

Key Points
PROTAC design based on crystal structures of JAK2 kinase domain in complex with ruxolitinib and baricitinib.
PROTACs targeting JAKs are efficacious in vivo in CRLF2r ALL; the most effective degrade multiple targets, including JAKs, IKZF1, and GSPT1.

Abstract
CRLF2-rearranged (CRLF2r) acute lymphoblastic leukemia (ALL) accounts for more than half of Philadelphia chromosome-like (Ph-like) ALL and is associated with a poor outcome in children and adults. Overexpression of CRLF2 results in activation of Janus kinase (JAK)-STAT and parallel signaling pathways in experimental models, but existing small molecule inhibitors of JAKs show variable and limited efficacy. Here, we evaluated the efficacy of proteolysis-targeting chimeras (PROTACs) directed against JAKs. Solving the structure of type I JAK inhibitors ruxolitinib and baricitinib bound to the JAK2 tyrosine kinase domain enabled the rational design and optimization of a series of cereblon (CRBN)-directed JAK PROTACs utilizing derivatives of JAK inhibitors, linkers, and CRBN-specific molecular glues. The resulting JAK PROTACs were evaluated for target degradation, and activity was tested in a panel of leukemia/lymphoma cell lines and xenograft models of kinase-driven ALL. Multiple PROTACs were developed that degraded JAKs and potently killed CRLF2r cell lines, the most active of which also degraded the known CRBN neosubstrate GSPT1 and suppressed proliferation of CRLF2r ALL in vivo, e.g. compound 7 (SJ988497). Although dual JAK/GSPT1-degrading PROTACs were the most potent, the development and evaluation of multiple PROTACs in an extended panel of xenografts identified a potent JAK2-degrading, GSPT1-sparing PROTAC that demonstrated efficacy in the majority of kinase-driven xenografts that were otherwise unresponsive to type I JAK inhibitors, e.g. compound 8 (SJ1008030). Together, these data show the potential of JAK-directed protein degradation as a therapeutic approach in JAK-STAT–driven ALL and highlight the interplay of JAK and GSPT1 degradation activity in this context.
Introduction
Philadelphia chromosome-like (Ph-like) acute lymphoblastic leukemia (ALL) remains 1 of the most clinically problematic forms of ALL.1 It is characterized by a gene expression profile similar to BCR-ABL1 ALL, as well as a range of chromosomal rearrangements, deletions, and mutations that activate kinase signaling pathways.2,3 These diverse alterations fall into 4 main groups: those activating Janus kinase (JAK)-STAT signaling, rearrangements of ABL1 and similar kinases (ABL1-class alterations), alterations activating Ras signaling, and infrequent alterations activating NTRK3, FLT3, LYN, PTK2B, and other kinases.4 In vitro modeling and preclinical studies show that these lesions typically induce constitutive kinase signaling that is abrogated by the use of tyrosine kinase inhibitors (eg, JAK inhibitors, such as ruxolitinib for JAK-STAT–driven Ph-like ALL, and imatinib/dasatinib for ABL1-class Ph-like ALL).5 The clinical efficacy of these agents has been observed in case reports2,6,7 and is now being prospectively evaluated in clinical trials.
CRLF2-rearranged (CRLF2r) ALL represents the largest subtype of Ph-like ALL. CRLF2 is located in the pseudoautosomal region 2 of chromosome Xp/Yp and encodes cytokine receptor-like factor 2 (also known as thymic stromal lymphopoietin receptor), which, with interleukin 7 receptor α (IL-7RA) forms a heterodimeric receptor for thymic stromal lymphopoietin. In 50% to 60% of patients with Ph-like and Down syndrome–associated leukemia, CRLF2 is deregulated and overexpressed as a result of rearrangement of the IGH enhancer (IGH-CRLF2) or P2R8Y promoter (P2RY8-CRLF2) or, less commonly, a missense mutation that results in receptor dimerization (CRLF2 p.Phe232Cys).8-10 The majority of CRLF2r cases have concomitant mutations in genes regulating signaling pathways, particularly those that facilitate signaling through the JAK-STAT pathway, such as mutations at/near JAK2 p.Arg683 in the pseudokinase domain, and, less commonly, in the pseudokinase and kinase domains on JAK1, JAK2, and JAK3. Notably, the JAK2 p.Val617Phe mutation that is characteristic of myeloproliferative neoplasms is rare in CRLF2r ALL, likely as a result of the disease-specific role of the different mutations mediating physical associations between JAK2 and CRLF2/EPOR. CRLF2r leukemic cells also commonly harbor mutations that facilitate leukemogenesis and are associated with poor outcome, including alteration of B lymphoid transcription factor genes, such as IKZF1 (Ikaros), and the tumor suppressor loci CDKN2A/CDKN2B (encoding INK4/ARF).
In engineered cell lines, CRLF2 and JAK2 p.Arg683Gly expression results in cytokine-independent proliferation and activation of the JAK-STAT signaling pathway, both of which are abrogated by type I JAK inhibitors, such as ruxolitinib.10CRLF2r leukemic cells characteristically exhibit activation of downstream signaling pathways that is accentuated by TSLP, including the JAK-STAT and phosphatidylinositol-3-kinase (PI3K) pathways.11 Despite the genetic and cell line evidence supporting a critical role for CRLF2-mediated JAK-STAT signaling in this form of ALL, the response of CRLF2r leukemic cells to ruxolitinib is variable in preclinical human ALL xenograft models11 and with variable activity of ruxolitinib in abrogating signaling or reducing leukemic burden when given as monotherapy or with chemotherapy in pretreated relapsed and refractory ALL.12,13 Resistance to type I JAK inhibitors has been attributed to persistent phosphorylation of JAK2 and activation of parallel signaling pathways, which can be attenuated with type II JAK inhibitors.14,15
Targeted protein degradation, using molecular glues (MGs) and proteolysis-targeting chimeras (PROTACs), has emerged as a promising approach to selectively degrade oncogenic dependencies for therapeutic effect.16,17 Both MGs and PROTACs rely on recruiting the activity of E3 ubiquitin ligases, such as CRBN (cereblon), to redirect nonphysiological neosubstrates for proteasomal degradation. MGs, such as the immunomodulatory drugs (IMiDs) thalidomide and derivatives, result in CRBN-dependent degradation of several proteins, including IKZF1, IKZF3, and casein kinase 1α.18 PROTACs are hetero-bifunctional molecules that couple a ligand- and E3 ligase–interacting small molecules using a linker, enabling engagement and degradation of a target with a known ligandable domain. PROTACs have been developed to target a number of kinase signaling targets and pathways relevant to leukemia, including BCLXL,19 BCR-ABL1,20 BRD4,21,22 BTK,23,24 CDK4/6,25-27 FLT3,28 MCL1,29 MDM2,30 SMARCA2/4,31 and STAT3.32 Important determinants of PROTAC activity include the nature of the target-engaging ligand, the length and composition of the linker, the E3 ligase–interacting moiety, and the choice of E3 ligase itself. Importantly, recently developed CRBN modulators, such as CC-885, change neosubstrate specificity to include GSPT1, the degradation of which has antitumor effects;33 existing data suggest that simultaneous degradation of the PROTAC targets and IMiD substrates is required for optimal antitumor effect.24
As a result of the presumed centrality of JAK-STAT signaling in the pathogenesis of CRLF2r ALL, as well as the variable activity of existing type I/II JAK inhibitors in inhibiting leukemic cell proliferation, we sought to develop PROTACs to target JAKs in JAK-STAT–activated and CRLF2r ALL.
Methods
PROTAC synthesis
Methods for synthesis of PROTAC compounds and intermediates, JAK2 protein purification and crystallography, proteomic analyses, KINOMEscan and equilibrium dissociation constant (Kd) measurements, plasmids and lentiviral transduction, cell line genome editing, ex vivo cytotoxicity immunoblotting, phosphoflow cytometry, and pharmacokinetic methods are described in full in supplemental Methods (available on the Blood Web site).
Cell lines and xenografts
The 18 cell lines used in this study include the B-progenitor ALL cell lines MHH–CALL-49,10,34 (CRLF2r; JAK2 I682F), KOPN4935 (CRFL2r; JAK2 R683G), NALM-6 (DUX4-rearranged),36,37 MHH–CALL-2 (near haploid),34 KOPN75 (PAX5-ETV6),35 697 (TCF3-PBX1),38 SUP-B15 and TOM-1 (BCR-ABL1), and Reh (ETV6-RUNX1); the T-ALL cell lines CCRF-CEM C7 (STIL-TAL1 and NKX2.5-BCL11B), DND-41 (BCL11B-TLX3, IL-7R mutation) and ALL-SIL (TLX1-TRD, NUP214-ABL1); the megakaryoblastic cell line SET-2 (JAK2-V617F); the non-Hodgkin lymphoma cell line Karpas 1106P; and 4 Hodgkin lymphoma cell lines (HDLM-2, SUP-HD1, L-1236, and KM-H2).39 All of the cell lines were confirmed as Mycoplasma spp. free using the Universal Mycoplasma Detection Kit (American Type Culture Collection, Manassas, VA). Cell lines were maintained in RPMI 1640 medium supplemented with 10% or 20% fetal bovine serum (HyClone), penicillin/streptomycin (100 U/mL), and glutamine (100 µM), with the exception of SUP-HD1 cells, which were maintained in McCoy’s 5A medium supplemented with 20% fetal bovine serum, penicillin/streptomycin (100 U/mL), and glutamine (100 µM). Cell identity was checked by short tandem repeat profiling using a PowerPlex Fusion System (Promega) and confirmed using the American Type Culture Collection and German Collection of Microorganisms and Cell Cultures GmbH (DSMZ) short tandem repeat (STR) databases. Cell lines were subject to whole genome and transcriptome sequencing, as previously described.40 Human (h)CD34+ cells were purified from cord blood using CD34 MicroBeads (Miltenyi Biotec) and expanded.41 Normal human peripheral blood mononuclear cells were isolated by Ficoll-Paque density gradient centrifugation from apheresis rings obtained from anonymous healthy blood donors. The donors provided informed consent for the research use of leftover specimens, which was approved by the Institutional Review Board of St Jude Children’s Research Hospital. All xenograft samples were profiled by whole genome and transcriptome sequencing to confirm their ALL subtype and genomic alterations. The xenograft SJBALL047370 (IGH-CRLF2, KRAS G12D, JAK2 wild-type) was used for in vivo preclinical studies; overexpression of CRLF2 was confirmed by flow cytometry using CRLF2-PE antibodies (#1205499-42; eBioscience).
Cytotoxicity assay
A total of 2 × 105 cells was seeded at 100 μL per well in 96-well assay plates (Corning). Assays were performed in triplicate; compounds to be screened were added to assay plates from DMSO stock solutions by pin transfer using 50SS pins (V&P Scientific). The assay plates were incubated at 37°C in 5% CO2 for 72 hours. Cells were then incubated for 4 hours with resazurin (Sigma) solution and read on a Synergy HT plate reader (BioTek Instruments, Winooski, VT). High-throughput assay data were analyzed using our in-house Robust Interpretation of Screening Experiments (RISE) application written in Pipeline Pilot (Biovia, v17.2.0) and the R program. Selected data were plotted and analyzed with GraphPad Prism software v7 using nonlinear regression curve fitting. The area under the drug-response curve per compound calculated from their dose-response curve utilizing the area under a fitted hill curve to the data, and a trapezoidal numeric integral of the actual data provided. In general, a high area under the drug-response curve implies high sensitivity to the drug.
Xenograft and in vivo preclinical studies
Female NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice aged 8 to 12 weeks were used for xenotransplantation. All experimental work was carried out according to Office of Laboratory Animal Welfare guidelines and was approved by the Institutional Animal Care and Use Committee of St Jude Children’s Research Hospital. Mice aged 8 to 12 weeks were inoculated by tail vein injection with 1 × 106 leukemic cells transduced with a lentiviral vector containing the firefly luciferase and yellow fluorescent protein genes (CL20SF2-Luc2aYFP).42 Leukemia burden was determined by weekly bioluminescence imaging using a Xenogen IVIS-200 system and Living Image software (Caliper Life Sciences). Treatment was commenced when luminescence averaged ∼1 × 108 photons per second. Mice were dosed intraperitoneally with compound 7 (30 mg/kg per day). Ruxolitinib was delivered by oral gavage (50 mg/kg twice daily).43 Studies were designed to quantitate and compare leukemia burden by bioluminescent imaging after 1 month of treatment. At the end of the study, cells from spleen, bone marrow, and blood were harvested and stained with mouse CD45-APC-Cy7, human CD45-BV405, or human CD19-PE antibodies and analyzed by flow cytometry to determine the percentage of hCD45+ cells. For western blot, spleen leukemia cells were obtained by purification with hCD19 MicroBeads (130-050-301; Milenty Biotec), per the manufacturer’s protocol. Efficacy was determined by linear regression and 2-way analysis of variance using Prism (GraphPad Software, version 8.4.2).
For pharmacodynamics studies, after engraftment, mice with >50% hCD45 cells in peripheral blood were dosed intraperitoneally with 10, 30, or 100 mg/kg of compound 7 twice daily. One hour after the second dose, mice were euthanized, and cells were collected from bone marrow and spleen. For phosphoflow cytometry analysis, 0.75 × 106 bone marrow cells in 3 mL of medium were fixed by adding 1 mL of 10% formaldehyde and incubating at room temperature for 10 minutes. The cells were spun down at 450g for 5 minutes, resuspended in cold 95% methanol, and stored at −20°C. For immunoblotting, splenic leukemia cells were purified by hCD19 MicroBeads (Miltenyi Biotec). Cells were pelleted, and total protein was extracted using RIPA buffer supplemented with Halt Protease Inhibitor Cocktail (ThermoFisher).
Results
Structure-based design of JAK- and CRBN-directed PROTACs
In view of the suboptimal efficacy of existing type I (eg, ruxolitinib [compound 1] and baricitinib [compound 2]) and type II (eg, CHZ-868) JAK inhibitors in CRLF2 ALL (Table 1), we sought to directly degrade JAKs using JAK-directed PROTACs based on derivatives of ruxolitinib or baricitinib conjugated via a linker to CRBN-binding molecules, including derivatives of lenalidomide and pomalidomide. To inform the design of JAK-directed PROTACs, we determined the crystal structures of the human JAK2 JH1 (kinase) domain in the presence of ruxolitinib and baricitinib to 1.8 Å and 1.7 Å resolution, respectively, providing the structures of these drugs bound to their primary target (Figure 1; supplemental Figure 1; supplemental Table 1, available on the Blood Web site). The previously reported structures of ruxolitinib (bound to chicken c-Src;63 PDB code 4U5J) and of baricitinib (bound to human BMP2K;64 PDB code 4W9X) were used for comparison. As is common for type I JAK inhibitors, both compounds bind to the JAK2-JH1 ATP binding site in the active “DFG-in” conformation, within a cleft between the N- and C-terminal lobes of the protein (Figure 1A). This structure also shows the location of the JAK2 kinase domain mutations observed in CRLF2r B-cell ALL (B-ALL), which are likely to disrupt the compact conformation of the kinase domain, leading to constitutive activation and enhanced STAT5 phosphorylation,44 consistent with their known transforming effect.10,45 Each compound forms 2 direct hydrogen bonding interactions between the pyrimidine ring and the protein backbone atoms of Glu930 and Leu932 in the hinge domain that connects the N-terminal lobe to the C-terminal lobe (Figure 1B-C). In addition, compound binding is accommodated by numerous hydrophobic interactions with various residues lining its binding pocket and a hydrogen bonding interaction with up to 2 nearby water molecules (supplemental Figure 2). The overall conformation of ruxolitinib is flipped by ∼180° compared with its position in the previously reported SRC (c-Src) structure, where 2 pyrimidine nitrogens interact only with the backbone of the SRC residue that is equivalent to Leu932 in JAK2 (Figure 1B). The pyrimidine and pyrazole groups of baricitinib bind similarly to JAK2 and BMP2K,64 whereas the azetidine substituents are displaced; its nitrile group is rotated by ∼100°, aligning parallel to the DFG motif rather than pointing toward it, as seen in JAK2 for ruxolitinib and baricitinib (Figure 1C; supplemental Figure 3). Structurally, the overlap of the common core shared by ruxolitinib and baricitinib is high (Figure 2A; supplemental Figure 3), and both JAK2 structures align with an overall root mean square deviation <0.5 Å. Yet, the differences in the substitution at position 1 manifest in baricitinib’s bulkier substituent displacing the tip of the glycine-rich P-loop by up to 3.1 Å (supplemental Figure 3). With its ability to accommodate ligands of various sizes, the flexibility of the P-loop was one of the structural elements that could be exploited for PROTAC design. Differences in flexibility may also help to distinguish the 4 JAK family members (JAK1-3 and TYK2);46 however, the key insight for rational PROTAC design was the conformation of the pyrimidine ring.
Cytotoxicity of ruxolitinib- and baricitinib-based PROTACs in different ALL cells and hCD34+ cells
| Compounds | EC50 (nM) in ALL cells and normal cells | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MHH–CALL-4 | KOPN49 | MHH–CALL-2 | KOPN75 | NALM-6 | 697 | hCD34+ cells | ||||
| Parental | CRBN-KD | Baricitinib (1.5 µM) | Lenalidomide (30 µM) | |||||||
| Compound 5 | 1.5 | 21.6 | 1.2 | 4.0 | 8.3 | 5.8 | 53 | 8.6 | 4.2 | |
| Compound 6 | 0.4 | 44.3 | 0.3 | 1.1 | 1.8 | 2.0 | 89.4 | 28.4 | 3.9 | |
| Compound 7 | 0.4 | 3456.2 | 0.4 | >57 | 1.1 | 2.2 | 1.0 | 13.3 | 4.2 | 1.3 |
| Compound 8 | 5.4 | >3000 | 15.9 | >3000 | >3000 | 76.9 | 138.9 | >3000 | >3000 | |
| Ruxolitinib | >7 000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | ||
| Baricitinib | 1 482 | >6000 | >6000 | >6000 | >6000 | >6000 | >6000 | >6000 | ||
| CHZ868 | 180 | |||||||||
| Thalidomide | >7 000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | ||
| Lenalidomide | 34 480 | |||||||||
| Pomalidomide | >7 000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | ||
| Compounds | EC50 (nM) in ALL cells and normal cells | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MHH–CALL-4 | KOPN49 | MHH–CALL-2 | KOPN75 | NALM-6 | 697 | hCD34+ cells | ||||
| Parental | CRBN-KD | Baricitinib (1.5 µM) | Lenalidomide (30 µM) | |||||||
| Compound 5 | 1.5 | 21.6 | 1.2 | 4.0 | 8.3 | 5.8 | 53 | 8.6 | 4.2 | |
| Compound 6 | 0.4 | 44.3 | 0.3 | 1.1 | 1.8 | 2.0 | 89.4 | 28.4 | 3.9 | |
| Compound 7 | 0.4 | 3456.2 | 0.4 | >57 | 1.1 | 2.2 | 1.0 | 13.3 | 4.2 | 1.3 |
| Compound 8 | 5.4 | >3000 | 15.9 | >3000 | >3000 | 76.9 | 138.9 | >3000 | >3000 | |
| Ruxolitinib | >7 000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | ||
| Baricitinib | 1 482 | >6000 | >6000 | >6000 | >6000 | >6000 | >6000 | >6000 | ||
| CHZ868 | 180 | |||||||||
| Thalidomide | >7 000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | ||
| Lenalidomide | 34 480 | |||||||||
| Pomalidomide | >7 000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | >7000 | ||
EC50, 50% effective concentration; KD, knockdown.
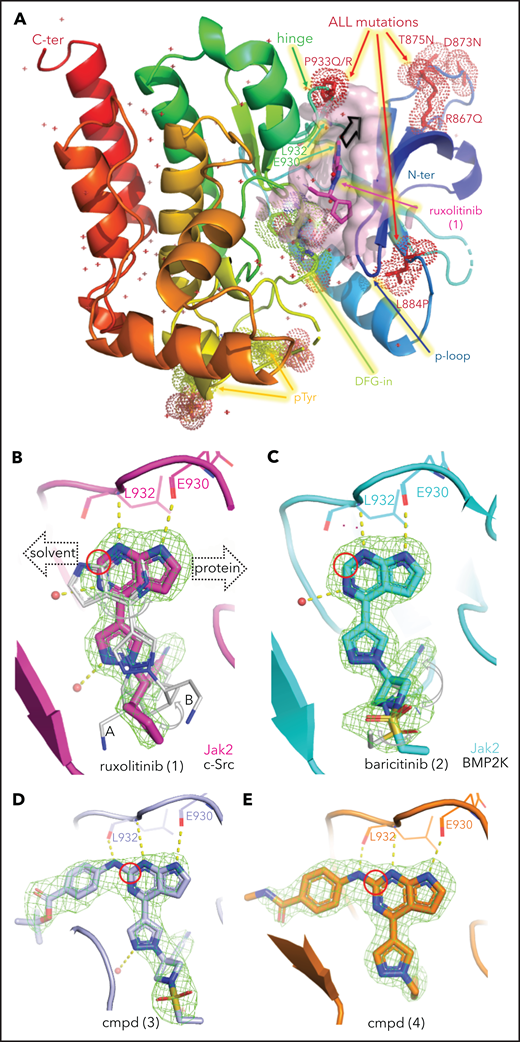
Structural analysis of JAK inhibitors and PROTACs bound to JAK2. (A) The JAK2 JH1 (kinase) domain assumes a “DFG-in” conformation upon binding ruxolitinib (pink sticks). Protein color is graded from blue at the N terminus (N-ter) to red at the C terminus (C-ter), with ALL mutations shown as red dots, the mobile P-loop in dark blue, and the ruxolitinib binding site shown as a pink surface close to the activation loop containing the DFG motif (green dots); 2 phosphorylated tyrosines (p-Tyr) are shown as yellow dots. The black arrow near ruxolitinib indicates the direction of linker extension for the design of PROTACs as shown in (B). (B) The orientation of ruxolitinib bound to human JAK2 (pink) is flipped with respect to its position in complex with SRC (4U5J, from chicken)63 shown in gray as conformation A and B for both molecules in the asymmetric unit, guiding the linker attachment point (red circle) for the PROTAC design toward solvent. Polder OMIT electron density maps are shown as green mesh at 3σ and hydrogen bonds are shown as yellow dotted lines for all structures in panels B-E. (C) Baricitinib bound to JAK2 shows a different placement of ligand moieties, such as the nitrile group, compared with BMP2-inducible kinase (4W9X).64 PROTAC precursor compounds (cmpd) 3 (D) and 4 (E) possess an additional H-bond interaction between the linker and the backbone of the hinge region and extend into solvent.
Structural analysis of JAK inhibitors and PROTACs bound to JAK2. (A) The JAK2 JH1 (kinase) domain assumes a “DFG-in” conformation upon binding ruxolitinib (pink sticks). Protein color is graded from blue at the N terminus (N-ter) to red at the C terminus (C-ter), with ALL mutations shown as red dots, the mobile P-loop in dark blue, and the ruxolitinib binding site shown as a pink surface close to the activation loop containing the DFG motif (green dots); 2 phosphorylated tyrosines (p-Tyr) are shown as yellow dots. The black arrow near ruxolitinib indicates the direction of linker extension for the design of PROTACs as shown in (B). (B) The orientation of ruxolitinib bound to human JAK2 (pink) is flipped with respect to its position in complex with SRC (4U5J, from chicken)63 shown in gray as conformation A and B for both molecules in the asymmetric unit, guiding the linker attachment point (red circle) for the PROTAC design toward solvent. Polder OMIT electron density maps are shown as green mesh at 3σ and hydrogen bonds are shown as yellow dotted lines for all structures in panels B-E. (C) Baricitinib bound to JAK2 shows a different placement of ligand moieties, such as the nitrile group, compared with BMP2-inducible kinase (4W9X).64 PROTAC precursor compounds (cmpd) 3 (D) and 4 (E) possess an additional H-bond interaction between the linker and the backbone of the hinge region and extend into solvent.
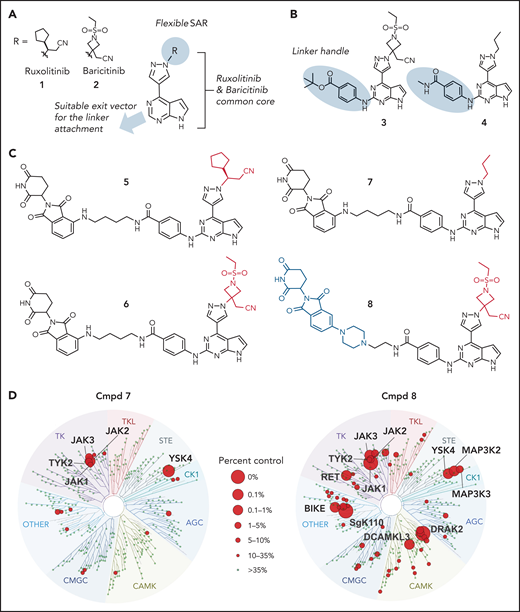
Design principles, molecular structure and properties of JAK PROTACs. (A) PROTAC design strategy. (B) Structures of key intermediates 3 and 4, containing the linker handle. (C) Structures of PROTACs 5 through 8. (D) TREEspot visualization of the kinome selectivity profile of compound (Cmpd) 7 (left panel) and Cmpd 8 at 100 nM concentration (Kinome Scan).
Design principles, molecular structure and properties of JAK PROTACs. (A) PROTAC design strategy. (B) Structures of key intermediates 3 and 4, containing the linker handle. (C) Structures of PROTACs 5 through 8. (D) TREEspot visualization of the kinome selectivity profile of compound (Cmpd) 7 (left panel) and Cmpd 8 at 100 nM concentration (Kinome Scan).
Thus, we identified the solvent-oriented C2-carbon of the pyrimidine core present in ruxolitinib and baricitinib as the most promising linker attachment point for PROTAC design (Figure 2A), because the exit vector points into solvent and is amenable to chemical modification. In contrast, reliance on the SRC-bound ruxolitinib crystal structure would have misled PROTAC design, because the C2-carbon in the flipped pyrimidine points toward the protein rather than solvent (Figure 1B). Lacking a suitable functional group in this position, we introduced a 4-amino-benzamide moiety as a synthetically amenable handle for linker incorporation via amide bond formation (Figure 2B-C). This structural modification was well tolerated, and we were able to generate structures of the related intermediate compounds 3 and 4 cocrystallized with JAK2. The electron density maps of both structures show the handle moiety unambiguously pointing toward solvent (Figure 1D-E), whereas the linker nitrogen forms another H-bond interaction with the backbone carbonyl of Leu932, forming an alternating H-bond donor and acceptor pattern with the backbone in the hinge region. Although this interaction stabilizes the linker within the binding pocket, the electron density map is less defined for the tert-butyl group of compound 3 compared with the carboxamide moiety in compound 4. The enhanced flexibility is evident across the protein in complex with compound 4, most notably at the tip of the P-loop.
In light of these findings, we synthesized the corresponding ruxolitinib- and baricitinib-based PROTAC compounds 5 and 6, respectively, containing the 4-amino-benzamide handle attached to a 4-carbon chain-linked pomalidomide as the E3 ligase recruiting element (Figure 2C). Based on the reportedly flexible structure-activity relationship around the pyrazole region, we also synthesized N-propyl analog compound 7. This structural adjustment was made to minimize the PROTAC molecular size and polar surface area and, thus, improve its cellular permeability. Indeed, the lower molecular weight (688.29 Da) and topological polar surface area (189.06 Å2) of PROTAC 7 resulted in a considerably higher cell permeability in the Caco-2 assay (the apparent permeability coefficient (Papp) = 19.12 nm/s; supplemental Table 2). Utilizing previously reported structure-activity relationship data,47 we designed baricitinib-based compound 8 containing a piperazine-linked thalidomide moiety with the goal of avoiding off-target degradation of GSPT1. Importantly, compounds 5, 6, 7, and 8 retained the high affinity for JAK2 (distribution coefficient = 0.63, 0.11, 0.3, and 0.095 nM respectively; supplemental Table 3) and the kinome-wide selectivity of compounds 7 and 8, similar to the parent inhibitors (Figure 2D; supplemental Tables 4 and 5).
JAK-directed PROTACs potently kill CRLF2r ALL cell lines
Consistent with prior data, we observed weak activity of existing type I JAK inhibitors in cell line and xenograft models of CRLF2r ALL (supplemental Figure 4). In contrast, multiple JAK-directed PROTACs exhibited potent activity, with several exhibiting >10 000-fold potency over the parent JAK inhibitor (ruxolitinib or baricitinib) or the CRBN-interacting moiety (thalidomide, lenalidomide, or pomalidomide; Table 1; supplemental Figure 4B; supplemental Table 6). These compounds were most active in the CRLF2r cell lines MHH–CALL-4 and KOPN49, with less potency in MHH–CALL-2 (hypodiploid ALL) and KOPN75 (PAX5-ETV6) cells and the least efficacy in 697 (TCF3-PBX1) and NALM-6 (DUX4-rearranged ALL) cells. Treatment with the parent JAK inhibitor, baricitinib (1.5 µM), resulted in partial attenuation of this activity for compound 8 only, showing that activity was not merely due to JAK inhibition (Table 1). In contrast, the activity of compound 7 was abolished by cotreatment with 30 µM lenalidomide or CRISPR/Cas9 genome editing–based knockdown of CRBN, indicating that E3 ligase activity was required for activity (Table 1).
PROTAC activity is influenced by JAK and GSPT1 degradation
To determine the mechanistic basis of cell killing by JAK-directed PROTACs, we performed time- and dose-dependent analyses of protein degradation by immunoblotting and global proteomic analysis. This demonstrated the ability of multiple PROTACs to degrade multiple JAKs (JAK1 > JAK3 > JAK2 >> TYK2) that was also abolished by knockdown of CRBN (Figure 3B-G; supplemental Figure 4C-G). In addition to JAKs, the known IMiD-induced CRBN neosubstrates IKZF1 and GSPT148 were degraded (Figure 3B-D; supplemental Figure 4C-D). Thus, PROTAC-induced cell killing occurred despite the loss of IKZF1, which otherwise promotes leukemic cell fitness in B-ALL.49-53 Correlative analysis of the extent of degradation of GSPT1 and JAK2 and killing of MHH–CALL-4 cells demonstrated that compounds with minimal activity on either target (eg, 59 and 80) had minimal activity, compounds resulting in preferential JAK2 (eg, 8, 56) or GSPT1 degradation (eg, 53 or 61) were intermediately effective, and those compounds with the greatest degradation of both targets resulted in the greatest cellular cytotoxicity (Figure 3H; supplemental Figure 5; supplemental Table 7). Phosphoflow cytometry of MHH–CALL-4 cells starved of cytokines overnight and treated with ruxolitinib or PROTAC (5, 6, or 7) for 1 hour before stimulation, with or without administration of the CRLF2 ligand TSLP for 30 minutes, showed that ruxolitinib and PROTACs resulted in comparable JAK inhibition (Figure 3I). This is consistent with the notion that degradation, rather than simple inhibition of JAK2 activity, was required for activity.
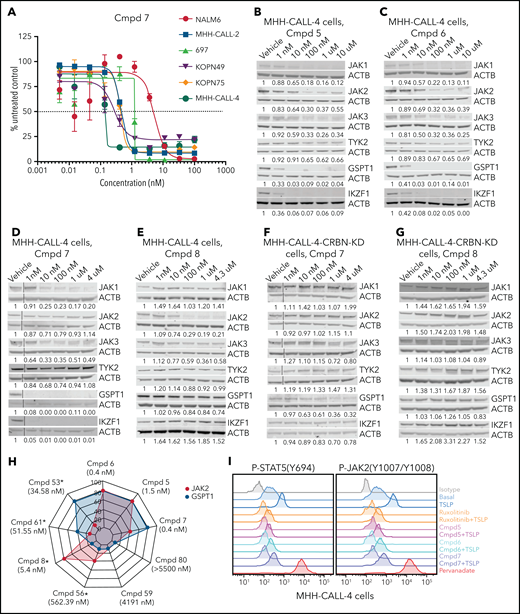
Protein degradation, cytotoxicity, and effect of PROTAC compounds on JAK-STAT signaling pathway in MHH–CALL-4 cells. (A) Cytotoxicity of compound (Cmpd) 7 in 6 ALL cell lines (72-hours incubation). Dose-dependent degradation of JAKs, GSPT1, and IKZF1 by Cmpd 5 (B), Cmpd 6 (C), Cmpd 7 (D), and Cmpd 8 (E) in MHH–CALL-4 cells. Protein degradation by Cmpd 7 (F) and Cmpd 8 (G) in MHH–CALL-4 cells with CRBN knockdown (KD). (H) Spider chart showing 50% effective concentration of 9 representative PROTACs and degradation of JAK2 and GSPT1 in MHH–CALL-4 cells. Numbers within the web are the percentages of protein degradation normalized to vehicle control. The structures of Cmpd 53, 56, 59, 61, and 80 are shown in supplemental Figure 5. *50% Effective concentration was determined based on partial cytotoxicity curve; detailed information is summarized in supplemental Table 7. (I) Phosphoflow analysis of JAK-STAT5 signaling pathway in MHH–CALL-4 cells treated or not with 25 ng/mL TSLP. For inhibition, cells were treated with 1 µM ruxolitinib or PROTACs for 1 hour before TSLP stimulation.
Protein degradation, cytotoxicity, and effect of PROTAC compounds on JAK-STAT signaling pathway in MHH–CALL-4 cells. (A) Cytotoxicity of compound (Cmpd) 7 in 6 ALL cell lines (72-hours incubation). Dose-dependent degradation of JAKs, GSPT1, and IKZF1 by Cmpd 5 (B), Cmpd 6 (C), Cmpd 7 (D), and Cmpd 8 (E) in MHH–CALL-4 cells. Protein degradation by Cmpd 7 (F) and Cmpd 8 (G) in MHH–CALL-4 cells with CRBN knockdown (KD). (H) Spider chart showing 50% effective concentration of 9 representative PROTACs and degradation of JAK2 and GSPT1 in MHH–CALL-4 cells. Numbers within the web are the percentages of protein degradation normalized to vehicle control. The structures of Cmpd 53, 56, 59, 61, and 80 are shown in supplemental Figure 5. *50% Effective concentration was determined based on partial cytotoxicity curve; detailed information is summarized in supplemental Table 7. (I) Phosphoflow analysis of JAK-STAT5 signaling pathway in MHH–CALL-4 cells treated or not with 25 ng/mL TSLP. For inhibition, cells were treated with 1 µM ruxolitinib or PROTACs for 1 hour before TSLP stimulation.
To further examine the relative roles of degradation of GSPT1 and JAK in PROTAC activity, we introduced a degradation-resistant mutant of GSPT1, G575N,33,54 into MHH–CALL-4 cells by lentiviral transduction. To distinguish endogenous GSPT1 from exogenous GSPT1, a FLAG tag was added to the C terminus of GSPT1. Wild-type and mutant GSPT1 were highly expressed in MHH–CALL-4 cells (Figure 4A-B). Compared with wild-type GSPT1, GSPT1-G575N was not degraded within 24 hours when treated with compound 6 (Figure 4C). GSPT1-G575N conferred partial resistance to compound 6, which degrades JAK2 and GSPT1 (Figure 4D), but it did not have any effect on sensitivity to compound 8, a JAK2-specific degrader (Figure 4E). Furthermore, MHH–CALL-4 cells expressing GSPT1-G575N, but not wild-type GSPT1, showed complete resistance to the GSPT1 degrader CC-885 (Figure 4F). Thus, GSPT1 degradation, in part, mediates cytotoxicity only for those PROTACs degrading both GSPT1 and JAKs.
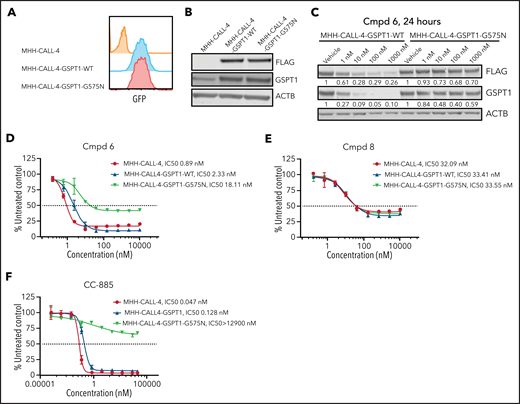
GSPT1 partially contributes to cytotoxicity of PROTACs in MHH–CALL-4 cells. (A) Flow cytometry confirmation of sorted cells expressing GFP. (B) Western blot analysis of exogenous expression of FLAG-tagged wild-type GSPT1 and GSPT1-G575N in MHH–CALL-4 cells. (C) Dose-dependent degradation of GSPT1 24 hours after compound (Cmpd) 6 treatment in MHH–CALL-4 cells expressing wild-type GSPT1 or GSPT1-G575N. Cytotoxicity of Cmpd 6 (D), Cmpd 8 (E), and CC-885 (F) in MHH–CALL-4 cells and MHH–CALL-4 cells expressing wild-type GSPT1 or GSPT1-G575N.
GSPT1 partially contributes to cytotoxicity of PROTACs in MHH–CALL-4 cells. (A) Flow cytometry confirmation of sorted cells expressing GFP. (B) Western blot analysis of exogenous expression of FLAG-tagged wild-type GSPT1 and GSPT1-G575N in MHH–CALL-4 cells. (C) Dose-dependent degradation of GSPT1 24 hours after compound (Cmpd) 6 treatment in MHH–CALL-4 cells expressing wild-type GSPT1 or GSPT1-G575N. Cytotoxicity of Cmpd 6 (D), Cmpd 8 (E), and CC-885 (F) in MHH–CALL-4 cells and MHH–CALL-4 cells expressing wild-type GSPT1 or GSPT1-G575N.
In vivo PROTAC activity
We next examined the pharmacokinetics, antitumor activity, and pharmacodynamics of compound 7 in vivo. Pharmacokinetics analysis of 30- and 100-mg/kg doses showed that sustained in vivo exposure could be achieved after intraperitoneal dosing, with the plasma concentration well above the cellular 50% effective concentration value after 24 hours (Figure 5A; supplemental Table 8). Doses of 10, 30, and 100 mg/kg were used for pharmacodynamics studies in which CRLF2r/KRAS G12D/JAK2 wild-type leukemic cells harvested from NSG mice marked with a lentiviral vector expressing CL20SF2-Luc2aYFP were subjected to phosphoflow cytometry and immunoblotting for JAKs and GSPT1. This showed a dose-dependent inhibition of JAK2 and STAT5 phosphorylation and JAK/GSPT1 degradation that was near maximal at the 30-mg/kg dose (Figure 5B-C). In vivo, ruxolitinib alone did not attenuate leukemic cell proliferation, whereas monotherapy with compound 7 administered at 30 mg/kg intraperitoneally daily resulted in a significant reduction in leukemia burden in bone marrow, blood, and spleen, as well as measured by total body bioluminescent imaging over a 28-day dosing period (Figure 5D-G; supplemental Figure 6). Compound 7 was well tolerated with no weight loss or perturbation in blood counts over a 28-day period (Figure 5H; data not shown).
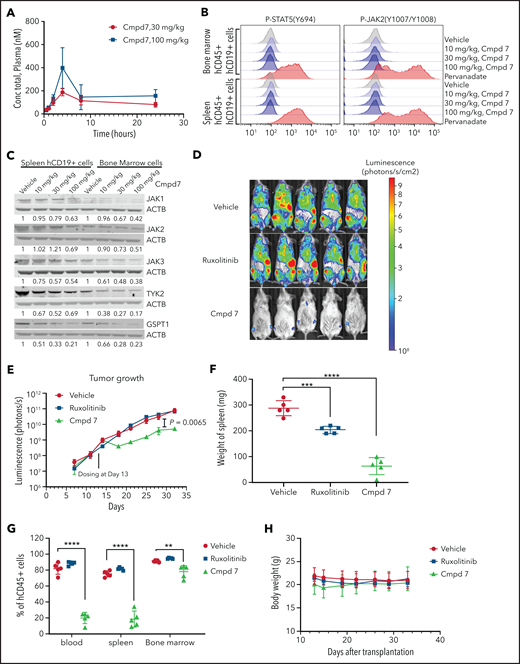
In vivo pharmacokinetics, pharmacodynamics, and efficacy studies of ruxolitinib-based PROTAC compound 7. (A) Plasma concentration of compound (Cmpd) 7 in a mouse injected intraperitoneally with a single dose. (B) Phosphoflow analysis of Cmpd 7’s effect on the JAK-STAT signaling pathway in vivo. (C) Degradation of JAK family kinases and GSPT1 by Cmpd 7 in vivo. (D-E) Cmpd 7, but not ruxolitinib, slowed leukemia growth in a patient-derived xenograft. (D) Bioluminescence imaging at day 32 after transplantation, 2 days before the end of the study. (E) Leukemia burden during the study. Cmpd 7 reduced spleen size (F) and tumor burden (G) in a patient-derived xenograft model. (H) Weight of NSG mice dosed with compounds and vehicle in an in vivo efficacy study. Xenograft of SJBALL047370 (CRLF2r/KRAS G12D/JAK2 wild type) was used in this in vivo study. **P < .01, ***P < .001, ****P < .0001.
In vivo pharmacokinetics, pharmacodynamics, and efficacy studies of ruxolitinib-based PROTAC compound 7. (A) Plasma concentration of compound (Cmpd) 7 in a mouse injected intraperitoneally with a single dose. (B) Phosphoflow analysis of Cmpd 7’s effect on the JAK-STAT signaling pathway in vivo. (C) Degradation of JAK family kinases and GSPT1 by Cmpd 7 in vivo. (D-E) Cmpd 7, but not ruxolitinib, slowed leukemia growth in a patient-derived xenograft. (D) Bioluminescence imaging at day 32 after transplantation, 2 days before the end of the study. (E) Leukemia burden during the study. Cmpd 7 reduced spleen size (F) and tumor burden (G) in a patient-derived xenograft model. (H) Weight of NSG mice dosed with compounds and vehicle in an in vivo efficacy study. Xenograft of SJBALL047370 (CRLF2r/KRAS G12D/JAK2 wild type) was used in this in vivo study. **P < .01, ***P < .001, ****P < .0001.
Ex vivo testing of a panel of PROTACs and leukemia genotypes
Because the activity of compound 7 may be attributable to GSPT1 and JAK2 degradation, and because a range of genetic alterations and signaling perturbations activate JAK-STAT signaling and complement CRLF2 rearrangements in Ph-like ALL,55-57 we next compared the ex vivo activity of 4 PROTACs of varied GSPT1/JAK activity (compounds 5-8, supplemental Table 7), 2 JAK inhibitors, and 2 IMiDs in 11 patient-derived ALL xenografts (CRLF2r, EPOR rearranged, JAK2 rearranged, IL7R/SH2B3 mutated; supplemental Tables 9 and 10). The majority of tumors were sensitive to JAK/GSPT1-active compounds 5, 6, and 7 but were insensitive to the parent compounds and, commonly, the type II inhibitor CHZ-868 (Figure 6A-F; supplemental Table 9). In contrast, responses to compound 8, a JAK2-specific PROTAC that lacks GSPT1 activity, varied. Two samples (1 of 2 EPOR rearranged and 1 of 6 CRLF2r) were unresponsive to type I inhibitors but showed partial responsiveness (<50% cell killing) to compound 8 (Figure 6A-B), several samples exhibited intermediate responsiveness (comparable to CHZ-868 but less than compounds 5, 6, and 7; Figure 6C-D), and several samples, including those with both CRLF2 and JAK2 rearrangements, were highly responsive (Figure 6E-F; supplemental Table 9). The JAK-STAT signaling pathway was inhibited by compound 8 in 1 such sample (SJBALL003145, P2RY8-CRLF2, PAX5-ZNF521; Figure 6G), with corresponding near-complete degradation of JAK2 but not GSPT1 (Figure 6H). Thus, our data show that CRBN-mediated selective degradation of JAK2 can be achieved without affecting the level of GSPT1, while retaining antileukemic efficacy that markedly exceeds that of the parental inhibitors in the majority of tumors.
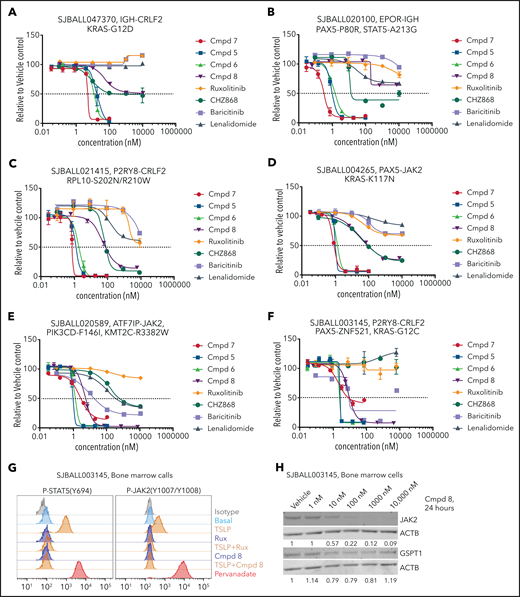
Ex vivo cytotoxicity and protein degradation by PROTACs in patient-derived xenograft ALL cells. (A-F) Response of multiple ALL xenografts of different genotypes to PROTACs in stromal cell–based ex vivo cytotoxicity assays. (G) Ex vivo phosphoflow analysis of xenograft SJBALL021415 cells harvested from bone marrow of NSG mice. (H) Ex vivo dose-dependent degradation of JAK2 and GSPT1 protein by compound 8 in xenograft SJBALL021415 cells. Cmpd, compound.
Ex vivo cytotoxicity and protein degradation by PROTACs in patient-derived xenograft ALL cells. (A-F) Response of multiple ALL xenografts of different genotypes to PROTACs in stromal cell–based ex vivo cytotoxicity assays. (G) Ex vivo phosphoflow analysis of xenograft SJBALL021415 cells harvested from bone marrow of NSG mice. (H) Ex vivo dose-dependent degradation of JAK2 and GSPT1 protein by compound 8 in xenograft SJBALL021415 cells. Cmpd, compound.
Discussion
Clinically, CRLF2r ALL is the most problematic form of B-ALL. CRLF2r ALL accounts for the majority of Ph-like ALL cases and is consistently associated with high-risk features and poor outcome with conventional therapy.1,58 In view of the extensive genetic and preclinical data indicating the importance of CRLF2 overexpression and concomitant lesions driving activation of JAK-STAT and parallel signaling pathways as drivers of leukemogenesis in this entity,8-11 abrogation of JAK-STAT signaling is intuitively attractive. Although type I JAK inhibitors have shown promise in preclinical models, data from human models (eg, patient-derived xenografts) and emerging clinical data have shown that ruxolitinib has variable or poor activity in CRLF2r ALL, at least when administered as a single agent.12 Although the basis for this is unclear, it has been attributed to activation of JAK1/3 as the result of stabilization of phosphorylated JAK2 with type I inhibitors and/or activation of PI3K and ERK signaling pathways.
Here, we obtained crystal structures of JAK inhibitors bound to the JH1 kinase domain of JAK2 to inform the rational design and synthesis of multiple series of JAK-directed PROTACs utilizing different parent JAK inhibitors, CRBN-interacting IMiDs, and their derivatives. We identified solute-exposed regions of JAK inhibitors, as well as moieties amenable for modification and attachment of linkers. PROTACs generated from ruxolitinib and baricitinib showed potent antileukemic efficacy. Several such compounds showed remarkable efficacy (eg, compound 7 showed >70 000-fold activity in CRLF2r MHH–CALL-4 cells compared with each parent compound [ruxolitinib and pomalidomide]). This activity required CRBN-mediated protein degradation, because the compounds showed inhibition of JAK2 phosphorylation similar to ruxolitinib; efficacy was not abolished by competition with a parent JAK inhibitor, whereas activity was abolished by genetic disruption of CRBN. These results contrast with prior efforts to design JAK-directed PROTACs that have not been tested in tumors.59
Importantly, by comparing protein degradation patterns and the antileukemic activity of multiple series of compounds, we were able to examine the relative contribution of the degradation of key targets, including GSPT1 and JAKs. GSPT1 is a widely expressed translation termination factor and a neosubstrate of CRBN upon treatment with MGs, such as CC-885, that has shown efficacy in acute myeloid leukemia.33 We showed that PROTACs active in cellular assays had a broad degradation profile, including multiple JAKs (typically, JAK1, JAK2, and JAK3 more than TYK2), GSPT1, and the known CRBN neosubstrates IKZF1 and IKZF3. Although IMiD or PROTAC-based degradation of IKZF1/3 is of therapeutic benefit in mature B-cell malignancies, such as myeloma or chronic lymphocytic leukemia, degradation in pre–B-ALL may be expected to increase leukemic cell survival, because loss-of-function/dominant-negative IKZF1/3 alterations are common initiating or cooperating leukemogenic mutations in multiple subtypes of pre–B-ALL, including Ph-like ALL.49,50,60 Moreover, many somatic mutant forms of IKZF1 lack the second N-terminal zinc finger that interacts with the thalidomide–CRBN–E3 ligase complex; thus, IMiD or PROTAC degraders may selectively degrade wild-type, but not mutant, IKZF1 observed in B-ALL. Despite these potential caveats, direct degradation of IKZF1 in ALL lines with IMiDs, such as thalidomide, had minimal effect on ALL cell viability. In contrast, degradation of GSPT1 was induced by the most potent PROTACs (ie, compounds 5, 6, and 7). We were also able to develop PROTACs with minimal GSPT1 degradation that retained JAK2 activity, such as compound 8. This modification resulted in a relative reduction in potency in ALL, and a loss of activity in the JAK2 V617F SET-2 and JAK2 amplified lymphoma cell lines, testing of compound 8 ex vivo in a range of representative JAK-STAT–activating lesions showed significantly greater activity than did type I and II JAK2 inhibitors, which were usually inactive, as well as activity that was comparable to GSPT1-degrading PROTACs in several tumors. Thus, although genetic inactivation of JAK2 alone has been reported to be dispensable for leukemic cell maintenance,61 our data indicate that degradation of JAK2 is cytotoxic in many JAK-STAT–driven ALLs in a manner that is superior to type I and II inhibitors. It remains possible that the superior efficacy of compound 8 may reflect activity against additional targets, which may be revealed by additional proteo-genomic screens. Moreover, these data support the notion that combined targeting of JAK2 with other cellular vulnerabilities, such as BCL2,5,62 PI3K,11 MYC,62 and, in the present study, GSPT1, is required for optimal antitumor activity. Our results describing the structural and chemical principles for the rational design of JAK-specific PROTACs with “tunable” specificity provide the foundation for the evaluation of such compounds across a range of potentially susceptible malignancies.
Acknowledgments
The authors thank T. Coop and R. Heath (Protein Production Facility) and R. Thorne (Center for In Vivo Imaging and Therapeutics), and the Biomolecular X-ray Crystallography Center of St Jude Children’s Research Hospital. They also thank J. Gray and the Vector Core of St. Jude Children’s Research Hospital for providing the CL20SF2-Luc2aYFP lentiviral vector and for preparing lentiviral supernatants and J. Cools (Vlaams Instituut voor Biotechnologie Katholieke Universiteit (VIB-KU) Leuven Center for Cancer Biology) for providing T-cell ALL cell line transcriptome sequencing data.
This work was supported by a Translational Research Program award from the Leukemia and Lymphoma Society and the Snowdome Foundation, a National Institutes of Health, National Cancer Institute Outstanding Investigator Award (R35 CA197695), a St Baldrick’s Foundation Robert J. Arceci Innovation Award, and a St Jude Cancer Center Support Grant (CA021765) (C.G.M.), the National Institutes of Health, National Cancer Institute (U24-CA196172) (M.R.L. and E.P.), and the American Syrian Associated Charities of St Jude Children’s Research Hospital.
Authorship
Contribution: J.M., J.A.J., M.A., A.M., and L.J.A. designed and synthesized PROTACs; S.Y-C.B., S.N., and M.F. performed structural studies; D.M., L.R., R.L. and J.J.Y. performed ex vivo cytotoxicity assays; K.G.R. and Y.C. performed in vivo assays; L.Y. and D.C. performed bioanalytical and drug formulation studies; H.G., T.I., and K.A. provided the KOPN49 cell line; E.M.P. and M.R.L. provided the patient sample for xenografting; C.Q. performed bioinformatic analyses; B.S.H. and S.M.P.-M. performed cell line genome editing; A.S. was project manager; Y.C. performed in vitro assays; and M.F., Z.R. and C.G.M. designed and supervised all studies.
Conflict-of-interest disclosures: C.G.M. has received research funding from Loxo Oncology, Pfizer, and AbbVie and has received honoraria from Pfizer, Illumina, and Amgen. The remaining authors declare no competing financial interests.
Correspondence: Marcus Fischer, Department of Chemical Biology, and Therapeutics, St Jude Children's Research Hospital, 262 Danny Thomas Place, Mail Stop 1000, Memphis, TN 38105, email: marcus.fischer@stjude.org; Zoran Rankovic, Department of Chemical Biology and Therapeutics, St Jude Children's Research Hospital, 262 Danny Thomas Place, Mail Stop 1000, Memphis, TN 38105; email: zoran.rankovic@stjude.org; and Charles G. Mullighan, Department of Pathology, St Jude Children's Research Hospital, 262 Danny Thomas Place, Mail Stop 342, Memphis, TN 38105, email: charles.mullighan@stjude.org.
Protein structure data reported in this article have been deposited in the Protein Data Bank as 6WTN (JAK2+ruxolitinib), 6WTO (JAK2+baricitinib), 6WTP (JAK2+compound 3), and 6WTQ (JAK2+compound 4). During revision, structures of JAK2+ruxolitinib (PDB 6VGL) and JAK2+baricitinib (PDB 6VN8) were reported. These structures were not utilized in this study. Cell line RNA and genome sequencing data have been deposited in the Sequence Read Archive (accession number PRJNA629583). Xenograft genomic data has been deposited in the European Genome-Phenome Archive, accession EGAS00001005180. The CRLF2r xenograft is available at the Public Resource of Patient-Derived and Expanded Leukemias (propel.stjude.cloud).
Data sharing requests should be sent to Charles G. Mullighan (e-mail: charles.mullighan@stjude.org).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal