

In this issue of Blood, investigate the molecular etiology of cytotoxic cutaneous T-cell lymphoma (CTCL). Little is known about genetic events that lead to the malignant transformation of T cells in this collection of aggressive skin-associated lymphomas. Given the aggressive nature of cytotoxic CTCL and poor response to therapy, deeper understanding of this subclass of CTCL is needed. The study by Lee et al utilizes modern genomic approaches to tackle this challenge.1
Although histopathological differences exist between the different cytotoxic CTCL entities, little is known about the oncogenic fusions that distinguish the various types of cytotoxic CTCLs. The authors use targeted anchored multiplexed polymerase chain reaction, whole-genome sequencing, and RNA sequencing to gain insight into genetic differences underlying primary cutaneous γδ T-cell lymphomas, primary cutaneous CD8+ aggressive epidermotropic T-cell lymphoma, and a cytotoxic CTCL subtype negative for CD8 and γδ T-cell receptor with mixed histological features (CTCL-NOS; not otherwise specified), all distinct, poorly understood CTCL subtypes characterized by rapid progression and poor prognosis. The authors identify frequent gene fusions of Janus kinase (JAK)2 in CD8+ aggressive epidermotropic CTCL that lead to activation of the JAK/STAT pathway. Despite clinical similarity of multiple cytotoxic CTCL disease entities, JAK2 fusion genes were found to be restricted to cytotoxic CTCL of CD8+ T-cell origin, suggesting that it may be a defining feature and potential cancer-intrinsic vulnerability that can be exploited by small molecule inhibitors (see figure).
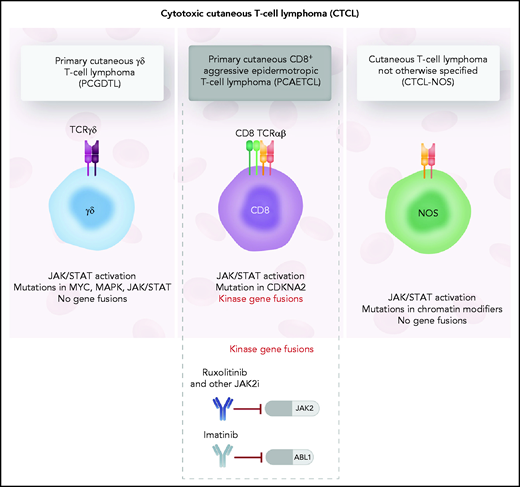
The 3 types of cytotoxic CTCL evaluated in the study by Lee et al. Although transcriptional analysis revealed upregulation of JAK/STAT3 signaling in all 3 cytotoxic CTCL entities, JAK2 fusions were found to be restricted to the CD8+ aggressive epidermotropic T-cell lymphoma. Professional illustration by Somersault18:24.
The 3 types of cytotoxic CTCL evaluated in the study by Lee et al. Although transcriptional analysis revealed upregulation of JAK/STAT3 signaling in all 3 cytotoxic CTCL entities, JAK2 fusions were found to be restricted to the CD8+ aggressive epidermotropic T-cell lymphoma. Professional illustration by Somersault18:24.
Aberrant JAK/STAT signaling is involved in the pathogenesis and progression of many T-cell lymphomas involving activating mutations, dysregulated cytokine signaling, or mutational changes in upstream regulators.2 Furthermore, an earlier effort using whole-exome and targeted Sanger sequencing has identified activating mutations in STAT3 and STAT5 in γδ T-cell lymphomas and in extranodal NK/T-cell lymphomas.3 Although not as common within other T-cell lymphomas as activating mutations, gene fusions involving JAK/STAT proteins, including JAK2 fusion proteins with constitutive kinase activity, have been implicated in multiple hematological malignancies.4
Adding on to the previous identification of JAK2 fusion proteins within a subset of CD8+ T-cell–derived cytotoxic CTCL,5 Lee et al investigate oncogenic gene fusions within 3 subgroups of cytotoxic CTCL. The authors establish the presence of fusion genes involving kinase proteins in all investigated CD8+ T-cell–derived cytotoxic CTCL samples and demonstrate that 8 of the 9 identified fusion products involve JAK2. For the 3 identified kinase fusion products that have not been previously characterized, Lee et al confirmed the constitutive kinase activity of the fusion proteins using the Ba/3F cell line system and show that their effect can be readily inhibited using US Food and Drug Administration–approved kinase inhibitors.
Intriguingly, the other cytotoxic CTCL entities studied by the authors, cytotoxic CTCL-NOS and primary cutaneous γδ T-cell lymphomas, had no detectable gene fusions. Nonetheless, transcriptional analysis of all 3 cytotoxic CTCL subtypes revealed activation of JAK/STAT3 signaling, and sequencing analysis revealed mutations in the JAK-STAT pathway alongside a host of other mutations associated with classical CTCL, such as mutations in chromatin remodeling and DNA damage response genes. These results add to growing evidence, that although individually recurring mutations in CTCL are rare, aberrant activation of the JAK/STAT pathway as well as interference with chromatin remodeling is a distinctive feature of this otherwise heterogeneous hematologic malignancy.6,7
This study adds to earlier work by the same group on primary cutaneous γδ T-cell lymphoma8 to advance our understanding of these rare cytotoxic CTCL entities and of the genetics of these aggressive cancers. The 3 cytotoxic CTCL entities are currently distinguished by virtue of histopathological presentation and lineage-defining markers, and as we gain better understanding of molecular etiology underlying each of these malignant diseases, the hope is that next-generation therapies can target the tumor vulnerabilities of individual subtypes of CTCL.
Although the authors elegantly show that the fusion proteins identified in this study confer interleukin 3 independence in the Ba/F3 cell line system, future studies should investigate how the recurrent JAK2 gene fusions affect primary CD8+ T cells from healthy or cytotoxic CTCL donors. In particular, it would be critical to determine whether JAK2 fusion proteins are sufficient to transform otherwise healthy CD8+ T cells and to what extent tumor cells remain dependent on JAK2 signaling. JAK2 inhibitors have shown tremendous promise in myeloproliferative disease,9 although durability of response remains a challenge. JAK2 fusions identified in primary cytotoxic CTCL of CD8+ origin in the study by Lee et al suggest that this malignancy may be highly responsive to JAK2 kinase inhibitors10 or to other drugs targeting the JAK/STAT signaling pathway.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal