targetable mutation in this gene in CTCLs.57 However, data by Caumont et al58 suggest that PLCG1 mutations are far less prevalent: only 3% to 5% of the CTCL tumor genomes (MF and Sz) harbor PLCG1 mutations.
McGirt et al49 identified an activating mutation of JAK3 in 1 patient with MF. The combined analysis of 18 MF tumor samples being part of large CTCL sequencing studies did not reveal recurrent mutations.26 The only reported recurrent mutations in MF came from Bastidas Torres et al,51 who showed recurrent pathogenic mutations in JAK3 and FGFR4 (Tables 4 and 5).
Mutations identified in Sz and MF
| Gene | Combined data from Park et al25 (total mutations in Sz; n = 186) | Combined data from Park et al25 (total mutations in MF; n = 25) | Data from Bastidas Torres et al51 (deleterious mutations in Mf; n = 9) |
|---|---|---|---|
| TP53 | 31 | 1 | 0 |
| PLCG1 | 18 | 4 | 0 |
| CARD11 | 12 | 1 | 0 |
| ARID1A | 12 | 3 | 0 |
| TRRAP | 11 | 0 | 0 |
| POT1 | 10 | 0 | 0 |
| DNMT3A | 10 | 0 | 0 |
| TET2 | 9 | 0 | 0 |
| RHOA | 9 | 1 | 0 |
| CD28 | 8 | 0 | 0 |
| ZEB1 | 8 | 0 | 0 |
| RLTPR | 7 | 0 | 0 |
| CREBBP | 7 | 0 | 0 |
| NCOR1 | 7 | 0 | 0 |
| STAT5B | 7 | 1 | 0 |
| KMT2D | 6 | 1 | 0 |
| BCOR | 6 | 0 | 0 |
| CCR4 | 6 | 1 | 0 |
| FAS | 6 | 0 | 0 |
| KMT2C | 5 | 0 | 2 |
| RARA | 5 | 0 | 0 |
| TNFRSF1B | 5 | 0 | 0 |
| PRKG1 | 4 | 1 | 0 |
| PTPRN2 | 4 | 1 | 0 |
| NFKB2 | 4 | 0 | 0 |
| ATM | 3 | 1 | 0 |
| SMARCB1 | 3 | 2 | 0 |
| CTCF | 3 | 0 | 0 |
| JAK3 | 3 | 2 | 2 |
| VAV1 | 3 | 1 | 0 |
| NF1 | 3 | 0 | 0 |
| ZNF365 | 2 | 0 | 0 |
| IRF4 | 2 | 0 | 1 |
| PDCD1 | 2 | 0 | 0 |
| PRKCQ | 2 | 0 | 0 |
| KDM6A | 2 | 0 | 0 |
| ARHGEF3 | 2 | 0 | 0 |
| RFXAP | 2 | 0 | 0 |
| CSNK1A1 | 2 | 0 | 0 |
| TNFAIP3 | 2 | 0 | 0 |
| PIK3R1 | 2 | 0 | 0 |
| SETDB2 | 1 | 0 | 0 |
| JAK1 | 1 | 1 | 0 |
| STAT3 | 1 | 1 | 1 |
| CD58 | 1 | 0 | 0 |
| PRKCB | 1 | 0 | 0 |
| BRAF | 1 | 0 | 1 |
| KRAS | 1 | 0 | 0 |
| NRAS | 1 | 0 | 0 |
| LATS1 | 1 | 0 | 0 |
| U2AF1 | 0 | 1 | 0 |
| MAPK1 | 0 | 3 | 1 |
| MAP2K1 | 0 | 1 | 0 |
| Gene | Combined data from Park et al25 (total mutations in Sz; n = 186) | Combined data from Park et al25 (total mutations in MF; n = 25) | Data from Bastidas Torres et al51 (deleterious mutations in Mf; n = 9) |
|---|---|---|---|
| TP53 | 31 | 1 | 0 |
| PLCG1 | 18 | 4 | 0 |
| CARD11 | 12 | 1 | 0 |
| ARID1A | 12 | 3 | 0 |
| TRRAP | 11 | 0 | 0 |
| POT1 | 10 | 0 | 0 |
| DNMT3A | 10 | 0 | 0 |
| TET2 | 9 | 0 | 0 |
| RHOA | 9 | 1 | 0 |
| CD28 | 8 | 0 | 0 |
| ZEB1 | 8 | 0 | 0 |
| RLTPR | 7 | 0 | 0 |
| CREBBP | 7 | 0 | 0 |
| NCOR1 | 7 | 0 | 0 |
| STAT5B | 7 | 1 | 0 |
| KMT2D | 6 | 1 | 0 |
| BCOR | 6 | 0 | 0 |
| CCR4 | 6 | 1 | 0 |
| FAS | 6 | 0 | 0 |
| KMT2C | 5 | 0 | 2 |
| RARA | 5 | 0 | 0 |
| TNFRSF1B | 5 | 0 | 0 |
| PRKG1 | 4 | 1 | 0 |
| PTPRN2 | 4 | 1 | 0 |
| NFKB2 | 4 | 0 | 0 |
| ATM | 3 | 1 | 0 |
| SMARCB1 | 3 | 2 | 0 |
| CTCF | 3 | 0 | 0 |
| JAK3 | 3 | 2 | 2 |
| VAV1 | 3 | 1 | 0 |
| NF1 | 3 | 0 | 0 |
| ZNF365 | 2 | 0 | 0 |
| IRF4 | 2 | 0 | 1 |
| PDCD1 | 2 | 0 | 0 |
| PRKCQ | 2 | 0 | 0 |
| KDM6A | 2 | 0 | 0 |
| ARHGEF3 | 2 | 0 | 0 |
| RFXAP | 2 | 0 | 0 |
| CSNK1A1 | 2 | 0 | 0 |
| TNFAIP3 | 2 | 0 | 0 |
| PIK3R1 | 2 | 0 | 0 |
| SETDB2 | 1 | 0 | 0 |
| JAK1 | 1 | 1 | 0 |
| STAT3 | 1 | 1 | 1 |
| CD58 | 1 | 0 | 0 |
| PRKCB | 1 | 0 | 0 |
| BRAF | 1 | 0 | 1 |
| KRAS | 1 | 0 | 0 |
| NRAS | 1 | 0 | 0 |
| LATS1 | 1 | 0 | 0 |
| U2AF1 | 0 | 1 | 0 |
| MAPK1 | 0 | 3 | 1 |
| MAP2K1 | 0 | 1 | 0 |
Minimal common regions (<3 Mb) identified in MF and Sz
| Cytogenetic band | Copy number alteration | Start | End | Size | Cancer gene (NCG 5.0) | Affected MF patients (%)51 | Affected Sz patients (%)14 | Start | End | Size |
|---|---|---|---|---|---|---|---|---|---|---|
| 9p21.3 | Loss | 21955000 | 21972000 | 17 kb | CDKN2A | 7 (78) | 3 (7.5) | 21958139 | 21998921 | 40 kb |
| 9q21.32 | Loss | 85800000 | 86650000 | 850 kb | KIF27, HNRNPK | 5 (56) | n.a. | |||
| 16p13.13 | Loss | 11050000 | 11355000 | 305 kb | SOCS1 | 5 (56) | n.a. | |||
| 1q31.3 | Loss | 198450000 | 198625488 | 175 kb | PTPRC | 4 (44) | n.a. | |||
| 13q14.3 | Loss | 50592381 | 51460719 | 868 kb | DLEU2/Mir-15a/16-1 locus | 4 (44) | n.a. | |||
| 1p36.11 | Loss | 26535118 | 27718137 | 1.2 Mb | ARID1A | 3 (33) | 7 (17.5) | 25163521 | 27933295 | 2.8 Mb |
| 10q23.31 | Loss | 89450000 | 90000000 | 550 kb | PTEN | 3 (33) | 2 (5) | 88803044 | 90764142 | 2.9 Mb |
| 10q23.33-10q24.1 | Loss | 96500000 | 97250000 | 750 kb | CYP2C9 | 3 (33) | n.a. | |||
| 9q21.31 | Loss | 81840000 | 82200000 | 360 kb | TLE4 | 3 (33) | n.a. | |||
| 20q13.13 | Loss | 48700000 | 50154295 | 1.4 Mb | PTPN1, PARD6B | 3 (33) | n.a. | |||
| 19p13.3 | Loss | 250000 | 2150000 | 1.9 Mb | C2CD4C, FSTL3, ABCA7, STK11, C19orf26, TCF3 | 3 (33) | 3 (7.5) | 232372 | 1597438 | 1.3 Mb |
| 5q15-5q21.1 | Loss | 96700000 | 98250000 | 1.5 Mb | CHD1 | 2 (22) | n.a. | |||
| 6p22.3-6p23 | Loss | 14200000 | 16750000 | 2.5 Mb | JARID2 | 2 (22) | 1 (5) | 152243466 | 152552262 | 9 kb |
| 13q14.11 | Loss | 40300000 | 41550000 | 1.2 Mb | FOXO1, MRPS31 | 2 (22) | 1 (5) | 40413041 | 42256311 | 43 kb |
| 16q24.3 | Loss | 88950000 | 89500000 | 550 kb | CBFA2T3, ANKRD11 | 2 (22) | n.a. | |||
| 5p13.3 | Gain | 32100000 | 32165000 | 65 kb | 3 (33) | n.a. | ||||
| 8q24.21 | Gain | 128747500 | 128757400 | 9.9 kb | MYC | 2 (22) | n.a. | |||
| 13q33.3-13q34 | Gain | 109350000 | 112250000 | 2.9 Mb | COL4A2 | 2 (22) | n.a. |
| Cytogenetic band | Copy number alteration | Start | End | Size | Cancer gene (NCG 5.0) | Affected MF patients (%)51 | Affected Sz patients (%)14 | Start | End | Size |
|---|---|---|---|---|---|---|---|---|---|---|
| 9p21.3 | Loss | 21955000 | 21972000 | 17 kb | CDKN2A | 7 (78) | 3 (7.5) | 21958139 | 21998921 | 40 kb |
| 9q21.32 | Loss | 85800000 | 86650000 | 850 kb | KIF27, HNRNPK | 5 (56) | n.a. | |||
| 16p13.13 | Loss | 11050000 | 11355000 | 305 kb | SOCS1 | 5 (56) | n.a. | |||
| 1q31.3 | Loss | 198450000 | 198625488 | 175 kb | PTPRC | 4 (44) | n.a. | |||
| 13q14.3 | Loss | 50592381 | 51460719 | 868 kb | DLEU2/Mir-15a/16-1 locus | 4 (44) | n.a. | |||
| 1p36.11 | Loss | 26535118 | 27718137 | 1.2 Mb | ARID1A | 3 (33) | 7 (17.5) | 25163521 | 27933295 | 2.8 Mb |
| 10q23.31 | Loss | 89450000 | 90000000 | 550 kb | PTEN | 3 (33) | 2 (5) | 88803044 | 90764142 | 2.9 Mb |
| 10q23.33-10q24.1 | Loss | 96500000 | 97250000 | 750 kb | CYP2C9 | 3 (33) | n.a. | |||
| 9q21.31 | Loss | 81840000 | 82200000 | 360 kb | TLE4 | 3 (33) | n.a. | |||
| 20q13.13 | Loss | 48700000 | 50154295 | 1.4 Mb | PTPN1, PARD6B | 3 (33) | n.a. | |||
| 19p13.3 | Loss | 250000 | 2150000 | 1.9 Mb | C2CD4C, FSTL3, ABCA7, STK11, C19orf26, TCF3 | 3 (33) | 3 (7.5) | 232372 | 1597438 | 1.3 Mb |
| 5q15-5q21.1 | Loss | 96700000 | 98250000 | 1.5 Mb | CHD1 | 2 (22) | n.a. | |||
| 6p22.3-6p23 | Loss | 14200000 | 16750000 | 2.5 Mb | JARID2 | 2 (22) | 1 (5) | 152243466 | 152552262 | 9 kb |
| 13q14.11 | Loss | 40300000 | 41550000 | 1.2 Mb | FOXO1, MRPS31 | 2 (22) | 1 (5) | 40413041 | 42256311 | 43 kb |
| 16q24.3 | Loss | 88950000 | 89500000 | 550 kb | CBFA2T3, ANKRD11 | 2 (22) | n.a. | |||
| 5p13.3 | Gain | 32100000 | 32165000 | 65 kb | 3 (33) | n.a. | ||||
| 8q24.21 | Gain | 128747500 | 128757400 | 9.9 kb | MYC | 2 (22) | n.a. | |||
| 13q33.3-13q34 | Gain | 109350000 | 112250000 | 2.9 Mb | COL4A2 | 2 (22) | n.a. |
Epigenetic alterations in MF
Histone/chromatin
Histone/chromatin modifications in MF have been studied solely indirectly (by determining alterations in genes encoding enzymes that modulate nucleosome structure and chromatin activity). In most of these studies, sporadic mutations were found (eg, in DNMT3A, TET2, and ARID1A26).
DNA (hyper)methylation
Few studies focused on DNA methylation in MF. Van Doorn et al59 used a genome-wide approach and identified hypermethylation of multiple tumor suppressor genes including BCL7a and CDKN2A/B in tumor stage MF. CDKN2A/B methylation was described for early and advanced MF disease and seems to be independent of disease stage. A specific DNA methylation profile that correlated with a high risk of disease progression in early-stage MF included PPARG, SOCS1, and NEUROG1.60 Sandoval et al61 found an inverse an inverse correlation between microRNA expression and DNA methylation in MF tumor stage. Table 6 summarizes the key findings in Sz and MF.
Methylation of gene promoter regions in most studied CTCL entities
| Gene name | Sz | MF | CD30+LPD | |||
|---|---|---|---|---|---|---|
| Hypo | Hyper | Hypo | Hyper | Hypo | Hyper | |
| PLS3 | x (39) | |||||
| GATA6 | x (40) | |||||
| TWIST1 | x (40) | |||||
| SAMHD1 | x (41) | |||||
| TMEM244 | x (119) | |||||
| CMTM2 | x (120) | |||||
| C2orf40 | x (120) | |||||
| G0S2 | x (120) | |||||
| HSBP6 | x (120) | |||||
| PROM1 | x (120) | |||||
| PAM | x (120) | |||||
| MLH1 | x (121) | |||||
| P15 | x (53,55,59, 122) | x (53) | ||||
| P16 | x (53,55,59, 122) | x (53) | ||||
| BCL7A | x (123) | |||||
| PTPRG | x (123) | |||||
| P73 | x (123) | |||||
| FAS | x (124) | |||||
| PPARG | x (60) | |||||
| NEUROG | x (60) | |||||
| SOCS1 | x (60) | |||||
| DUSP22 | x (125) | |||||
| 126 coding genes | x (120) | |||||
| miRNAgenes | ||||||
| miR-141/200c | x (61) | |||||
| miR-193b | x (61) | |||||
| miR-10b | x (61) | |||||
| miR-21 | x (61) | |||||
| miR-429 | x (61) | |||||
| miR-142 | x (61) | x (61) | ||||
| Gene name | Sz | MF | CD30+LPD | |||
|---|---|---|---|---|---|---|
| Hypo | Hyper | Hypo | Hyper | Hypo | Hyper | |
| PLS3 | x (39) | |||||
| GATA6 | x (40) | |||||
| TWIST1 | x (40) | |||||
| SAMHD1 | x (41) | |||||
| TMEM244 | x (119) | |||||
| CMTM2 | x (120) | |||||
| C2orf40 | x (120) | |||||
| G0S2 | x (120) | |||||
| HSBP6 | x (120) | |||||
| PROM1 | x (120) | |||||
| PAM | x (120) | |||||
| MLH1 | x (121) | |||||
| P15 | x (53,55,59, 122) | x (53) | ||||
| P16 | x (53,55,59, 122) | x (53) | ||||
| BCL7A | x (123) | |||||
| PTPRG | x (123) | |||||
| P73 | x (123) | |||||
| FAS | x (124) | |||||
| PPARG | x (60) | |||||
| NEUROG | x (60) | |||||
| SOCS1 | x (60) | |||||
| DUSP22 | x (125) | |||||
| 126 coding genes | x (120) | |||||
| miRNAgenes | ||||||
| miR-141/200c | x (61) | |||||
| miR-193b | x (61) | |||||
| miR-10b | x (61) | |||||
| miR-21 | x (61) | |||||
| miR-429 | x (61) | |||||
| miR-142 | x (61) | x (61) | ||||
MicroRNAs
The first study on miRNA profiling of MF used the same in house developed platform as described for Sz and compared MF tumors skin biopsies with benign inflammatory dermatoses.62 Further validation of identified differences was achieved using Q-PCR and biopsies of an independent cohort of patients and controls. It was found that, compared with benign controls in tumor stage MF, the majority (30 of 49) of the differentially expressed miRNAs are upregulated. Several upregulated miRNAs (miR-93, miR-155, and miR-17-92) have been validated functionally as oncomirs,63-65 and aberrant expression of many of these identified dysregulated miRs was described previously in cancer. In part based on these results, a Q-PCR–based classifier consisting of miR-155, miR-203, and miR-205 was developed by Ralfkiaer et al,66 enabling the ability to distinguish CTCLs (including Sz) from benign skin disorders. Close inspection of the data of Qin et al36 showed that miR-203 and -205 are indeed lowly expressed in Sz but are absent in healthy T cells. Moreover, miR-203 and -205 were previously described as being characteristic for keratinocytes,67-69 which suggests that loss of expression of these miRs in CTCL skin biopsies simply reflects the amount of skin tissue over T cells (low in tumor skin biopsies and high in benign controls) rather than downregulation of these miRs in tumor T cells. This also shows that RNA expression results from biopsies (both mRNA and miRNA) should be interpreted with care because biopsies may fail to detect changes in expression because of the relative paucity of MF tumor cells in the collected tissue. Validation by results from genomic studies and/or tissue with a high tumor cell content can circumvent this. The oncogenic role of miR-155 in MF was confirmed by different groups,70-72 and inhibitors of miR-155 are currently tested in clinical trials.73 Recently an interesting link between miR dysregulation and chromatin modifications was postulated by Kohnken et al,74 showing an inverse relationship between expression of miR-29b and the epigenetic reader bromodomain-containing protein 4 (BRD4) in CTCL.
JAK-STAT signaling in MF
Like in Sz, aberrant JAK-STAT signaling also appears to be pivotal in MF. Starting with aberrant activation of the JAK-STAT pathway because of inflammation of the skin, a protumorigenic inflammatory environment can emerge. An impressive example of malignant inflammation most likely caused by Staphylococcus aureus infection was demonstrated by Lindahl et al.75 It is postulated that this initiates the oncogenic process resulting in activation of STAT3 as is frequently seen in advanced stages of MF.76 In contrast to Sz, however, copy number gains of STAT3/5B or gain-of-function mutations are less prevalent in MF In particular, SOCS1 emerges as a candidate culprit gene in MF explaining enhanced JAK-STAT activity in early and late stage MF.76 Loss of SOCS1, either to deletion or unbalanced translocation is observed in approximately 35% of the cases (and can already be found in early stage MF51), whereas upregulation of miR-155, a microRNA targeting SOCS1 mRNA, is documented by many studies 61,62,66,72 across different laboratories. Furthermore, an independent study showed inactivation of the SOCS1 promoter by DNA methylation in 18% of MF cases.60 The SOCS (suppressor of cytokine signaling) family of proteins are negative-feedback inhibitors of signaling induced by cytokines that act via the JAK/STAT pathway. SOCS1, the most potent member of the family, inhibits JAK signaling directly by interfering with JAK1 or JAK3 but can also act as ubiquitin ligase by recruiting Cullin5 to ubiquitinate signaling components.77 The JAK-STAT pathway and aberrancies found in MF is depicted in Figure 3.
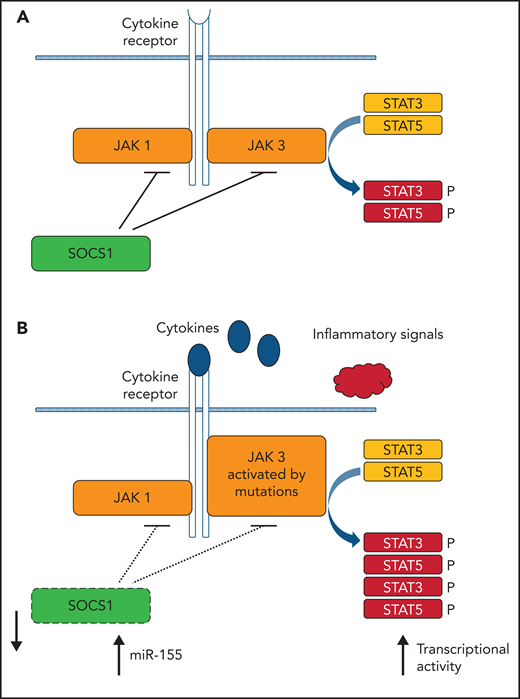
Central role of (aberrant) JAK/STAT signaling in MF. (A) Simplified scheme of JAK/STAT signaling. (B) Alterations found leading to increased transcriptional activity of gene promoters with STAT binding sites. Some features are shared with Sz (eg, malignant inflammation and JAK mutations), whereas others are typical for MF (loss of SOCS1 and upregulation of miR-155).
Central role of (aberrant) JAK/STAT signaling in MF. (A) Simplified scheme of JAK/STAT signaling. (B) Alterations found leading to increased transcriptional activity of gene promoters with STAT binding sites. Some features are shared with Sz (eg, malignant inflammation and JAK mutations), whereas others are typical for MF (loss of SOCS1 and upregulation of miR-155).
TCR clonality and sequencing
It is generally assumed that TCR genes are rearranged polyclonally in normal and reactively proliferating T cells (resulting in random rearrangements), whereas identically rearranged TCR genes are hallmarks for neoplastic T cells. Based on this principle, TCR sequencing is often used to determine clonality and hence the presence of tumor cells in skin lesions of early MF, leukemic involvement in MF/SS, and/or monitoring of residual disease. This assumption should be taken with care, and pros, cons, and pitfalls of this approach were recently reviewed by Walia and Yeung.78 Carried out in a proper way, high-throughput sequencing of the TCRb gene permits calculation of so-called tumor clone frequencies, for which De Masson et al79 showed that a tumor clone frequency > 25% at an early stage of MF has the ability to predict a poorer outcome than any other prognostic marker. With this technique, it was also demonstrated that low-dose radiotherapy can eradicate malignant T cells in MF.80 A combination of TCR sequencing with whole exome sequencing of microdissected tissue of MF lesions led to the identification of multiple T-cell clones within a single tumor cell fraction, with considerable variation between patients and between lesions from the same patient.81
Primary cutaneous CD30+ lymphoproliferative disease
Primary cutaneous CD30-positive lymphoproliferative disorders (pcCD30+LPDs) account for ∼20% of all cutaneous T-cell lymphomas. This group forms a spectrum, with primary cutaneous anaplastic large cell lymphoma (pcALCL) on 1 end, lymphomatoid papulosis (LyP) on the other, and borderline cases in between.82-84 In contrast to systemic ALCL, (sALCL), which appears to be driven by a variety of genetic alterations that (over)activate STAT3 signaling,85-90 the molecular pathogenesis and key drivers of pcALCL (and LyP) remain largely unknown.
CNAs were studied by van Kester et al91 using aCGH showing a pattern distinct from Sz and MF. Combining these data with array-based expression profiling revealed that the lower tendency of cALCL to disseminate to extracutaneous sites may be explained by higher expression of the skin-homing chemokine receptor genes CCR10 and CCR8.
Chromosomal rearrangements involving ALK, DUSP22, TP63, and TYK2, common in sALCL, have also been detected in pcALCL, albeit at much lower frequencies.92,93 ALK gene rearrangements have been reported initially in pediatric patients, all having an excellent prognosis,94,95 and recently in adults96,97 in approximately 2% of the cALCL cases. In 50% of these cases, a NPM1-ALK fusion was observed, whereas in the remaining cases, a translocation of ALK with TRAF1, ATIC, or TPM3 was observed.97
Rearrangements involving the DUSP22/IRF4 locus have been described in cALCL with an overall frequency of approximately 30%.98-101 This latter translocation is also found in a subgroup of LyP (5%) defining a distinct and novel subtype.102
In a large ALCL NGS study, Creszenzo et al86 included 44 pcALCL tumor biopsy specimens. Recurrent translocations/fusion transcripts were not reported, but a fraction of the samples (5%) displayed mutations in JAK1/STAT3, suggesting that the activation of the JAK-STAT signaling pathway might play a role in cutaneous CD30-positive lymphoproliferative disorders as well.
Epigenetic alterations in pcCD30+LPD
MiRs
Thus far, only 2 studies described the miRnome of cALCL. Using a combination of arrays and miRNA-Q-PCR, Benner et al103 identified statistically significantly differential expression of miR-155, miR-27b, miR-93, miR-29b, and miR-92a between tumor stage MF and cALCL. No differences in global expression between MF and cALCL were found by Sandoval et al,61 but an inverse correlation between microRNA expression and DNA methylation for both MF tumor and the CD30+ cALCL groups was detected.61 Furthermore, the expression of methylated microRNAs was restored by demethylating agents, supporting the role of DNA methylation on the regulation of the differentially expressed microRNAs in cALCL (and MF).
DNA methylation and chromatin modifications. SATB1 (special AT-rich sequence-binding protein 1) expression seems to be upregulated in most anaplastic cells in LyP (91.8%) and in approximately one-third of ALCL cases (38.1%). SATB1 is a chromatin organizer that is necessary for the development of thymocytes. A potential mechanism was described by Sun et al,104 showing SATB1 promoter demethylation in SATB1+ cutaneous anaplastic lymphoma.
Finally, the gene encoding the CD30 antigen (TNFRSF8) has been the subject of many studies; it is, however, still unclear whether CD30 expression plays a role in the pathogenesis of pcCD30+LPD or is merely a marker of the disease. Irrespective of its function, the specific and high expression of the protein offers excellent opportunities for treatment protocols using anti-CD30 antibodies (eg, brentuximab vedotin105).
Genetic alterations in primary cutaneous CD8 aggressive epidermotropic cytotoxic T-cell lymphoma
Primary cutaneous CD8 aggressive epidermotropic cytotoxic T-cell lymphoma (pcAECyTCL) is a rare CTCL variant, characterized by an abrupt onset and a highly aggressive clinical course.82,106 Malignant T cells typically express CD3, CD7, CD8, CD45RA, TCR-βF1, T-BET, and 1 or more cytotoxic markers (eg, granzyme B, perforin, and TIA-1), which strongly suggests that neoplastic cells in this lymphoma derive from CD8+ T cells.82,107
Thus far, the pathogenetic basis of this malignancy has been large unknown. Two clinical case reports included the evaluation of CNAs in single patients using array-based methods,108,109 and recently, a study performed on tumors from 20 patients defined the CNA profile of pcAECyTCL by using array-based comparative genomic hybridization.110 Recurrent CNAs uncovered by these studies include losses within 1p, 9p, 13q, and 16p and gains within 7q, 8q, and 17q, with the loss of the region containing CDKN2A/B the most frequent CNA.110
Early this year, the first high-resolution genomic analysis of pcAECyTCL using whole-genome sequencing and RNA sequencing was published by Bastidas Torres et al.111 From this study, it became clear that overactivation of JAK2 signaling because of oncogenic changes in JAK2 and SH2B3, 2 genes with key roles in this signaling pathway, predominantly underlie pcAECyTCL (Figure 4).
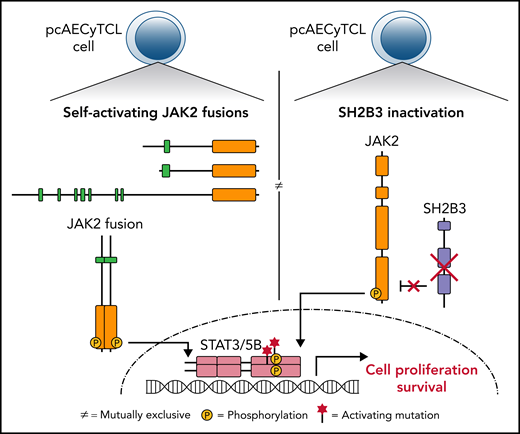
AECyTCL. Genes and pathways recurrently altered in AECyTCL. JAK2 and SH2B3, genes that govern the activation and termination of JAK2 signaling in normal hematopoietic cells, respectively, are mutually exclusively affected in 9 of 12 patients. The remaining 3 patients carry pathogenic indels/single nucleotide variations in other JAK-STAT pathway genes.
AECyTCL. Genes and pathways recurrently altered in AECyTCL. JAK2 and SH2B3, genes that govern the activation and termination of JAK2 signaling in normal hematopoietic cells, respectively, are mutually exclusively affected in 9 of 12 patients. The remaining 3 patients carry pathogenic indels/single nucleotide variations in other JAK-STAT pathway genes.
Genetic alterations in primary cutaneous γδ T-cell lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) originates from activated mature γδ T cells with a cytotoxic phenotype and is an extremely rare disease with an unfavorable prognosis. First knowledge on genetic alterations came from a single case report describing very complex cytogenetic alterations that include translocations involving breakpoints at 9p21, 14q11.2, 14q32.1, or 16q23.1.112 Very recently, a genome-wide DNA, RNA, and TCR sequencing study on 29 cutaneous γδ lymphomas revealed that PCGDTLs are not uniformly derived from Vδ2 cells. Instead, it was shown that the cell-of-origin depends on the tissue compartment from which the lymphomas are derived, and Vδ1 and Vδ2 lymphomas could be distinguished. The results also showed that TCR chain use is nonrandom, suggesting that Vδ1 and Vδ2 might have common antigens. Both types of PCGDTLs harbor similar genomic alterations with potentially targetable oncogenic mutations in the MAPK, MYC, JAK/STAT, and chromatin modification pathways.113
Genetic alterations in subcutaneous panniculitis-like T-cell lymphoma
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare primary cutaneous lymphoma of mature cytotoxic T cells responding very well to immunosuppressive therapy. A breakthrough in the understanding of this disease was made by the identification of germ-line mutations in the HAVCR2 gene encoding T-cell immunoglobulin mucin 3 (TIM3).114 TIM3 is a transmembrane protein expressed by CD8+ T cells and natural killer cells and acts as a negative immune checkpoint. The homozygous p.Y82C mutation turned out to be the most prevalent (10 of 13 patients115; 12 of 16 patients114). Less frequent is the p.I97M variant (3 of 16114). Compound heterozygous mutations (p.Y82C and p.T101I) were described by Polprasert et al115 for 1 patient (of 11 analyzed). The p.Y82C mutation in TIM3 induces protein misfolding and loss of expression of this molecule on the plasma membrane, resulting in loss of its function and promoting acceleration of immunity and inflammation in SPTCL. In addition to this germline predisposition, recurrent somatic mutations were identified in genes involved in epigenetic regulation and phosphatidylinositol 3-kinase/Ak strain transforming/mammalian target of rapamycin signal pathway, suggesting association with the pathogenesis of SPTCL.115,116
Genetic alterations in primary cutaneous peripheral T-cell lymphoma, not otherwise specified
Only 1 study aimed at a genome-wide analysis of primary cutaneous peripheral T-cell lymphoma, not otherwise specified.91 The overall pattern of CNA is distinct from those found for cALCL, MF, and Sz but did not show apparent similarities with peripheral T-cell lymphoma of noncutaneous origin.117 It was suggested that overexpression of the signaling protein encoded by PRKCQ might contribute to the clinical aggressiveness seen in this lymphoma entity.91
Conclusions and perspectives
The combination of state-of-art techniques (initially arrays, subsequently followed by NGS) and clinically well-defined material (according to the WHO-EORTC classification) accelerated the knowledge on the pathogenesis of CTCLs. These studies identified molecular alterations possibly underlying initiation and progression of different CTCL entities and improved therapy.
In a next phase, several developments can be expected. (1) More detailed genomic data also of rare CTCL entities. (2) Combination of single cell sequencing of RNA, DNA, and (surface) proteins of (isolated) tumor cells for in-depth determination of tumor heterogeneity (eg, Eccite technique and derivatives thereof).118 (3) Subdivision and/or reclassification of CTCL based on molecular features as was recently performed for B-cell lymphoma.119 (4) Functional studies aiming to delineate the precise contribution of identified genes and pathways and development of (novel) therapies. These steps will eventually lead to improved diagnosis, prognosis and (precision) treatment of this hematologic malignancy in the skin.
Acknowledgments
Authorship
Contribution: C.P.T., K.D.Q., and M.H.V. designed research, performed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Maarten H. Vermeer, Rm B1-91, Albinusdreef 2, 2333 ZA Leiden, The Netherlands; e-mail: m.h.vermeer@lumc.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal