

Key Points
VWF released during acute TBI was hyperadhesive in mediating extracellular vesicles to active endothelial cells and platelets.
Recombinant VWF A2 domain prevented TBI-induced coagulopathy by selectively blocking the exposed A1 domain of the hyperadhesive VWF.

Abstract
Traumatic brain injury-induced coagulopathy (TBI-IC) causes life-threatening secondary intracranial bleeding. Its pathogenesis differs mechanistically from that of coagulopathy arising from extracranial injuries and hemorrhagic shock, but it remains poorly understood. We report results of a study designed to test the hypothesis that von Willebrand factor (VWF) released during acute TBI is intrinsically hyperadhesive because its platelet-binding A1-domain is exposed and contributes to TBI-induced vascular leakage and consumptive coagulopathy. This hyperadhesive VWF can be selectively blocked by a VWF A2-domain protein to prevent TBI-IC and to improve neurological function with a minimal risk of bleeding. We demonstrated that A2 given through intraperitoneal injection or IV infusion reduced TBI-induced death by >50% and significantly improved the neurological function of C57BL/6J male mice subjected to severe lateral fluid percussion injury. A2 protected the endothelium from extracellular vesicle-induced injury, reducing TBI-induced platelet activation and microvesiculation, and preventing a TBI-induced hypercoagulable state. A2 achieved this therapeutic efficacy by specifically blocking the A1 domain exposed on the hyperadhesive VWF released during acute TBI. These results suggest that VWF plays a causal role in the development of TBI-IC and is a therapeutic target for this life-threatening complication of TBI.
Introduction
Uncontrolled hemorrhage accounts for 30% to 50% of trauma fatalities1,2 and is caused by vascular injury and secondary coagulopathy. Trauma-induced coagulopathy is caused by significant blood loss (hemorrhagic shock); hemodilution and hypothermia from fluid resuscitation; metabolic acidosis caused by tissue ischemia; dysfunctional platelets; and hyper-fibrinolysis.3-7 However, coagulopathy is equally common in patients with isolated traumatic brain injury (TBI),8-10 who do not suffer from significant blood loss, are restricted on fluid resuscitation, and rarely develop hypothermia or systemic metabolic acidosis. This suggests that TBI-induced coagulopathy (TBI-IC) differs mechanistically from coagulopathy arising after extracranial trauma and hemorrhagic shock. Although it is often presented as secondary intracranial hemorrhage, TBI-IC is a systemic condition because it can be detected in peripheral blood of patients, raising the critical question as how a localized cerebral injury is disseminated systemically to alter the hemostatic system, which is not directly affected as it is in patients with extracranial trauma and hemorrhagic shock.
We recently demonstrated that traumatically injured brains release brain-derived extracellular vesicles (BDEVs) that express procoagulant anionic phospholipids and tissue factor,11 inducing a systemic hypercoagulable state that is rapidly turned into consumptive coagulopathy.12 We further suggested that the adhesive ligand von Willebrand factor (VWF) could mediate extracellular vesicle (EV)-induced endothelial injury and activate platelets to propagate TBI-IC.13 VWF has been studied extensively for causing bleeding when it is deficient (eg, von Willebrand disease) and microvascular thrombosis when it is hyperadhesive (eg, thrombotic thrombocytopenic purpura [TTP]). However, despite the fact that VWF is an acute phase reactant associated with poor outcomes of trauma, including TBI,14-19 the causal role of VWF in TBI-IC remains poorly understood.
VWF initiates hemostasis by tethering platelets to the subendothelium exposed at sites of vascular injury. It is synthesized in megakaryocytes and endothelial cells (ECs) as single-chain propolypeptides20 that form multimers of various molecular masses.21-28 Newly synthesized VWF multimers are either constitutively released into circulation or stored in the Weibel-Palade bodies of ECs and the α-granules of megakaryocytes and platelets, where multimerization continues.29,30 VWF multimers secreted from the storage granules of ECs (eVWF) are not only large but intrinsically hyperadhesive, able to spontaneously bind platelets and other cells in solution.22,31,32 By contrast, VWF multimers found in the plasma of circulating blood (pVWF) bind platelets poorly unless they are immobilized onto the subendothelial matrix or exposed to high fluid shear stress.33-35 A key structural difference between the 2 forms of VWF is that the platelet-binding A1-domain is hidden in the globular structure of pVWF but exposed on the surface of eVWF multimers.36,37
The A1-domain can be hidden in pVWF by forming a high affinity complex with the adjacent A2-domain.38-40 Disrupting this A1-A2 complex activates pVWF by exposing the A1-domain. A recombinant A2 protein (A2) binds to the A1-domain of plasma VWF multimers and inhibits flow-dependent platelet adhesion.38 These observations led us to hypothesize that VWF multimers released during acute TBI are intrinsically activated because the A1-domain is exposed and can be selectively blocked by A2 to prevent TBI-IC and improve the outcome of TBI. Here, we present data from a study designed to test these hypotheses.
Materials and methods
Study design
This case-control study was designed to test the hypotheses that: (1) TBI-induced acute phase reaction results in the release of intrinsically hyperadhesive VWF that causes vascular injury and consumptive coagulopathy, and (2) the recombinant A2-domain blocks the VWF to prevent TBI-IC and improve the outcome of TBI.
We took several steps to control experimental biases. First, the mouse study had 2 controls: mice undergoing sham surgery and those subjected to TBI, but received the vehicle buffer. Second, mice were randomized to receive either A2 or the vehicle control, and the results were analyzed without prior knowledge of experimental conditions. Third, we collected longitudinal samples from mice so that the baseline samples could serve as self-controls. Fourth, individual experiments were performed in multiple laboratories on coded samples. Finally, multiple and complementary assays were used to evaluate key outcomes.
Expression and purification of VWF-A2 protein
A2 (G1481-R1668) from human VWF was expressed in Escherichia coli as a fusion protein with an N-terminal 6× His-tag.39,41 Transformed E coli were incubated with 0.5 mM isopropyl β-d-thiogalactopyranoside (Roche Diagnostics, Indianapolis, IN) overnight at 4°C to induce protein expression. The cells were then sonicated (Misonix, Farmingdale, NY) and centrifuged at 15 000g for 30 minutes. The supernatant was loaded onto a HisPur Cobalt Spin Column (Thermo Fisher Scientific, Carlsbad, CA) to purify A2, following the manufacturer's protocol. The purified A2 (90% to 95% purity; supplemental Figure 1 on the Blood Web site) was dialyzed overnight at 4°C against Tris-buffered saline containing 0.05% Tween-20 (TBST, 25 mM Tris-HCl, 150 mM NaCl, pH 7.5) and quantified by optical density at 280 nm on a NanoDrop spectrometer (Thermo Fisher Scientific). A commercial assay (GenScript, Piscataway, NJ) was used to ensure the A2 was endotoxin-free.
Mouse model of TBI
Adult male C57BL/6J mice (12-16 weeks and 22-25 g; The Jackson Laboratory, Bar Harbor, ME) were subjected to lateral fluid percussion injury (supplemental Methods).11,13 The mice received either a single dose of A2 (4 mg/kg) or an equal volume of the vehicle TBST through intraperitoneal (IP) injection 30 minutes after TBI. For validation, mice in a separate group received A2 (3.2 mg/kg) through IV infusion, which achieved the maximal plasma concentration faster than the IP delivery did (supplemental Figure 2). Blood samples were collected at baseline and at 3 and 6 hours after TBI for laboratory measurements. The mice were monitored for 3-day survival and evaluated using a modified neurological severity score (mNSS), which examines surviving mice for neurological deficits. The mNSS data should be considered alongside the accumulative survival of the mice because the data may have bias against death that occurred to mice with the most severe injuries. In a subset of the experiments, mice deficient in the metalloprotease ADAMTS-13,13 which cleaves VWF to reduce its adhesive activity,42 were also subjected to TBI to measure the effect of constitutively hyperadhesive VWF on TBI-induced neurological deficits and death. This mouse study was approved by the Institutional Animal Care and Use Committee of the Bloodworks Research Institute.
Flow cytometry
For platelet activation, blood samples collected in sodium citrate (0.32% final concentration) were centrifuged at 120g for 20 minutes at room temperature (RT) to obtain platelet-rich plasma (PRP), which was incubated with a phycoerythrin-conjugated monoclonal rat-anti-mouse CD62p (eBioscience, San Diego, CA) for 20 minutes at RT and analyzed using an LSR II flow cytometer (Beckon Dickinson, San Jose, CA).11,13 To measure VWF-bound EVs, PRP was centrifuged at 1500g for 20 minutes at RT to obtain platelet-poor plasma (PPP), which was incubated for 30 minutes at RT with a fluorescein isothiocyanate-conjugated polyclonal sheep-anti-mouse VWF antibody (Abcam, Cambridge, MA) and either allophycocyanin-conjugated monoclonal rat-anti-mouse CD41a (eBioscience) or Alexa Fluor 647-conjugated monoclonal rabbit-anti-mouse CD31 antibody (Abcam). EVs were detected first by their size (<1.0 µm) using Megamix standard microbeads (BioCytex, Marseille, France), and then by their expression of the cell markers. All buffers used for EV analyses by flow cytometry were filtered with a 0.1-μm filter (EDM Millipore, Billerica, MA) to reduce small particle contamination.
Vascular permeability
The effect of A2 on TBI-induced vascular permeability was measured in vivo by fluorescein isothiocyanate (FITC)-dextran leakage and Evans blue dye extravasation,11 and also by measurement of the water content in the brains (supplemental Methods). These in vivo measurements were validated in a transwell culture system (supplemental Methods).43,44 The impact of A2 on the TBI-induced disruption of the EC junction was visualized directly.43 For this experiment, human umbilical cord vein endothelial cells (HUVECs; Lonza, Allendale, NJ) were grown on glass slides coated with fibronectin (Sigma-Aldrich, St. Louis, MO; 50 µg/mL, 2 hours at 37°C) until confluent and then incubated for 3 hours at 37°C with PPP from sham mice and TBI mice receiving A2 or TBST. After being washed with phosphate-buffered saline, the cells were fixed in 4% paraformaldehyde for 20 minutes at RT, blocked with 3% bovine serum albumin, and permeabilized with 0.1% Triton X-100 for 30 minutes at 37°C. They were then incubated with the polyclonal rabbit-anti-human CD31 antibody (Abcam) overnight at 4°C, followed by incubation with Alexa Fluor 594-conjugated goat anti-rabbit immunoglobulin G (Invitrogen, Carlsbad, CA) for 1 hour at RT and counterstained with 4′6-diamidino-2-phenyl-indole (Abcam). Images were captured under an inverted fluorescence microscope (Olympus IX81, Waltham, MA).
Cerebral blood flow
Cortical cerebral blood flow was monitored at the baseline and at 3, 24, and 72 hours post-TBI using the noninvasive laser speckle imager with a 70-mW built-in laser and a 1388 × 1038-pixel CCD camera (PeriCam PSI System, Perimed, Sweden; supplemental Methods), as we recently described.45 The probe scanned a 2 × 2 cm area covering ∼80% to 90% of an adult mouse head.
VWF measurements
We used several complementary techniques to globally evaluate VWF release and adhesive-activity. First, plasma VWF antigen (VWF:Ag) was measured with a commercial enzyme-linked immunosorbent assay (ELISA) kit (Lifespan Biosciences, Seattle, WA). To measure VWF release from cultured ECs, confluent HUVECs were incubated with serum-free endothelial basal medium-2 (EBM-2; Lonza) for 2 hours and then with 3x104/μL of EVs (supplemental Methods) pre-incubated with A2 (40, 80, or 160 μg/mL) or TBST for 3 hours at 37°C in the presence of 300 000 platelets/μL. VWF:Ag in the conditioned media was then measured.13 A2 at the dosages used for the study did not interfere with VWF:Ag detection using ELISA (supplemental Figure 3A).
Second, VWF binding to collagen (VWF:CB) was used to define the ability of VWF to bind the subendothelial matrix13 and the activity of the VWF-cleaving ADAMTS-13.46 Type III collagen from human placenta (Sigma-Aldrich) was incubated overnight at 4°C in 96-well plates (200 ng/well) and blocked with 2.5% bovine serum albumin for 2 hours at 37°C. Diluted PPP from sham or TBI mice was incubated with collagen for 2 hours at 37°C. After being washed, horseradish peroxidase-conjugated rabbit polyclonal anti-human VWF antibody known to cross-react with mouse VWF (Dako, Carpinteria, CA) was incubated for 2 hours at RT and the bound antibody was recognized with 3,3′,5,5′-Tetramethylbenzidine at optical density (OD) 450 nm in a plate reader (Molecular Device, Sunnyvale, CA). The VWF:CB-to-VWF:Ag ratio was calculated to define the intrinsic VWF adhesive activity after VWF:Ag was adjusted.
Third, a commercial kit (Helena Laboratories, Beaumont, TX) was used to measure A2’s effects on VWF:Rco in human samples, but not mouse samples because mouse VWF is not activated by ristocetin.
Fourth, shear-induced platelet aggregation (SIPA) was measured in PRP diluted with homologous PPP to a final platelet count of 1×105/μL.47 The PRP was incubated with either A2 or TBST for 10 minutes at RT and then exposed to a shear stress of 100 dynes/cm2 (shear rate: 10 000−s, viscosity: ∼1 cp) for 1 minute at RT on a cone-and-plate viscometer (HAAKE RheoStress 1; Thermo Fisher Scientific).
Fifth, we used a reverse inhibitor assay to measure the selective binding of A2 to A1-exposed (activated) VWF. A2 (5 μg/mL) was captured to nickel-coated microtiter plates through His-tag for 60 minutes at RT. After washing, pVWF multimers purified from human cryoprecipitate were incubated with captured A2 for 60 minutes at 37°C in the presence and absence of 1 mg/mL of ristocetin (Helena Laboratories). The plates were washed extensively with TBST, and A2 was detected by an affinity-purified goat-anti-human A2 antibody (Bethyl Laboratories, Houston, TX) followed by a horseradish peroxidase-conjugated rabbit anti-goat immunoglobulin G (Thermo Fisher Scientific).
Sixth, the formation of an A2-VWF complex in TBI mice receiving A2 was determined using coimmunoprecipitation (supplemental Methods). The kinetic interaction of A2 with a recombinant A1 protein (D1261-T1468; U-Protein Express, Utrecht, The Netherlands) was measured by surface plasmon resonance (supplemental Methods). The patterns of VWF multimers before and after TBI were measured by nonreducing sodium dodecyl sulfate-1% agarose gel electrophoresis.48
Coagulation tests
The TBI-induced hypercoagulable and hyperfibrinolytic states were defined by 4 complementary tests. Plasma levels of fibrinogen and D-dimer were measured using commercial ELISA.11 For thrombin generation, EVs were purified from plasma of TBI mice by ultracentrifugation at 100 000g for 60 minutes at 4°C (twice) and resuspended in phosphate-buffered saline.11 An 80-μL EV suspension was mixed with 20 μL of the MP-reagent in a 96-well plate. After prewarming to 37°C in a Fluoroskan Ascent (Thermo Fisher Scientific), thrombin generation was monitored for 60 minutes after the addition of 20 μL FluCa (Thrombinoscope BV, Maastricht, The Netherlands) and quantified using the Thrombinoscope software. Freshly purified EVs from plasma of TBI and sham mice were also examined for their ability to induce phospholipid-free porcine plasma to clot at 37°C on a CoaScreener Coagulation Analyzer (American Labor Corp, Durham, NC), using a STA-Procoag-PPL Kit (Diagnostica Stago, Asnieres, France).11 To examine the effect of VWF on EV-induced coagulation, the coagulation test was performed either after VWF+EVs were depleted by the polyclonal VWF antibody (Dako) using a magnetic cell separation system (Thermo Fisher Scientific) or in the presence of 5 μg/mL of a polyclonal rabbit anti-human VWF antibody (Dako), which blocks VWF-dependent EVs binding to ECs13 and inhibited VWF-induced platelet aggregation under 100 dynes/cm2 of shear stress (supplemental Figure 4).
Tissue histology
Mice subjected to various experimental conditions were euthanized by CO2 under anesthesia 24 hours after TBI. The brains, lungs, and kidneys were dissected and fixed with 4% paraformaldehyde for 24 hours. Paraffin-embedded tissue blocks were sectioned and stained for hematoxylin and eosin or phosphotungstic acid hematoxylin (PTAH).11 Cerebral hemorrhage was examined and quantified (supplemental Methods).
Statistical analysis
The data were presented as means ± standard error of the mean for continuous variables and as percentages for categorical variables. The quantitative data were first analyzed with the Shapiro-Wilk normality test for data distribution. The Student t test was then used to compare 2 groups of quantitative variables, and 1-way analysis of variance (ANOVA) or 1-way ANOVA on ranks was used for Bonferroni post hoc comparisons. The cumulative survival was analyzed by Kaplan-Meier plot. A value of P < .05 was considered statistically significant. All analyses were performed using Sigma plot (v.11.2, SYSTAT Software, San Jose, CA).
Results
A2 improved outcomes in mice subjected to severe TBI
C57BL/6J mice exposed to TBI at 1.9 ± 0.2 atm had a mortality of ∼40%, with all deaths occurring within 12 hours of injury; survival was significantly improved when mice received 4 mg/kg of A2 through IP injection 30 minutes after the injury (Figure 1A, Tab. 1). The surviving mice developed neurological deficits defined by mNSS that were most severe in the first 6 hours and progressively improved thereafter (Figure 1B). The neurological deficits were significantly reduced in TBI mice receiving A2 (Figure 1B). The TBI mice developed subdural hematoma at the site of injury (Figure 1C-D) and focal intracerebral hemorrhage (Figure 1E), which were reduced in TBI mice receiving A2 (Figure 1F-G; supplemental Figure 5), as shown through tissue histology and quantified by the amount of hemoglobin in brain homogenates (Figure 1I). Consistent with this effect on bleeding arrest, A2 reduced TBI-induced vascular leakage, as defined by the transvascular leakage of FITC-dextran (Figure 1J-M) and validated using the Evans blue dye extravasation assay (supplemental Figure 6). A2 protected vascular integrity by reducing EC activation because plasma levels of EC-derived EVs were significantly reduced in TBI mice receiving A2, in comparison with the controls (Figure 1N). A2 also reduced TBI-induced pulmonary perivascular bleeding and tissue edema (supplemental Figure 7), consistent with clinical observations that more than 60% of TBI patients develop pulmonary dysfunction.49 By protecting the endothelial integrity, A2 significantly reduced TBI-induced cerebral edema measured by brain water content (Figure 2A). This protective effect remained detectable 3 days after TBI (supplemental Figure 8). Because of the cerebral edema and resultant high intracranial pressure, the cerebral blood flow decreased drastically within 3 hours postinjury and improved over time, but remained lower in TBI mice receiving TBST (Figure 2B-J). A2 improved cerebral blood flow measured at 3 and 24 hours postinjury (Figure 2B-J). Although A2 given through IV reached the maximal plasma level faster than that given through IP injection (supplemental Figure 2), A2 achieved a similar efficacy at 3.2 mg/kg IV and 4 mg/kg IP (Figure 1 vs supplemental Figure 9).
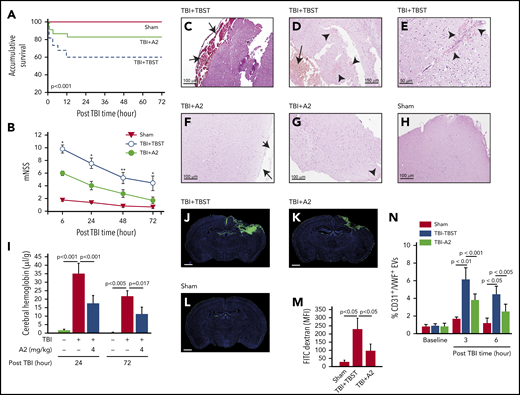
A2 improved survival and neurological function of TBI mice. C57BL/6J male mice were subjected to TBI and received 4 mg/kg of A2 through IP 30 minutes after injury. (A,B) Survival (n = 95) (A) and mNSS (n = 59) (B) were compared between mice receiving A2 and those receiving TBST (1-way ANOVA on rank, *P < .001 and **P < .005 vs untreated). (C-E) Hematoxylin and eosin brain histology on brain tissue collected 24 hours after TBI shows subdural hematoma (C,D, arrow) and multiple intracerebral hemorrhage (D,E, arrowhead) loci of mice receiving TBST. (F-H) The subdural (F, arrow) and intracerebral bleeding (G, arrow) were observed less frequently in TBI mice receiving A2 (H, the brain of a sham mouse as control). The images are representative of 36 mice examined. (I) Hemoglobin in brain homogenates at 24 and 72 hours after TBI (n = 9, 1-way ANOVA). (J-L) Coronal views of the brains from TBI mice receiving TBST (J) or A2 (K) and sham mice (L) infused with FITC-dextran (3 hours after TBI) and counterstained with DAPI (scale bars, 1 mm, representative of 9 brains reviewed). (M) FITC fluorescein intensity of these brains (1-way ANOVA). (N) Plasma levels of endothelial EVs (CD31+/VWF+) in TBI mice receiving A2 or TBST and sham mice (n = 21, 1-way ANOVA).
A2 improved survival and neurological function of TBI mice. C57BL/6J male mice were subjected to TBI and received 4 mg/kg of A2 through IP 30 minutes after injury. (A,B) Survival (n = 95) (A) and mNSS (n = 59) (B) were compared between mice receiving A2 and those receiving TBST (1-way ANOVA on rank, *P < .001 and **P < .005 vs untreated). (C-E) Hematoxylin and eosin brain histology on brain tissue collected 24 hours after TBI shows subdural hematoma (C,D, arrow) and multiple intracerebral hemorrhage (D,E, arrowhead) loci of mice receiving TBST. (F-H) The subdural (F, arrow) and intracerebral bleeding (G, arrow) were observed less frequently in TBI mice receiving A2 (H, the brain of a sham mouse as control). The images are representative of 36 mice examined. (I) Hemoglobin in brain homogenates at 24 and 72 hours after TBI (n = 9, 1-way ANOVA). (J-L) Coronal views of the brains from TBI mice receiving TBST (J) or A2 (K) and sham mice (L) infused with FITC-dextran (3 hours after TBI) and counterstained with DAPI (scale bars, 1 mm, representative of 9 brains reviewed). (M) FITC fluorescein intensity of these brains (1-way ANOVA). (N) Plasma levels of endothelial EVs (CD31+/VWF+) in TBI mice receiving A2 or TBST and sham mice (n = 21, 1-way ANOVA).
A2 improved survival of TBI mice
| Post-TBI time (h) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.75 | 1 | 3 | 6 | 12 | 24 | 48 | 72 | |
| Sham | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| TBI+TBST | 35 | 32 | 26 | 24 | 23 | 21 | 21 | 21 | 21 |
| TBI+A2 | 30 | 29 | 27 | 26 | 26 | 25 | 25 | 25 | 25 |
| Post-TBI time (h) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.75 | 1 | 3 | 6 | 12 | 24 | 48 | 72 | |
| Sham | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| TBI+TBST | 35 | 32 | 26 | 24 | 23 | 21 | 21 | 21 | 21 |
| TBI+A2 | 30 | 29 | 27 | 26 | 26 | 25 | 25 | 25 | 25 |
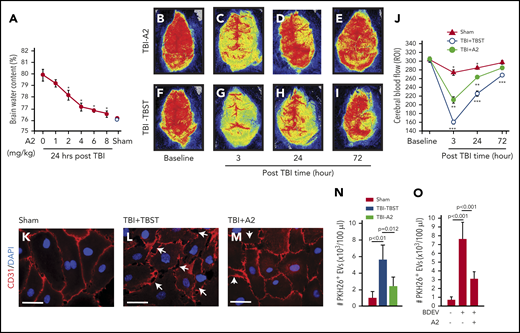
A2 prevented BDEV-induced vascular leakage. (A) Water content of the brains from TBI mice receiving A2 or TBST, showing a dose-dependent reduction of brain water content in A2-treated TBI mice (red dots), compared with that of untreated mice (white dot, n = 12, 1-way ANOVA, P < .01 vs untreated mice as control). (B-I) Laser speckle images from TBI mice treated with either A2 or TBST (n = 8). (J) Blood flow of the contusional and pericontusional cortex in the region of interest (ROI) from TBI mice receiving either A2 or TBST and sham mice (n = 9, 1-way ANOVA, *P < .01 among 3 groups at 3 and 24 hours, **P < .01 vs baseline in TBI-A2 mice, ***P < .001 vs baseline in TBI-TBST mice). (K-M) HUVECs were cultured to confluence and then treated with plasma from sham and TBI mice (K) receiving TBST (L) or A2 (M) for 3 hours at 37°C. The plasma samples were obtained 3 hours post-TBI. The cells were stained with a CD31 antibody (red) to mark the tight junction (scale bars, 100 μm; arrows: disrupted junctions, representative of 3 separate experiments). (N) PKH26-labeled EVs purified from equal volumes of plasma from sham mice and TBI mice treated with A2 or TBST were incubated with HUVECs for 3 hours at 37°C in the presence of 3 × 105/μL of human platelets. The labeled EVs were quantified in the bottom chamber using flow cytometry (n = 12, 1-way ANOVA). (O) The same experiments as described in panel N were used to measure the transendothelial migration of PKH26-labeled BDEVs (n = 12, 1-way ANOVA).
A2 prevented BDEV-induced vascular leakage. (A) Water content of the brains from TBI mice receiving A2 or TBST, showing a dose-dependent reduction of brain water content in A2-treated TBI mice (red dots), compared with that of untreated mice (white dot, n = 12, 1-way ANOVA, P < .01 vs untreated mice as control). (B-I) Laser speckle images from TBI mice treated with either A2 or TBST (n = 8). (J) Blood flow of the contusional and pericontusional cortex in the region of interest (ROI) from TBI mice receiving either A2 or TBST and sham mice (n = 9, 1-way ANOVA, *P < .01 among 3 groups at 3 and 24 hours, **P < .01 vs baseline in TBI-A2 mice, ***P < .001 vs baseline in TBI-TBST mice). (K-M) HUVECs were cultured to confluence and then treated with plasma from sham and TBI mice (K) receiving TBST (L) or A2 (M) for 3 hours at 37°C. The plasma samples were obtained 3 hours post-TBI. The cells were stained with a CD31 antibody (red) to mark the tight junction (scale bars, 100 μm; arrows: disrupted junctions, representative of 3 separate experiments). (N) PKH26-labeled EVs purified from equal volumes of plasma from sham mice and TBI mice treated with A2 or TBST were incubated with HUVECs for 3 hours at 37°C in the presence of 3 × 105/μL of human platelets. The labeled EVs were quantified in the bottom chamber using flow cytometry (n = 12, 1-way ANOVA). (O) The same experiments as described in panel N were used to measure the transendothelial migration of PKH26-labeled BDEVs (n = 12, 1-way ANOVA).
The in vivo effect of A2 on endothelial integrity was reproduced in vitro, which allowed us to examine the effect of A2 in a more controlled environment without the confounding influence of trauma. Plasma from TBI mice (Figure 2L), but not from sham mice (Figure 2K), disrupted the tight junction of confluent ECs in culture, and the plasma from TBI mice receiving A2 was less disruptive to ECs (Figure 2M). As a result, ECs treated with plasma from TBI mice receiving A2 were significantly less permeable to the larger EVs (0.2-1 μm11 ) collected from TBI mice (Figure 2N), and to BDEVs released from mouse brains subjected to freeze-thaw injury (Figure 2O).13 These data suggest that A2 protected mice against TBI-induced hemorrhage and vascular leakage.
A2 blocked VWF hyperadhesive activity
BDEVs, on which a significant amount of VWF was detected (supplemental Figure 10), activated HUVECs to release VWF and their effect was blocked by A2 in a dose-dependent manner (Figure 3A). Furthermore, plasma from TBI mice receiving TBST (TBI-PPP), but not that from sham mice (sham-PPP), induced a significant trans-endothelial diffusion of FITC-dextran after 3 hours of coincubation at 37°C (Figure 3B). The endothelial leakage was less severe when HUVECs were treated with plasma from TBI mice receiving A2 (TBI-PPP+A2). More importantly, when plasma from TBI mice was separated into EV and EV-free plasma fractions, this endothelial-disrupting activity was detected almost entirely in the EV fraction and was blocked by A2 or the VWF-blocking antibody13 (Figure 3B). In the control, neither culture medium nor purified VWF multimers induced the endothelial permeability (Figure 3B). These data suggest that: (1) VWF bound to EVs mediated EV-EC interaction to induce vascular leakage and (2) A2 blocked the vascular leakage by preventing EVs from interacting with ECs through VWF. Consistent with this notion, both VWF:Ag (Figure 3C) and VWF:CB (Figure 3D-E) were significantly increased in the blood samples from TBI mice, without significant change in VWF multimer distributions (supplemental Figure 3B). The VWF:CB-to-VWF:Ag ratio, which measures VWF adhesive activity after the high VWF:Ag was adjusted, was also increased (Figure 3F), suggesting that, unlike plasma VWF found in noninjured mice, VWF in the plasma of TBI mice was intrinsically hyperadhesive, independent of VWF multimer size. A single dose of A2 given 30 minutes after TBI reduced plasma VWF:Ag by 19.8 ± 2.6% but reduced VWF:CB by 56.6 ± 11.2% and reduced the VWF:CB-to-VWF:Ag ratio by 37.5 ± 7.6%, suggesting that A2 not only protected ECs from being activated by EVs to release hyperadhesive eVWF, but also blocked its adhesive activity directly. The effect of A2 on VWF:CB was reversed by an A2 antibody (Figure 3D), which blocked the interaction between A2 and ristocetin-activated VWF (supplemental Figure 11). Platelet activation (CD62p expression) during acute TBI13,44 was also reduced in mice receiving A2, and this A2 effect was again reversed by the A2 blocking antibody (Figure 3G). A2 blocked VWF-induced platelet activation to reduce platelet microvesiculation, as shown by low levels of platelet-derived and VWF-bound EVs (Figure 3H) and increasing platelet counts (supplemental Figure 12) in TBI mice receiving A2, without changing the hematocrit (supplemental Figure 13). These data suggest that TBI induced the systemic release of hyperadhesive eVWF, which mediated EV-induced endothelial injury and activated platelets that could bind the activated endothelium or lost to microvesiculation. This conclusion was supported by the finding that mice deficient in the metalloprotease ADAMTS-13, which cleaves eVWF to reduce its hyperadhesive activity, developed more severe neurological deficits and suffered more deaths than their wild-type littermates did (supplemental Figure 14).
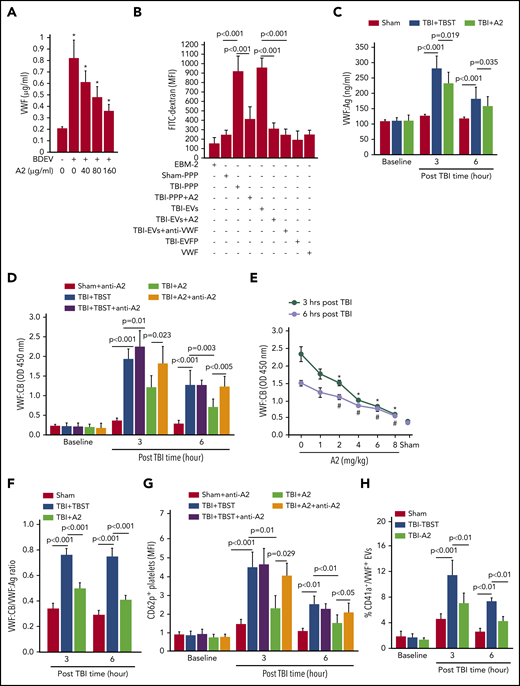
A2 blocked activated VWF in TBI mice. (A) HUVECs were incubated with BDEVs for 3 hours at 37°C with increasing doses of A2. The supernatants were collected and analyzed for VWF using ELISA (n = 25, 1-way ANOVA, *P < .01 vs untreated). (B) The culture medium (EBM-2), sham plasma (sham-PPP), plasma from TBI mice receiving TBST (TBI-PPP) or A2 (TBI-PPP+A2), purified EVs (TBI-EVs and TBI-EVs+A2), EV-free plasma (EVFP) from the TBI mice, and purified VWF were incubated with confluent HUVECs for 3 hours at 37°C. The HUVECs were washed and incubated with FITC-dextran (100 μg/mL) for 1 hour at 37°C to quantify FITC intensity in the bottom chamber (n = 56, 1-way ANOVA). (C,D) VWF:Ag (C) and VWF:CB (D) in plasma samples collected at 3 and 6 hours post-TBI (n = 36, 1-way ANOVA). (E) VWF:CB measured in mice receiving increasing doses of A2 (n = 12, 1-way ANOVA). (F) The VWF:CB-to-VWF:Ag ratio calculated for the 3 groups of mice (n = 21, 1-way ANOVA). (G) The expression of CD62p on platelets from sham mice and TBI mice receiving TBST or A2 (n = 21, 1-way ANOVA). (H) Plasma levels of platelet EVs with surface-bound VWF (CD41a+/VWF+, n = 21, 1-way ANOVA).
A2 blocked activated VWF in TBI mice. (A) HUVECs were incubated with BDEVs for 3 hours at 37°C with increasing doses of A2. The supernatants were collected and analyzed for VWF using ELISA (n = 25, 1-way ANOVA, *P < .01 vs untreated). (B) The culture medium (EBM-2), sham plasma (sham-PPP), plasma from TBI mice receiving TBST (TBI-PPP) or A2 (TBI-PPP+A2), purified EVs (TBI-EVs and TBI-EVs+A2), EV-free plasma (EVFP) from the TBI mice, and purified VWF were incubated with confluent HUVECs for 3 hours at 37°C. The HUVECs were washed and incubated with FITC-dextran (100 μg/mL) for 1 hour at 37°C to quantify FITC intensity in the bottom chamber (n = 56, 1-way ANOVA). (C,D) VWF:Ag (C) and VWF:CB (D) in plasma samples collected at 3 and 6 hours post-TBI (n = 36, 1-way ANOVA). (E) VWF:CB measured in mice receiving increasing doses of A2 (n = 12, 1-way ANOVA). (F) The VWF:CB-to-VWF:Ag ratio calculated for the 3 groups of mice (n = 21, 1-way ANOVA). (G) The expression of CD62p on platelets from sham mice and TBI mice receiving TBST or A2 (n = 21, 1-way ANOVA). (H) Plasma levels of platelet EVs with surface-bound VWF (CD41a+/VWF+, n = 21, 1-way ANOVA).
A2 prevented a TBI-induced hypercoagulable state
Consistent with previous reports,11,13,43,44 TBI mice developed the acute hypercoagulable and hyperfibrinolytic states defined by fibrinogen depletion (Figure 4A), elevated D-dimer (Figure 4B), thrombin generation (Figure 4C), shortened clotting time (Figure 4D), and elevated levels of anionic phospholipid-expressing EVs from the brain, ECs, and platelets (supplemental Figure 15). This systemic hypercoagulable state was also indicated by the extensive fibrin deposition in the renal vasculature of TBI mice (Figure 4E). The coagulation induced by EVs from TBI mice was significantly reduced when VWF+EVs were depleted or blocked by the polyclonal VWF-blocking antibody (Figure 4F), which blocked VWF-induced platelet aggregation under high fluid shear stress (supplemental Figure 4). As a result, A2 corrected the consumptive coagulopathy developed in TBI mice (Figure 4G) without significantly impairing the baseline hemostasis of noninjured mice (supplemental Figure 16).
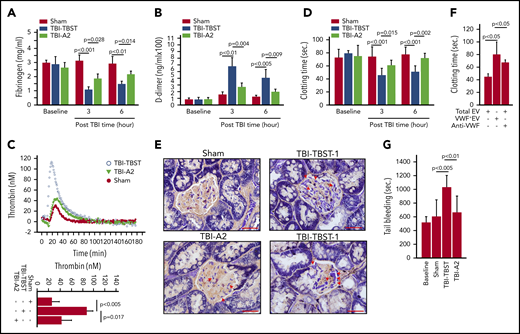
A2 reduced TBI-induced hypercoagulable and hyperfibrinolytic states. (A,B) Fibrinogen (A) and D-dimer (B) were measured in plasma from sham and TBI mice receiving either A2 or buffer control. (C) Thrombin generation was induced in plasma collected 3 hours after TBI (top: representative traces; bottom: summary of 9 experiments). (D) The time to coagulate anionic phospholipid-depleted plasma by EVs from sham mice and TBI mice receiving A2 or TBST (A-D, n = 9-15; 1-way ANOVA). (E) The PTAH stain of kidney tissue sections shows extensive fibrin deposition in the glomerular microvasculature (TBI-TBST-1, red arrows; scale bars, 100 μm) and in perivascular interstitial tissue (TBI-TBST-2, red arrows) from mice subjected to TBI, but not from sham mice (sham). The PTAH stain is less extensive in TBI mice receiving A2 (TBI-A2, red arrows). Images are representative of 12 mice/group. (F) EVs from TBI were tested for inducing clot formation before and after VWF+EVs were depleted; the number of remaining VWF− EVs was adjusted to the predepletion level, or a VWF-blocking antibody was added (n = 12, 1-way ANOVA on ranks). (G) The tail bleeding time of sham and TBI mice receiving A2 or TBST, measured 3 hours after injury (n = 21, 1-way ANOVA).
A2 reduced TBI-induced hypercoagulable and hyperfibrinolytic states. (A,B) Fibrinogen (A) and D-dimer (B) were measured in plasma from sham and TBI mice receiving either A2 or buffer control. (C) Thrombin generation was induced in plasma collected 3 hours after TBI (top: representative traces; bottom: summary of 9 experiments). (D) The time to coagulate anionic phospholipid-depleted plasma by EVs from sham mice and TBI mice receiving A2 or TBST (A-D, n = 9-15; 1-way ANOVA). (E) The PTAH stain of kidney tissue sections shows extensive fibrin deposition in the glomerular microvasculature (TBI-TBST-1, red arrows; scale bars, 100 μm) and in perivascular interstitial tissue (TBI-TBST-2, red arrows) from mice subjected to TBI, but not from sham mice (sham). The PTAH stain is less extensive in TBI mice receiving A2 (TBI-A2, red arrows). Images are representative of 12 mice/group. (F) EVs from TBI were tested for inducing clot formation before and after VWF+EVs were depleted; the number of remaining VWF− EVs was adjusted to the predepletion level, or a VWF-blocking antibody was added (n = 12, 1-way ANOVA on ranks). (G) The tail bleeding time of sham and TBI mice receiving A2 or TBST, measured 3 hours after injury (n = 21, 1-way ANOVA).
A2 selectively bound activated VWF
The data presented in Figures 1-4 suggest that A2 improved the outcome of TBI by blocking hyperadhesive VWF from EV-induced activation of ECs and from activating platelets. We then used several complementary assays to investigate whether A2 afforded this protection by selectively blocking the A1 domain exposed on activated VWF.
First, when infused into TBI mice, A2 formed a complex with circulating VWF because A2 was immunoprecipitated from TBI mice receiving A2 using a VWF antibody (Figure 5A) and VWF was immunoprecipitated by an antibody against His-tag fused into the A2 (Figure 5B). A2 also bound to VWF in plasma collected from TBI mice in vitro, but not in plasma from sham mice (Figure 5C, left), and this binding was blocked by the isolated A1 (Figure 5C, middle). As a control, A2 also bound VWF from a patient with congenital TTP, which is known to have activated VWF resulting from ADAMTS-13 deficiency,34,50 but it did not bind VWF from normal subjects (Figure 5C, right). Second, A2 bound the A1-domain in vitro with a high affinity (Figure 5D) and also bound pVWF treated with ristocetin (Figure 5E), which activates VWF to bind the platelet GP Ib-IX-V complex.51 Finally, A2 blocked SIPA (Figure 5F) and ristocetin cofactor activity (VWF:Rco; Figure 5G), in which pVWF was activated by either high shear stress or ristocetin to aggregate platelets.51,52 These results demonstrate that A2 blocked hyperadhesive VWF by binding to the exposed A1-domain.
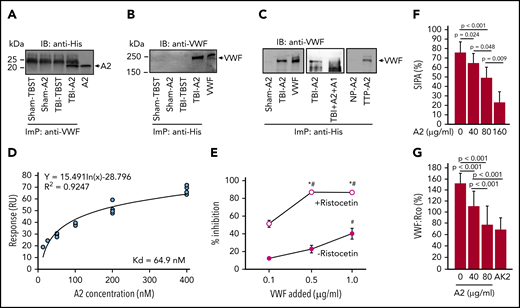
A2 blocked A1 exposed on activated VWF. (A,B) An anti-VWF antibody immunoprecipitated (ImP) A2, which was detected by an anti-His antibody in immunoblots (IB) (A) and an anti-His antibody immunoprecipitated VWF (B), which was detected by an anti-VWF antibody in TBI mice receiving A2. (C) A2 was incubated with plasma from TBI and sham mice and from TTP patients and healthy controls (NP, normal plasma) for 60 minutes at 37°C. Plasma VWF was then immunoprecipitated by an anti-His antibody and probed with a polyclonal VWF antibody through IB. As control, A2 was first treated with an equal molar concentration of the isolated A1-domain before incubation with plasma VWF from TBI mice (C, center; A-C, representatives from 6 individual experiments). (D) The binding kinetics of A2 to a recombinant human A1 measured by surface plasmon resonance after the nonspecific background binding was subtracted (n = 15). (E) Plasma VWF treated with 1 mg/mL of ristocetin blocked the anti-A2 antibody from binding to nickle-captured A2 in a dose-dependent manner; untreated plasma VWF did not do the same (n = 12, 1-way ANOVA, *P < .01 vs no ristocetin; #P < .001 vs the lowest concentration of VWF). (F,G) A2 blocked SIPA (F) and VWF:Rco (G) of human platelets in a dose-dependent manner. The GPIbα antibody AK2, which blocked VWF-induced platelet aggregation,51 was tested as a control for VWF:Rco (G, n = 9, 1-way ANOVA).
A2 blocked A1 exposed on activated VWF. (A,B) An anti-VWF antibody immunoprecipitated (ImP) A2, which was detected by an anti-His antibody in immunoblots (IB) (A) and an anti-His antibody immunoprecipitated VWF (B), which was detected by an anti-VWF antibody in TBI mice receiving A2. (C) A2 was incubated with plasma from TBI and sham mice and from TTP patients and healthy controls (NP, normal plasma) for 60 minutes at 37°C. Plasma VWF was then immunoprecipitated by an anti-His antibody and probed with a polyclonal VWF antibody through IB. As control, A2 was first treated with an equal molar concentration of the isolated A1-domain before incubation with plasma VWF from TBI mice (C, center; A-C, representatives from 6 individual experiments). (D) The binding kinetics of A2 to a recombinant human A1 measured by surface plasmon resonance after the nonspecific background binding was subtracted (n = 15). (E) Plasma VWF treated with 1 mg/mL of ristocetin blocked the anti-A2 antibody from binding to nickle-captured A2 in a dose-dependent manner; untreated plasma VWF did not do the same (n = 12, 1-way ANOVA, *P < .01 vs no ristocetin; #P < .001 vs the lowest concentration of VWF). (F,G) A2 blocked SIPA (F) and VWF:Rco (G) of human platelets in a dose-dependent manner. The GPIbα antibody AK2, which blocked VWF-induced platelet aggregation,51 was tested as a control for VWF:Rco (G, n = 9, 1-way ANOVA).
Discussion
We have presented experimental data showing that hyperadhesive VWF plays multiple roles in initiating and propagating TBI-IC and a recombinant A2 protein selectively blocks this VWF hyperadhesive activity with a minimal impact on basal hemostasis. A2 affords the protection primarily by reducing the VWF-mediated activation of endothelial cells induced by extracellular vesicles and by blocking induced platelet activation and microvesiculation to prevent the TBI-induced hypercoagulable state and resultant consumptive coagulopathy, thus improving the outcome of TBI in mouse models. The key activities associated with hyperadhesive VWF are schematically shown in Figure 6. We made several novel observations that have identified the underlying mechanism as how hyperadhesive VWF promotes TBI-IC.
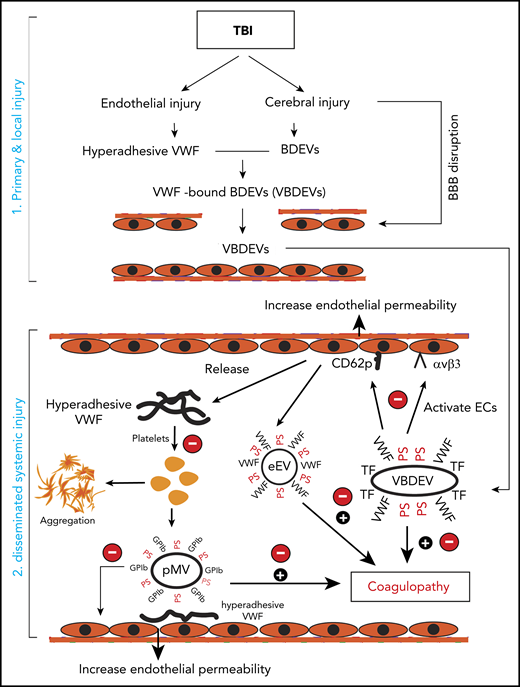
The role of VWF in TBI-IC and its blockage by A2. TBI-IC can be considered as a 2-step process of initiation and dissemination. The first step occurred at the site of injury, and trauma promotes endothelial cells to release hyperadhesive VWF and disrupts the blood–brain barrier, and injures cerebral cells to release BDEVs. The hyperadhesive VWF binds BDEVs, likely through integrins on BDEVs and the RGD sequence in the C domain of VWF, to become VWF-bound BDEVs (VBDEVs). In the second step, VBDEVs released into circulation bind and activate endothelial cells at vascular beds remote from the injury, using VWF as the adhesive ligand. The activated endothelial cells release procoagulant EVs (eEVs) and more hyperadhesive VWF multimers that can be either EC-bound or a soluble form. The EC-bound VWF tethers platelets, leukocytes, and EVs to the endothelium to propagate endothelial injury and permeability. The soluble VWF binds and activates platelets to express PS and release platelet EVs (pEVs). Together, activated endothelial cells and platelets, and PS+EVs induce consumptive coagulopathy (black circle with a plus sign), which can be inhibited by A2 at key action points by blocking hyperadhesive VWF (red circle with a minus sign).
The role of VWF in TBI-IC and its blockage by A2. TBI-IC can be considered as a 2-step process of initiation and dissemination. The first step occurred at the site of injury, and trauma promotes endothelial cells to release hyperadhesive VWF and disrupts the blood–brain barrier, and injures cerebral cells to release BDEVs. The hyperadhesive VWF binds BDEVs, likely through integrins on BDEVs and the RGD sequence in the C domain of VWF, to become VWF-bound BDEVs (VBDEVs). In the second step, VBDEVs released into circulation bind and activate endothelial cells at vascular beds remote from the injury, using VWF as the adhesive ligand. The activated endothelial cells release procoagulant EVs (eEVs) and more hyperadhesive VWF multimers that can be either EC-bound or a soluble form. The EC-bound VWF tethers platelets, leukocytes, and EVs to the endothelium to propagate endothelial injury and permeability. The soluble VWF binds and activates platelets to express PS and release platelet EVs (pEVs). Together, activated endothelial cells and platelets, and PS+EVs induce consumptive coagulopathy (black circle with a plus sign), which can be inhibited by A2 at key action points by blocking hyperadhesive VWF (red circle with a minus sign).
First, A2 at a single bolus dosing through IP or IV 30 minutes postinjury reduced intracranial bleeding and improved neurological function and survival in mice subjected to severe TBI (Figures 1 and 2). A2 afforded this protection by preventing hyperadhesive VWF from mediating EVs to bind and activate ECs, thus reducing vascular leakage, coagulopathy, and cerebral edema. A2 also prevented the VWF from binding and activating platelets to expose procoagulant anionic phospholipids and to produce VWF-bound and phosphatidylserine-expressing EVs that propagate systemic hypercoagulable state initiated by BDEVs and subsequent consumptive coagulopathy (Figures 3G and 4). In this regard, the actual number of VWF+EVs found in the peripheral blood of TBI mice was probably underestimated because many EVs had adhered to the endothelium and circulating platelets, and thus were not detected in plasma samples. In addition, the A1 exposed on VWF bound to EVs could also bind platelets through GP Ibα to form EV-platelet complexes. These PS+ membrane EVs can then fuse with platelet membrane to increase PS exposure on platelets.
Second, injured ECs released a large quantity of hyperadhesive VWF multimers during acute TBI (Figure 3). However, there is no significant increase in ADAMTS-13 release because there is no intracellular storage pool of ADAMTS-13 and its transcription was not increased during the acute phase reaction.53-55 As a result, the balance between VWF and ADAMTS-13 activities was disrupted, resulting in insufficient VWF cleavage and the accumulation of hyperadhesive VWF multimers on the endothelium and in the circulation, as indicated by the high VWF:CB (Figure 3). This notion is supported by the finding that exogenous ADAMTS-13 prevents TBI-IC in ADAMTS-13 efficient mice.13 Furthermore, oxidative stress associated with TBI56,57 could potentially oxidize VWF and ADAMTS-13 during acute TBI, making the former excessively adhesive by resisting proteolysis23,24,58 and the latter inactive to cleave VWF.59
Third, plasma VWF multimers in circulation bind poorly to platelets because the platelet-binding A1-domain forms a complex with the adjacent A2-domain.38,39 This cross-inhibitory A1-A2 complex can be formed when the 2 vicinal cysteine residues (C1669-C1670) in the A2-domain are reduced, but it disassociates when these cysteines are oxidized.40 We engineered the A2 to exclude these 2 vicinal cysteines so that it would not lose its A1 binding ability in the oxidative environment of acute TBI. We found that A2 selectively blocks A1-exposed VWF in TBI mice because: (1) A2 formed a complex with VWF from TBI mice and the isolated A1-domain protein prevented the A2 binding (Figure 5A-C). (2) A2 bound the isolated A1-domain with a Kd of 64.9 nM (Figure 5D). (3) Plasma VWF multimers bound A2 only after they were activated by ristocetin (Figure 5E). (4) A2 blocked SIPA and VWF:Rco, both of which require VWF to be activated (Figure 5F-G). (5) A2 prevented consumptive coagulopathy in TBI mice but did not induce severe bleeding in noninjured mice (Figure 4). The therapeutic potential of A2 in selectively targeting activated VWF is supported by the recent US Food and Drug Administration approval of the A1-blocking nanobody caplacizumab.50,60 Caplacizumab is indicated for acquired TTP, which is characterized by microvascular thrombosis and consumptive coagulopathy induced by uncleaved VWF multimers.61 We also found that A2 bound VWF in the plasma of a TTP patient (Figure 5C).
Fourth, VWF-mediated and EV-induced vascular injury is multifactorial because VWF multimers alone did not activate ECs (Figure 3B) but mediated the adhesion of EVs to ECs (Figure 3B). Both A2 and a polyclonal VWF antibody blocked endothelial permeability induced by extracellular vesicles, but we have yet to identify the molecules that capture VWF-bound EVs to the endothelium. CD62p,62 integrin αvβ3,63 and vimentin64 on endothelial cells are known to interact with VWF, but CD62p is unlikely to be the anchor because it is expressed primarily on activated endothelial cells. Integrin αvβ3 is constitutively expressed on resting endothelial cells, but how VWF-αvβ3 interaction is blocked by A2 remains to be investigated. VWF binding to vimentin through its A2 domain so that an isolated A2 can block the binding, but it is not known whether vimentin is constitutively expressed on resting endothelial cells.
Finally, A2 prevented the development of a TBI-induced hypercoagulable state (Figure 4). The finding that the procoagulant activity of EVs from TBI mice was significantly reduced when VWF-bound EVs were depleted (Figure 4F) highlights the importance of VWF in regulating EV-induced coagulation.65 However, the direct role of VWF in coagulation requires further investigation because A2 also changes the rate of fibrin formation.66 One limitation of this study is that it was conducted with mouse models and in vitro experiments; whether the results are applicable to the pathogenesis of TBI-IC in patients has yet to be investigated.
In summary, we have shown that a recombinant VWF-A2 protein prevented TBI-IC, improved neurological function, and increased survival in TBI mice by blocking the A1-domain exposed on the surface of activated VWF and thus preventing VWF-mediated EV-induced activation of ECs during the systemic propagation of TBI and VWF-induced platelet activation. These observations suggest that VWF may be a therapeutic target for TBI. A2 may also be used to detect intrinsically hyperadhesive (A1-exposed) VWF in samples from conditions, in which VWF is hyperadhesive and disease-causing.
All data associated with this study are available in the main text or the supplementary Materials. For original data, please contact jfdong@bloodworksnw.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke Grants NS087296 (J.-f.D.) and NS094280 (M.A.C.), National Heart, Lung, and Blood Institute Grants HL152200 and HL125957 (J.-f.D.), and National Institutes of General Medical Sciences Grant GM112806 (M.A.C.); Natural Science Foundation of China State Key Program Grants 81330029 and 81930031 (J.N.Z.); and Research Grants 81271361 (J.N.Z.) and 81672399 (M.L.).
Authorship
Contribution: X.X. conducted experiments, analyzed data, and wrote the manuscript; C.W. performed tissue histology and analyzed data; Y.W. performed experiments; K.H. performed experiments; T.H. performed experiments; A.Z. performed experiments; X.W. designed flow cytometry experiments, analyzed the data, and wrote the manuscript; C.H. performed mouse experiments and analyzed data; M.Y. performed experiments; W.Y. performed SPR experiments; F.-D.S. wrote the manuscript; M.S. wrote the manuscript; M.A.C. developed and provided A2; M.L. supervised tissue pathology analyses of TBI mice, analyzed data, and wrote the manuscript; J.Z. formulated hypothesis, designed the study, and wrote manuscript; and J.-f.D. formulated the hypothesis, designed the study, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jing-fei Dong, Bloodworks Research Institute, 1551 Eastlake Ave East, Seattle, WA 98102; e-mail: jfdong@bloodworksnw.org; Jianning Zhang, Department of Neurosurgery, Tianjin Medical University General Hospital, 154 Anshan Rd, Tianjin 300052, China; e-mail: jianningzhang@hotmail.com; Min Li, Institute of Pathology, Lanzhou University School of Basic Medical Sciences, No. 199 Donggang West Rd, Lanzhou 730000, China; e-mail: limin@lzu.edu.cn; or Miguel A. Cruz, Cardiovascular Research Section, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030; e-mail: miguelc@bcm.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal