

Key Points
NOTCH/PI3K-AKT signaling axis and cell cycle regulators are the key players that drive T-LBL.
Mutations in KMT2D, an epigenetic modifier, are associated with poor prognosis in T-LBL.

Abstract
T-cell lymphoblastic lymphoma (T-LBL) is a heterogeneous malignancy of lymphoblasts committed to T-cell lineage. The dismal outcomes (15%-30%) after T-LBL relapse warrant establishing risk-based treatment. To our knowledge, this study presents the first comprehensive, systematic, integrated, genome-wide analysis including relapsed cases that identifies molecular markers of prognostic relevance for T-LBL. NOTCH1 was identified as the putative driver for T-LBL. An activated NOTCH/PI3K-AKT signaling axis and alterations in cell cycle regulators constitute the core oncogenic program for T-LBL. Mutated KMT2D was identified as a prognostic marker. The cumulative incidence of relapse was 47% ± 17% in patients with KMT2D mutations, compared with 14% ± 3% in wild-type KMT2D. Structural analysis of the mutated domains of KMT2D revealed a plausible impact on structure and functional consequences. These findings provide new insights into the pathogenesis of T-LBL, including high translational potential. The ongoing LBL 2018 trial (www.clinicaltrials.gov #NCT04043494) allows for prospective validation and subsequent fine tuning of the stratification criteria for T-LBL risk groups to improve survival of pediatric patients.
Introduction
T-cell lymphoblastic lymphoma (T-LBL) represents the second most frequent subtype of non-Hodgkin lymphoma (NHL) in children and adolescents. T-LBL displays a large overlap with acute T-cell lymphoblastic leukemia (T-ALL) in morphological, clinical, and immune-phenotypic features.1,2 Hence, it is often debated whether T-LBL and T-ALL are different entities or represent manifestations of the same disease.3 T-LBL presumably develops from a multistep process wherein aberrant genetic and epigenetic alterations result in increased cell proliferation, survival, self-renewal, and arrest of differentiation of immature T-cell progenitors.4 Gain-of-function mutations in NOTCH1 have been extensively reported and characterized in T-ALL,5-15 and recent studies have reported a similar frequency of NOTCH1 mutations in T-LBL.1,16-18 Loss-of-function (LOF) mutations in PTEN result in constitutive activation of PI3K-AKT in T-ALL19 and T-LBL.18 Interestingly, cross talk between NOTCH and PI3K-AKT signaling is reported in T-ALL. Blocking activity of NOTCH1 results in upregulation of PTEN and inhibition of PI3K-AKT pathway.19,20 Most of the identified mutations for pediatric T-ALL can be assigned to NOTCH, PI3K/AKT/mTOR, and JAK/STAT signaling pathways and epigenetic modifiers.21-23 BCL11b is associated with improved overall survival in adult T-ALL, but not in pediatric T-ALL.24,25 So far, BCL11b has not been investigated in T-LBL. Furthermore, chromosome 9p deletions have been identified in both T-LBL and T-ALL, although they were not associated with prognostic relevance.1,26
Epigenetic modifications play an important role in establishing T-cell identity.27 KMT2D is frequently mutated in NHL and other cancers.28-30 LOF mutations in KMT2C and KMT2D promote tumorigenesis by dysregulation of enhancer- and superenhancer-related genes.31-33 The precise order of acquiring genetic and epigenetic lesions during the initiation of T-LBL is largely unknown, because T-LBL molecular data are sparse. Genome-wide information is lacking except for limited reports of whole-exome sequencing (WES),34,35 and there are few studies of gene expression profiles,1,2 methylation profiles, and copy number alterations (CNAs).1,36 Therefore, little is known about markers with prognostic relevance for T-LBL.16-18,34,37 The lack of valid prognostic markers and the poor chance of surviving relapse underline the medical need to learn more about the pathogenesis and the molecular characteristics in T-LBL. The key findings from the current analysis include identification of recurrent genetic alterations, additional potential hotspots in previously identified molecular markers, altered pathways, and potential prognostic markers for T-LBL.
Patients and methods
Patient samples
From 1995 through 2018, 668 pediatric and adolescent patients with T-LBL were registered in the NHL-BFM (non-Hodgkin lymphoma-Berlin-Frankfurt-Muenster) study center. Informed consent was obtained from parents or guardians. Patients were treated uniformly according to NHL-BFM treatment protocol for LBL, as described previously.38 Clinical data for patient characteristics, diagnostics, treatment, and outcome were obtained from the clinical trial databases. For the current study, lymphoma samples from 131 patients referred to as the “extended cohort” were included, of which 127 samples were evaluable (supplemental Table 1; supplemental Figure 1, available on the Blood Web site). In addition, corresponding germline samples from 29 patients were included.
WES and targeted sequencing
WES was performed on a “limited cohort” of 16 patients with T-LBL, including 10 paired samples (10 germline and 10 diagnostic/primary samples), and 6 triplet samples, including corresponding relapse samples (6 germline, 6 primary, and 6 relapse). “Primary samples” refers to those collected at the time of diagnosis before the start of therapy from patients who did or did not experience relapse; “relapse samples” refers to samples collected from patients after relapse but before the commencement of relapse therapy. One of the samples was excluded from the WES analysis due to tumor cell content in the corresponding germline sample. WES was performed (mean depth, ∼300×) with the Sureselect XT Human All Exon V6+COSMIC panel. The WES data were validated by high coverage, targeted sequencing of ∼5000× for germline and ∼6000× (with duplicates) for primary and relapse samples with a capture-based technique from Agilent Technologies. The targeted panel for T-LBL was designed with 76 to 80 genes, the probes were designed for complete coding regions of the genes, except in the case of 5 large genes, for which probes were included only for specific exons (supplemental Table 2). Further details are provided in the supplemental Materials and Methods.
SNP arrays and methylation arrays
Single nucleotide polymorphism (SNP) analyses were performed on arrays from Infinium Omni2.5Exome-8 v1.3 for the limited cohort and on arrays from Infinium Omni2.5Exome-8 v1.4 for the TP53-mutated samples (n = 6; variant allele frequency [VAF] cutoff, 1). Methylation analyses were performed with arrays from the Infinium MethylationEPIC BeadChip Kit39 on samples from the limited cohort. Data were analyzed by Illumina GenomeStudio 2.0.3. All samples passed the quality control quality control filters with genotyping rates ≥0.99 and no conspicuous control probes. Copy number variations [CNVs] were called by using cnvPartition 3.2.0, with a minimum probe count of 10. Output from GenomeStudio was manually inspected in comparison with the called CNVs. Details of the analysis of the methylation arrays and genotyping are provided in the supplemental Materials and methods.
Multiplex ligation-dependent probe amplification
LOH6q analysis was performed with SALSA multiplex ligation-dependent probe amplification (MLPA) P200 Reference-1 in combination with custom-made probes and the SALSA MLPA EK1 reagent kit-FAM (MRC Holland, Amsterdam, The Netherlands), as described by Rohde et al.40 The SALSA MLPA probemix P383T-A2 T-ALL (MRC-Holland, The Netherlands) was used to screen the PTEN deletions. Details are provided in the supplemental Materials and methods.
Absence of biallelic TCR-γ deletion analysis
Single-tube multiplex T-cell receptor γ chain gene (TRG) polymerase chain reaction was performed as described by Derrieux et al.41 Details of the analysis are provided in the supplemental Materials and methods.
Statistical analysis
For the analysis of targeted sequencing data, only mutations in primary samples from both nonrelapse and relapse cases, with a VAF cutoff of 10% were included. Synonymous mutations in NOTCH1 and FBXW7 were excluded. To maintain consistency with earlier published data,16,17 only data for mutations of NOTCH1 and FBXW7 in the reported hotspots were included. In addition, analysis was performed that included all the mutations of NOTCH1 and FBXW7. Statistical analysis was also performed as described previously17 for CNAs for genes included in MLPA probemix P383T-A2 T-ALL. Cumulative incidence functions for relapse (CIR) were constructed by the method of Kalbfleisch and Prentice and compared by using Gray’s test. Differences in distribution of individual parameters among patient subsets were analyzed by χ2 test or Fisher’s exact test. Multivariate analysis of the CIR was performed according to the Fine and Gray model.42
Simulation of structures and modeling of the mutations in KMT2D
The primary amino acid sequence of KMT2D was analyzed by the VSL2 (VSL2-M1) predictor tool to predict disordered regions.43 Phyre244 was used in intensive mode to model the PHD7, FYR, and SET domains and also structural changes after the mutated residues were inserted. The structures of the FYRN and FYRC motifs of KMT2D were modeled using the crystal structure of TBRG1,45 and zinc molecules (Zn2+) were modeled into the structure using PyMOL. The crystal structure of the SET domain of KMT2D (PDB: 4Z4P)46 was used to model the mutation identified in the SET domain.
Results
Genome-wide screening for recurrent genetic and epigenetic alterations in T-LBL
WES revealed 1168 somatic mutations, of which 70% were nonsynonymous and included 49 indels. The majority of the samples (18 of 20) demonstrated a low tumor mutational burden of 0.39/Mb (supplemental Table 3), except for 2 hypermutated relapse samples (TR14 and TR15) with a mutation rate of 7.93 and 2.89/Mb, respectively. Both cases featured mutations in MLH1 (Figure 1A; supplemental Figure 2) involved in mismatch repair.47 NOTCH1 (73%) and FBXW7 (33%) were the most recurrently mutated genes. NOTCH1 was identified as the putative driver gene (supplemental Table 4) by MutSigCV (P = .0028).48 About 27% of genes from the published WES data34 overlapped with those in the current data. Bioinformatic analysis49 revealed altered key pathways, namely the NOTCH, JAK-STAT, PI3K-AKT, Ras, and P53 signaling pathways, and also cell-cycle regulators, epigenetic modifiers, and transcription factors involved in T-cell development that presumably drive T-LBL. Besides somatic mutations in NOTCH1 and FBXW7, hypermethylation of NCOR2, a regulator of NOTCH signaling,50 and of FOXP1, a key transcriptional regulator for T cells,51 was observed in comparison with their corresponding germline samples (Figure 1B-C). Although no major difference in methylation was observed between primary and relapse samples or between primary samples from patients who relapsed (primary-relapse+) and those who did not (primary-relapse−), a small number of genes were hypomethylated in primary-relapse+ cases in comparison with primary-relapse− (supplemental Figure 3A-C). Interestingly, some of these genes, such as SOX2OT, PEPD, and WBSCR17, were reportedly involved in tumor progression, aggressive phenotype, and treatment resistance in several cancers.52-54 Frequent deletions in the chromosome 9p region, including the CDKN2A/2B locus encoding p16/INK4A and p14/ARF tumor suppressors involved in cell cycle regulation, were identified in primary-relapse− cases, and amplifications in chromosome 8 were seen in primary-relapse+ cases and in their corresponding relapse samples. Four of 16 cases revealed deletions in chromosome 17 and 1 of them also revealed amplification in chromosome 17 (Figure 1D). LOH for TP53 was identified in only 1 case, in both the primary sample and in the corresponding relapse sample (TP16 and TR16). In STAT5B, a duplication event in 1 of the primary samples (TP15) and LOH in the corresponding relapse sample (TR15) were confirmed. Results from 16 SNP arrays were validated alterations: deletions in CDKN2A/2B (n = 16), LEF1 (n = 3), MLLT3 (n = 3), MTAP (n = 2), SUZ12 (n = 2), and NF1 (n = 1) and gains in EZH2 (n = 2), CASP8AP2 (n = 1) and MYB (n = 1) were confirmed.
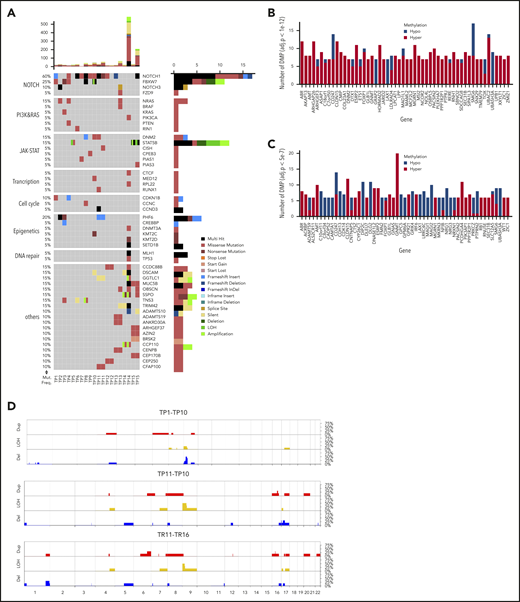
Genome-wide screening for recurrent genetic and epigenetic alterations in T-LBL. (A) Overview of the top 50 mutated genes identified by WES in 15 pediatric T-LBL samples including 5 corresponding relapse samples. The relative mutation frequencies (sample-wise) are indicated to the left and the pathways to the far left. Gene names and types of mutation are to the right. The number of mutations is displayed at the top in a bar plot. (B) The top 55 differentially methylated genes enriched by significant CpGs in primary samples in comparison with their corresponding germline samples (TG vs TP 1e−4). (C) The top 55 differentially methylated genes enriched by significant CpGs in relapse samples in comparison with their corresponding germline samples (TG vs TR 5e−7). Array used: Infinium MethylationEPIC BeadChip 850K. (D) The frequency of CNAs detected in 22 T-LBL samples including select primary and matched relapse samples. CNAs in the analyzed samples are displayed against the chromosomal numbers and position. The CNA profiles for primary relapse− samples (TP1-10), for primary relapse+ samples (TP11-16), and matched relapse samples (TR11-16) are presented separately. Array used: Infinium Omni2.5Exome-8 v1.3. Results validated independently by MLPA for specific regions in Chr 1p, 4q, 6q, 9p, 10q, 11p, and 17q. Red: amplifications; blue: deletions; yellow: loss of heterozygosity.
Genome-wide screening for recurrent genetic and epigenetic alterations in T-LBL. (A) Overview of the top 50 mutated genes identified by WES in 15 pediatric T-LBL samples including 5 corresponding relapse samples. The relative mutation frequencies (sample-wise) are indicated to the left and the pathways to the far left. Gene names and types of mutation are to the right. The number of mutations is displayed at the top in a bar plot. (B) The top 55 differentially methylated genes enriched by significant CpGs in primary samples in comparison with their corresponding germline samples (TG vs TP 1e−4). (C) The top 55 differentially methylated genes enriched by significant CpGs in relapse samples in comparison with their corresponding germline samples (TG vs TR 5e−7). Array used: Infinium MethylationEPIC BeadChip 850K. (D) The frequency of CNAs detected in 22 T-LBL samples including select primary and matched relapse samples. CNAs in the analyzed samples are displayed against the chromosomal numbers and position. The CNA profiles for primary relapse− samples (TP1-10), for primary relapse+ samples (TP11-16), and matched relapse samples (TR11-16) are presented separately. Array used: Infinium Omni2.5Exome-8 v1.3. Results validated independently by MLPA for specific regions in Chr 1p, 4q, 6q, 9p, 10q, 11p, and 17q. Red: amplifications; blue: deletions; yellow: loss of heterozygosity.
Mutational spectrum and recurrently altered pathways of T-LBL
Integrated analysis was performed for the extended cohort including targeted sequencing (Figure 2, VAF cutoff, >10%; supplemental Figure 2, VAF cutoff, >1%) of a panel of 76 to 80 genes, validation of CNAs, and analysis of additional markers. Constitutive activation of NOTCH signaling (68%) along with deletion of the CDKN2A (75%) and CDKN2B (61%) locus in the chromosome 9p region, as previously reported for T-LBL26 and T-ALL,4,21 emerged as main oncogenic lesions in T-LBL. NOTCH1 (63%) and FBXW7 (25%) mutations were identified at known hotspots7,16,17 and were analogous to hotspots in T-ALL.5,15,55 Additional somatic mutations were identified in other exons besides the reported hotspots both in NOTCH1 and FBXW7 (supplemental Figure 4A-C). Interestingly, another candidate, NOTCH3 from the NOTCH signaling pathway, was mutated in 8% of cases. Four of 10 NOTCH3muts cooccurred with NOTCH1mut mutations. BCL11b mutated in 9% of cases in our current analysis, similar to its mutation rate in pediatric T-ALL (supplemental Figure 5A).25
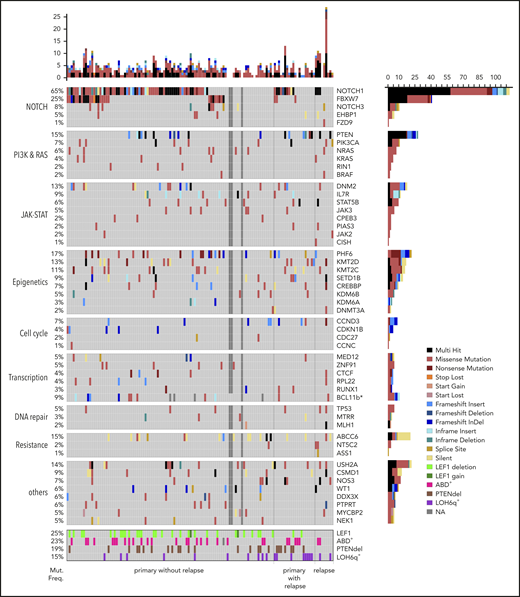
Mutational spectrum of T-LBL. An overview of recurrently mutated genes (VAF cutoff, >10%) and alterations in T-LBL. The samples are sorted into 3 sections: primary samples from relapse− patients, primary samples from relapse+ patients, and relapsed cases, as indicated at the bottom of the panel. The frequency of mutations (Mut. Freq.) and name of the pathways is indicated to the left. The names of the genes and the type of mutations are indicated on the right. The number of mutations identified by targeted sequencing is displayed at the top of the plot as bar plots. TRG rearrangements (ABD), PTEN deletions (PTENdel), and LOH6q alterations are displayed in a subpanel below. *BCL11b was not part of the targeted panel, but was analyzed by Sanger sequencing. Samples that failed in targeted sequencing are represented in dark gray boxes.
Mutational spectrum of T-LBL. An overview of recurrently mutated genes (VAF cutoff, >10%) and alterations in T-LBL. The samples are sorted into 3 sections: primary samples from relapse− patients, primary samples from relapse+ patients, and relapsed cases, as indicated at the bottom of the panel. The frequency of mutations (Mut. Freq.) and name of the pathways is indicated to the left. The names of the genes and the type of mutations are indicated on the right. The number of mutations identified by targeted sequencing is displayed at the top of the plot as bar plots. TRG rearrangements (ABD), PTEN deletions (PTENdel), and LOH6q alterations are displayed in a subpanel below. *BCL11b was not part of the targeted panel, but was analyzed by Sanger sequencing. Samples that failed in targeted sequencing are represented in dark gray boxes.
PI3K-AKT was identified as the second most affected pathway. Mutations identified in PTEN (15%), a negative regulator of PI3K-AKT pathway, and in PIK3CA (7%) were slightly higher than previously reported (6%)18 and could result in activation of the PI3K-AKT pathway.56,57 Interestingly, all PTEN mutations were nonsynonymous and mostly located at the hotspot reported in T-ALL and T-LBL,56-58 with a few in other exons (supplemental Figure 5B). In addition, deletions in PTEN (PTENdel) were identified in 19% of cases, and of those, 9 cases displayed both mutations and deletions. In PIK3CA, mutations were identified at hotspots in exons 10 and 21, as reported earlier,18 and in other exons (namely, exons 1, 8, and 18). The JAK-STAT pathway was identified as the third most affected pathway. STAT5B, a mutation often reported to be associated with an inferior response, mutated in 6% of the cases.59,60 Some of the mutations identified in STAT5B, JAK3, and IL7R in the current study reportedly have an activating effect on the JAK-STAT pathway or IL7R signaling in T-ALL and other cancers.59,61-66 Besides the activating mutation N642H,59,61 G698V was identified as a novel mutation in STAT5B (supplemental Figure 5C) next to Y699, which is crucial for DNA binding and transcriptional activity.67 JAK3 mutations occurred at known hotspots reported in T-cell neoplasms with an activating affect.21,65,68 Five of 6 mutations occurred at hotspot M511I. JAK2 mutated at a low frequency (2%). Interestingly, I1051T and M1064I were novel mutations. IL7R (8%) mutations occurred at the hotspots reported in pediatric T-ALL (supplemental Figure 5D).66 Reportedly, IL7 signaling induces phosphorylation of JAK1 and JAK3, which in turn phosphorylates STAT5B.69 Notably, cooccurrence of IL7R mutations with NOTCH1 accelerated the development of leukemia in mice.70 In the current analysis, STAT5Bmut frequently cooccurred with NOTCH1mut (supplemental Figure 6). RAS/MEK/ERK signaling is activated through mutations in NRAS, KRAS, and BRAF.21 Mutations were identified in these genes in 2% to 7% of cases. In NRAS and KRAS, the mutations occurred in the hotspots reported in T-ALL and T-LBL.18,23 Addition, novel mutations, V14L and Y64N, were identified in KRAS.
The epigenetic modifiers SETD1B, KMT2C, KMT2D, DNM2, and PHF6 mutated at a frequency of 9% to 17%. Unlike the KMT2Cmuts, the KMT2Dmuts were twice as frequent in primary samples from relapse+ cases. Furthermore, KMT2Dmut rarely co-occurred with KMT2Cmut. Genes related to transcription and DNA repair included in the panel mutated at a low frequency (2% to 7%). Of the 94% of mutations from the targeting sequencing that displayed VAF <75%, ∼6% of mutations displayed VAF >75%. Some of the interesting candidates that displayed VAF >75% were TP53, STAT5B, PHF6, DDX3X, FBXW7, CDKN1B, and MED12.
Furthermore, other markers, such as loss of heterozygosity in chromosome 6q (LOH6q; 15%) and incidence of absence of the biallelic TRG deletion (ABD; 23%), which had been reported earlier in T-LBL,16,17,37 were also analyzed for the extended cohort. Within the LOH6q+ cohort, 8 of 18 LOH6q+ occurred in primary-relapse+ cases and of those, 6 were NOTCH1wt. In primary-relapse− cases, 5 of 10 LOH6q+ were NOTCH1wt, whereas the remaining 5 were NOTCH1mut, displaying a higher number than previously reported.17
Prognostic relevance of the most significant candidate genes
The clinical characteristics of the patients in the current extended cohort were comparable to those of the cohort in the EURO-LB02 trial, with the exception that a higher proportion of patients in the current analysis were in poor general condition (Table 1).71 The results of the current study for the frequency and prognostic relevance of the published markers NOTCH1 and/or FBXW7 mutations (N/Fmut), PTENmut, and LOH6q were consistent, as reported earlier.1,7,16-18,37,72 PTENmut and LOH6q+ were associated with an unfavorable prognosis. Furthermore, ABD+ cases were not associated with any outcome in the current analysis, contradicting an earlier report.16 Intriguingly, PTENdel was not associated with an inferior outcome, in contrast to PTENmut (supplemental Figure 7A-F).
Clinical characteristics of patients from the extended cohort and the trial EURO-LB02
| Targeted sequence (n = 122) n | % | T-LBL cohort from EURO-LB 02 (n = 279) n | % | P (targeted cohort vs T-LBL cohort) | |
|---|---|---|---|---|---|
| Total patients | 122 | 279 | |||
| Sex | .5 | ||||
| Male | 90 | 73.8 | 214 | 76.7 | |
| Female | 32 | 26.2 | 65 | 23.3 | |
| Age, y | .6 | ||||
| <10 | 64 | 52.5 | 154 | 55.2 | |
| 10-14 | 41 | 33.6 | 95 | 34.1 | |
| ≥15 | 17 | 13.9 | 30 | 10.8 | |
| Stage | .4 | ||||
| II | 2 | 1.8 | 9 | 3.9 | |
| III | 84 | 77.1 | 164 | 70.7 | |
| IV | 23 | 21.1 | 59 | 25.4 | |
| Bone marrow involvement | .9 | ||||
| No | 84 | 80.8 | 227 | 81.4 | |
| Yes | 20 | 19.2 | 52 | 18.6 | |
| Mediastinal tumor | .2 | ||||
| No | 12 | 9.8 | 18 | 6.5 | |
| Yes | 110 | 90.2 | 261 | 93.5 | |
| CNS involvement | .1 | ||||
| No | 116 | 98.3 | 266 | 95.3 | |
| Yes | 2 | 1.7 | 13 | 4.7 | |
| General condition | .03 | ||||
| Good | 92 | 80.7 | 247 | 88.8 | |
| Poor (Karnofsky ≤20%) | 22 | 19.3 | 31 | 11.2 | |
| Status | |||||
| Relapse | 20 | 16.4 | 34 | 12.2 |
| Targeted sequence (n = 122) n | % | T-LBL cohort from EURO-LB 02 (n = 279) n | % | P (targeted cohort vs T-LBL cohort) | |
|---|---|---|---|---|---|
| Total patients | 122 | 279 | |||
| Sex | .5 | ||||
| Male | 90 | 73.8 | 214 | 76.7 | |
| Female | 32 | 26.2 | 65 | 23.3 | |
| Age, y | .6 | ||||
| <10 | 64 | 52.5 | 154 | 55.2 | |
| 10-14 | 41 | 33.6 | 95 | 34.1 | |
| ≥15 | 17 | 13.9 | 30 | 10.8 | |
| Stage | .4 | ||||
| II | 2 | 1.8 | 9 | 3.9 | |
| III | 84 | 77.1 | 164 | 70.7 | |
| IV | 23 | 21.1 | 59 | 25.4 | |
| Bone marrow involvement | .9 | ||||
| No | 84 | 80.8 | 227 | 81.4 | |
| Yes | 20 | 19.2 | 52 | 18.6 | |
| Mediastinal tumor | .2 | ||||
| No | 12 | 9.8 | 18 | 6.5 | |
| Yes | 110 | 90.2 | 261 | 93.5 | |
| CNS involvement | .1 | ||||
| No | 116 | 98.3 | 266 | 95.3 | |
| Yes | 2 | 1.7 | 13 | 4.7 | |
| General condition | .03 | ||||
| Good | 92 | 80.7 | 247 | 88.8 | |
| Poor (Karnofsky ≤20%) | 22 | 19.3 | 31 | 11.2 | |
| Status | |||||
| Relapse | 20 | 16.4 | 34 | 12.2 |
Characteristics of the extended cohort (targeted sequencing) and of the EURO-LB02 trial as the representative reference cohort. All cases for which the corresponding clinical data are available in the NHL-BFM databases are included.
The current analysis identified KMT2D as a putative novel marker associated with inferior outcomes. KMT2D mutations were predominantly localized in 2 hotspots in exons 31 and 48. In primary-relapse+ cases and relapse samples, most mutations were identified in exon 48, located before the SET domain (Figure 3). KMT2Dmut was associated with a significantly higher incidence of relapse compared with KMT2Dmut (47% ± 17% vs 14% ± 3%; P = .0064; Figure 4A). The combination of KMT2D and PTEN was identified in a subgroup with significantly higher incidence of relapse (Figure 4B). A KMT2D and/or a PTEN mutation was detected in 27 cases representing 22% of the analyzed cohort. Almost half of all relapses occurred in these 22% of cases. Furthermore, the mutational status of NOTCH1 and FBXW7 had significant impact on the prognostic relevance of KMT2Dmut and/or PTENmut (Figure 4C). Among cases with NOTCH1wt and FBXW7wt, the relapse incidence was 67% ± 15% in cases with KMT2Dmut and/or PTENmut status, compared with 11% ± 6% in cases with KMT2Dwt/PTENwt status (P < .0001). Interestingly, KMT2Dmut (n = 4) and PTENmut (n = 7) in this subgroup did not overlap, except in 1 additional case, suggesting that KMT2D and PTEN are independent parameters of prognostic relevance (supplemental Table 5). Furthermore, multivariate analysis suggested that, in patients with NOTCH1wt and FBXW7wt, both PTENmut and KMT2Dmut are independent parameters that are associated with a higher incidence of relapse (Table 2).
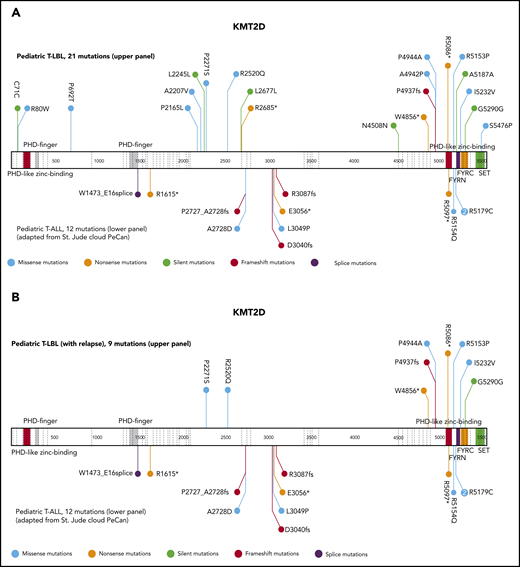
Schematic display of localization and frequencies of mutations in KMT2D and prognostic value of the most relevant candidate genes. Schematic display of localization and frequencies of mutations identified for KMT2D in total T-LBL samples (VAF cutoff, >1%) (A), KMT2D in relapse+ samples (B). The data for mutations (A-B) in pediatric patients with T-ALL were imported from the St Jude Pediatric Cancer Genomic data portal and are displayed in the lower part of the schematic structure, and the mutations identified in the current T-LBL project are displayed in the upper part.
Schematic display of localization and frequencies of mutations in KMT2D and prognostic value of the most relevant candidate genes. Schematic display of localization and frequencies of mutations identified for KMT2D in total T-LBL samples (VAF cutoff, >1%) (A), KMT2D in relapse+ samples (B). The data for mutations (A-B) in pediatric patients with T-ALL were imported from the St Jude Pediatric Cancer Genomic data portal and are displayed in the lower part of the schematic structure, and the mutations identified in the current T-LBL project are displayed in the upper part.
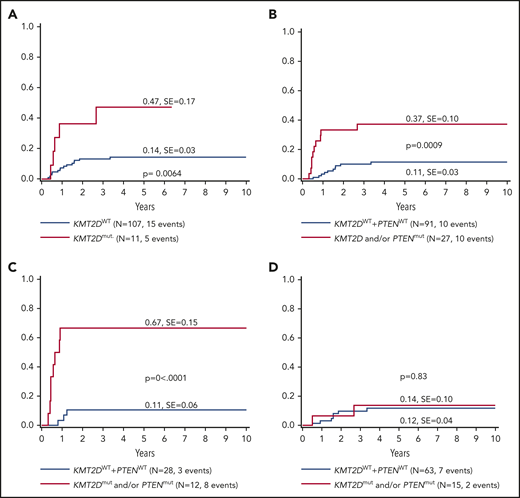
Five-year cumulative incidence of relapse.KMT2D mutational status (A), KMT2D and PTEN mutational status (B), KMT2D and PTEN mutational status on an N/Fwt background (C) and KMT2D and PTEN mutational status on an N/Fmut background (D). A VAF cutoff of >10% and only nonsynonymous mutations were included for analysis. In case of N/Fmut, only mutations from the hotspots (exons 26, 27, and 34 for NOTCH1 and exons 9, 10, and 12 for FBXW7) were included.
Five-year cumulative incidence of relapse.KMT2D mutational status (A), KMT2D and PTEN mutational status (B), KMT2D and PTEN mutational status on an N/Fwt background (C) and KMT2D and PTEN mutational status on an N/Fmut background (D). A VAF cutoff of >10% and only nonsynonymous mutations were included for analysis. In case of N/Fmut, only mutations from the hotspots (exons 26, 27, and 34 for NOTCH1 and exons 9, 10, and 12 for FBXW7) were included.
Multivariate analysis of the cumulative incidence of relapse using a Fine and Gray model
| Parameters | Hazard ratio | 95% CI | P |
|---|---|---|---|
| FBXW7/NOTCH1 | 1.2 | 0.25-5.5 | .84 |
| PTEN | 1.8 | 0.20-15.7 | .61 |
| KMT2D | 1.4 | 0.17-11.7 | .76 |
| LOH6q | 3.2 | 1.0-9.9 | .04 |
| Interaction: NOTCH1 or FBXW7 with PTEN | 0.4 | 0.04-5.7 | .53 |
| Interaction: NOTCH1 or FBXW7 with KMT2D | 0.86 | 0.06-11.8 | .90 |
| Interaction: NOTCH1 or FBXW7 with PTEN or KMT2D | 13.5 | 1.1-170.5 | .04 |
| Parameters | Hazard ratio | 95% CI | P |
|---|---|---|---|
| FBXW7/NOTCH1 | 1.2 | 0.25-5.5 | .84 |
| PTEN | 1.8 | 0.20-15.7 | .61 |
| KMT2D | 1.4 | 0.17-11.7 | .76 |
| LOH6q | 3.2 | 1.0-9.9 | .04 |
| Interaction: NOTCH1 or FBXW7 with PTEN | 0.4 | 0.04-5.7 | .53 |
| Interaction: NOTCH1 or FBXW7 with KMT2D | 0.86 | 0.06-11.8 | .90 |
| Interaction: NOTCH1 or FBXW7 with PTEN or KMT2D | 13.5 | 1.1-170.5 | .04 |
In a multivariate analysis with the factors NOTCH1mut or FBXW7mut, KMT2D, PTEN, LOH6q mutations and interaction terms between PTEN, KMT2D, PTEN or KMT2D and NOTCH1/FBXW7 (both WT); only LOH6q and the interaction term with PTEN or KMT2D were significant.
Importantly, in cases with NOTCH1mut and/or FBXW7mut, when either mutations in the hotspots (Figure 4D) or total mutations were included, the mutational status of KMT2D and PTEN was of no prognostic relevance. KMT2C, another member of Set1/COMPASS-like complexes,29,73 was not associated with prognostic relevance (supplemental Figure 8A). Mutations in other candidates of interest, such as NOTCH3, PHF6, BCL11b, and PIK3CA, as well as deletions in CDKN2A/CDKN2B, were not of prognostic relevance. Mutations or deletions in TP53 were observed in too few cases to draw a conclusion on association with outcome. Deletions in LEF1 (LEF1del) showed a trend toward favorable outcome (supplemental Figure 8B-F).
Impact of the identified mutations on the structure and function of KMT2D
KMT2D is a rather large protein (∼600 kDa), closely related to KMT2A74 and KMT2C.75 KMT2D features 7 PHD fingers, FY-rich domains (FYRN and FYRC), and a highly conserved C-terminal catalytic su(var)3-9, enhancer of zeste, trithorax (SET) domain.46 The PHD2 in KMT2D has an E3 ligase function similar to the PHD2 from MLL1,74 PHD4-PHD6 was reported to be associated with histone H4,76 and PHD6 was shown to have high selectivity for H4K16ac.77 The PHD7 and FYR region in KMT2D have not been found to be associated with any function. Interestingly, most mutations in KMT2D from the relapse− cases were located in the disordered regions, and, in relapse+ cases, most were located in the ordered regions (Figure 5A). The mutations R5153P, I5232V, and S5476P, identified in T-LBL, and R5154Q, found in pediatric T-ALL, located in the ordered regions, were simulated and modeled to determine their impact on the potential structure and molecular dynamics and their plausible functional impact. An additional mutation, R5179C, has been reported recently in pediatric T-ALL.78 The modeled structure of PHD7 revealed an atypical Cys4-His-Cys3-His motif. R5153 and R5154 were predicted to be located in a loop structure farther away from the zinc fingers. The R5153P or R5154Q mutations would disrupt interactions with regions of tandem Glu and Asp residues in FYRC domain (supplemental Figure 9A-B; Figure 5B). The FYRN and FYRC motifs are poorly characterized phenylalanine/tyrosine-rich regions common to all KMT2 proteins. The only available crystal structure of FYRN and FYRC motifs is that of the transforming growth factor-β regulator 1 (TBRG1) protein.45 The FYRN and FYRC motifs are adjacent to each other in KMT2D and in KMT2C, unlike their location in other members of the KMT2 family.46 To analyze the effect of these newly identified mutations on the FYR motif in KMT2D, the crystal structure of the TBRG1 motif was used (PDB: 2WZO). Phyre2 predicted a nearly identical structure via homology modeling, and Ile5232 was located in the core of the hydrophobic pocket. Mutation I5232V led to loss of a CH3 group that normally occupies part of the space in the hydrophobic pocket (Figure 5C-D) and results in destabilization of the domain and causes folding defects.
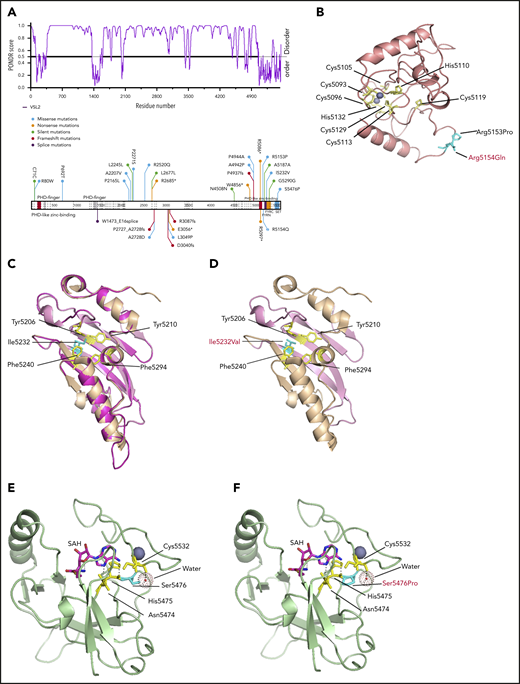
Modeling of KMT2D domain structures and comparison with the mutated domain structures. (A) Schematic overview of the predictive value of intrinsically disordered regions in KMT2D. The KMT2D gene is aligned with the PONDR (Predictor of Naturally Disordered Regions) chart to visualize mutations that lie in the predicted disordered region. (B) The domain structure of PHD7 of KMT2Dwt was generated by Phyre2. The schematic display of Arg residues (cyan) are represented in the stick structures near PHD7 of KMT2Dmut at the respective positions R5153P and R5154Q (cyan), the latter having been identified in T-ALL (red). (C) The crystal structure of the FYR domain, superimposition of Phrye2 depiction of the KMT2D (magenta), FYRN (pink), and FYRC (orange) motifs with the crystal structure of the TBRG1 motif (PDB: 2WZO).45 The residues forming the core hydrophobic pocket (yellow) are shown with stick structures and display of the Ileu5232 (cyan) in the FYRN motif. (D) The mutation Ileu5232Val (cyan) leads to loss of CH3, which could result in destabilizing of the domain. (E) Crystal structure of KMT2D SET domain (PDB: 4Z4P).46 The mutation Ser5476 (cyan) is located near the SAM binding cleft (SAH is modeled) and makes direct contact with a water molecule that supports Cys5532 (yellow, stick structure) located in the post-SET loop involved in zinc binding (purple). (F) The Ser5476Pro mutation impairs water binding, which could destabilize the Cys-zinc interaction required for proper folding.
Modeling of KMT2D domain structures and comparison with the mutated domain structures. (A) Schematic overview of the predictive value of intrinsically disordered regions in KMT2D. The KMT2D gene is aligned with the PONDR (Predictor of Naturally Disordered Regions) chart to visualize mutations that lie in the predicted disordered region. (B) The domain structure of PHD7 of KMT2Dwt was generated by Phyre2. The schematic display of Arg residues (cyan) are represented in the stick structures near PHD7 of KMT2Dmut at the respective positions R5153P and R5154Q (cyan), the latter having been identified in T-ALL (red). (C) The crystal structure of the FYR domain, superimposition of Phrye2 depiction of the KMT2D (magenta), FYRN (pink), and FYRC (orange) motifs with the crystal structure of the TBRG1 motif (PDB: 2WZO).45 The residues forming the core hydrophobic pocket (yellow) are shown with stick structures and display of the Ileu5232 (cyan) in the FYRN motif. (D) The mutation Ileu5232Val (cyan) leads to loss of CH3, which could result in destabilizing of the domain. (E) Crystal structure of KMT2D SET domain (PDB: 4Z4P).46 The mutation Ser5476 (cyan) is located near the SAM binding cleft (SAH is modeled) and makes direct contact with a water molecule that supports Cys5532 (yellow, stick structure) located in the post-SET loop involved in zinc binding (purple). (F) The Ser5476Pro mutation impairs water binding, which could destabilize the Cys-zinc interaction required for proper folding.
The crystal structure of the SET domain of KMT2D46 was used to model the S5476P mutation (PDB: 4Z4P). S5476 is located near the S-adenosylmethionine (SAM) binding cleft (S-adenosyl homocysteine [SAH] is modeled) and makes direct contact with a water molecule that supports Cys5532 (located in a post-SET loop), which is involved in zinc binding (Figure 5E). The S5476P mutation most likely has multiple effects on protein stability. First, the mutation could affect interaction with water binding, destabilizing the Cys-zinc interaction required for folding. In addition, substituting a Pro for a Ser residue may make the loop rigid and reduce the interaction of SAM to the backbones of Asn5474 and His5475 (Figure 5F). In both scenarios, the mutations would affect the function and structure of the protein.
Discussion
The regulation of Notch-mediated transcription depends on the fine balance of the dynamics between the 2 regulatory complexes, namely the NCoR repressive complex and the KMT2D activating complex, which compete to bind the SPOC (spen paralogue and ortholog C-terminal) domain of SHARP (SMRT/HDAC1-associated repressor protein).50,79 The findings from the current analysis suggest that this balance is plausibly disrupted in T-LBL (supplemental Figure 10A), predominantly by NOTCH1 and FBXW7 oncogenic alterations that could result in the prolonged activation of NOTCH signaling in >60% of cases in the current analysis and also as reported earlier in T-LBL1,7,16-18 and T-ALL.7,9-11,80 Furthermore, somatic mutations and epigenetic modifications, respectively, were identified in the key players KMT2D and NCoR2 of the Notch regulatory network. Hypermethylation of NCoR2 in primary samples plausibly results in reduced expression of NCoR2, affecting the stability of the NCoR repressive complex and providing an additional mechanism that aids in activation of the NOTCH target genes. In contrast, LOF mutations in KMT2D could alter the stability of the activator complex (supplemental Figure 10B), thus affecting the expression of tumor-suppressor genes and lineage-specific genes besides the NOTCH target genes. Furthermore, 1 of the downstream targets of hyperactive PI3K-AKT pathway is KMT2D. KMT2D is phosphorylated by AKT, resulting in attenuation of its methyltransferase activity,81-83 presumably having an impact similar to that of LOF mutations in KMT2D.
Similar to T-ALL, the mutated genes identified in the current analysis could be assigned to the major oncogenic pathways: NOTCH signaling and the PI3K-AKT, Ras, and JAK-STAT pathways, along with cell cycle regulators and epigenetic modifiers. NOTCH signaling (68%) and deletion of the tumor suppressors p16IKN4A and p14ARF encoded in the CDKN2A locus were the most frequent alterations (identified in >70% cases), comparable to previous reports on T-LBL and T-ALL,26,84 suggesting that deregulation of the cell cycle and prolonged activation of NOTCH signaling are predominantly the key oncogenic events leading to T-cell transformation in both T-ALL and T-LBL. Cross talk between NOTCH signaling and other pathways such as PI3K-AKT, JAK-STAT, and the epigenetic modifier KMT2D was suggested by the results of our analysis. In addition, LEF1del (23%) was identified. LEF1 inactivation presumably coordinates with NOTCH1 activation to activate MYC expression. Interestingly, LEF1del showed a trend toward favorable prognosis, as was reported for T-ALL.85
N/Fmut was associated with a good prognosis for T-LBL, as reported earlier.1,7,16-18 In the ongoing international clinical trial LBL 2018 (www.clinicaltrials.gov #NCT04043494) with 21 participating countries, the mutational status of N/F serves as a stratification criterion. One of the most important translational outcomes of the current study was the observation of the impact of KMT2D and PTEN mutations. The incidence of relapse for patients with N/Fwt status was 28% ± 7%. Among the N/Fwt cohort, 8 of 12 relapses occurred in the patients with PTEN and/or KMT2D mutations. Multivariate analysis suggested that, in patients with wild-type NOTCH1 and FBXW7, the independent prognostic parameters PTEN or KMT2D are associated with a higher incidence of relapse. Interestingly, in the N/Fmut cohort, the mutational status of KMT2D and PTEN did not have any prognostic relevance.
Furthermore, investigations were conducted into the effect of mutations on the individual domains of KMT2D. FYRN and FYRC motifs are thought to play a role in chromatin binding45 ; identified mutations in this region may affect the binding of KMT2D to its target genes. PHD domains in KMT2C/KMT2D have been reported to play an important role in homodimerization, recruitment of KMT2C/KMT2D complexes to their target genes, and methylation.75 KMT2D protein stability is tightly regulated by its H3K4 methyltransferase activity; mutations in the SET domain results in enzymatically dead KMT2D.86 Hence, the mutations identified in cases of T-LBL relapse may cause truncation leading to loss of the SET domain, resulting in enzymatically dead protein affecting expression of some of the target genes. Very little is known about KMT2D mutations in T-ALL, no association with prognostic relevance has been reported so far.21,22 In a mouse model, ablation of KMT2D in B cells promoted lymphoma, and brain-specific knockout of KMT2D induced medulloblastoma.32,87 Interestingly, its paralogue, KMT2C,88 was not associated with prognostic relevance in the current analysis, suggesting a diverse and nonredundant role of KMT2D in T-LBL. These results certainly require validation in larger and prospective cohorts, which will be achieved by the molecular analyses of patients enrolled in the LBL 2018 trial.
In contrast to T-ALL, 2 aspects are highly relevant to the interpretation and consequences of our current results. First, the treatment regimen for T-LBL, based on the EURO-LB02 trial, was internationally uniform.71 Second, no stratification criteria were applied for intensification of treatment of the analyzed cohort. These factors resulted in uniform treatment of the patients with T-LBL in the current study and sets a strong basis for translational research aimed at identifying molecular markers with prognostic relevance.
Similar to observations in T-ALL,21 only NOTCH1 and CDKN2A/B were mutated or deleted in >60% of the cases, whereas most of the genes analyzed were mutated in <10% of cases, suggesting the complexity and heterogeneity of events underlying the malignant transformation of T-LBL. Some of the limitations of this study were that the methylation arrays were performed only for the limited cohort and that 1 (TP79) of 122 analyzed cases did not feature any somatic mutations in the genes included in the targeted panel or other markers included in the study, suggesting the need to perform genome-wide analysis on larger cohorts to identify additional candidate genes and/or epigenetic alterations relevant to T-LBL.
The WES and targeted sequencing data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB36436 (https://www.ebi.ac.uk/ena/data/view/PRJEB36436). The methylation array has been deposited in the ArrayExpress database at EMBL-EBI under accession number E-MTAB-8762 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-8762). SNP array data have been deposited in the ArrayExpress database at EMBL-EBI under accession number E-MTAB-8763 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-8763).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the participating centers of the NHL-BFM Group that provided samples and clinical information; Harshana Sabharwal for technical help in preparing the samples for targeted sequencing; the members of the NHL-BFM study center in Muenster for feedback and constant help in organizing data and patient samples; and the family of Jonas Goeckener for their contribution, which was helpful in the preliminary experiments.
This work was supported by Deutsche Krebshilfe (DKH) grant 111347 (B.B., M.D.).
Authorship
Contribution: B.B., T.K., M.D., and M.Z. conceptualized and designed the project. B.B., S.M., H.H., I.O., W.K., and W.W. organized the patient and study material; T.K., S.S., J.S., C.R., M.Z., C.B., G.R., S.M., H.H., C.W., I.O., W.K., M.D., and B.B. collected and assembled the data; T.K., B.B., S.S., M.D., M.Z., J.S., C.R., M.Z., C.B., A.B.N., H.G., P.H., S.H., L.W., W.W., M.W., and M.D. analyzed and interpreted the data; T.K., B.B., S.S., M.D., H.G., and M.Z. wrote the manuscript; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Birgit Burkhardt, Pediatric Hematology and Oncology, University Hospital Muenster, 48149 Muenster, Germany; e-mail: birgit.burkhardt@ukmuenster.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal