

Key Points
AnxA1Ac2-26 plays a key role in mitigating neutrophil-dependent thromboinflammatory responses in sickle cell disease.
Targeting the AnxA1/Fpr2/ALX-pathway switches neutrophil phenotype, driving the resolution of thromboinflammation.

Abstract
Neutrophils play a crucial role in the intertwined processes of thrombosis and inflammation. An altered neutrophil phenotype may contribute to inadequate resolution, which is known to be a major pathophysiological contributor of thromboinflammatory conditions such as sickle cell disease (SCD). The endogenous protein annexin A1 (AnxA1) facilitates inflammation resolution via formyl peptide receptors (FPRs). We sought to comprehensively elucidate the functional significance of targeting the neutrophil-dependent AnxA1/FPR2/ALX pathway in SCD. Administration of AnxA1 mimetic peptide AnxA1Ac2-26 ameliorated cerebral thrombotic responses in Sickle transgenic mice via regulation of the FPR2/ALX (a fundamental receptor involved in resolution) pathway. We found direct evidence that neutrophils with SCD phenotype play a key role in contributing to thromboinflammation. In addition, AnxA1Ac2-26 regulated activated SCD neutrophils through protein kinase B (Akt) and extracellular signal–regulated kinases (ERK1/2) to enable resolution. We present compelling conceptual evidence that targeting the AnxA1/FPR2/ALX pathway may provide new therapeutic possibilities against thromboinflammatory conditions such as SCD.
Introduction
Neutrophils, the most abundant immune cells (contributing to 60%-70% of the leukocyte population) and one of the most important effector cells of the immune system, play crucial roles in resolution of inflammation. Inflammation is interconnected with thrombosis, with one begetting the other, leading to a prothromboinflammatory state1,2 that is associated with several diseases including sickle cell disease (SCD),3 diabetes,4 and cancer.5 These chronic thromboinflammatory diseases often result in severe acute cardiovascular complications (acute ischemic stroke [AIS], pulmonary embolism, and microvascular angiopathies6 ) that are the most frequent cause of morbidity and mortality worldwide.6,7
Sickle cell disease (SCD) is an inherited autosomal recessive disorder resulting from a single amino acid substitution in the hemoglobin β chain.8 The pathophysiology of SCD is characterized by a relentless prothromboinflammatory state that enables a heightened propensity for ischemic events such as AIS. These patients are more susceptible to ischemic events and have poorer outcomes after AIS, and the mechanisms remain poorly understood. However, these detrimental effects may be caused by inadequate class switching of endogenous proresolving mediators.9,10 Thus, SCD provides a unique model for the study of a defective resolution process to ascertain whether resolution pharmacology could be an optimal therapeutic approach to target thromboinflammation.11,12
Under homeostatic conditions, resolution of inflammation is orchestrated by a tightly controlled series of endogenous biosynthetic mediators (eg, annexin A1 [AnxA1] and its biologically active N-terminal domain, peptide AnxA1Ac2-26,13 aspirin-triggered lipoxin A4 [15(R)-epi-LXA4 (ATL)])14 and protective proresolution pathways (eg, the formyl peptide receptor [FPR] pathway),15 However, these endogenous biosynthetic circuits may be hampered in chronic thromboinflammatory diseases. Both AnxA1 and AnxA1Ac2-26 are known to exert their anti-inflammatory and proresololution actions in acute and chronic inflammation via FPR2/ALX.13,15,16 Thus, exploring AnxA1/FPR2/ALX pathways in disease states characterized by chronic uncontrolled thromboinflammation may provide a viable therapeutic strategy to promote resolution, thus affording protection.
Using genetic and pharmacological approaches, along with clinical samples, we present novel and compelling data that AnxA1Ac2-26 plays a key role in mitigating neutrophil-dependent thromboinflammatory responses in the cerebral microvasculature via the AnxA1/FPR2/ALX pathway. Furthermore, we demonstrate that exogenous administration of AnxA1Ac2-26 regulates prothrombotic neutrophil responses without affecting physiological responses, through protein kinase B (Akt) and extracellular signal-regulated kinases (ERK1/2),17,18 which act as molecular switches to transform the neutrophil phenotype from a pro-NETotic (neutrophil extracellular trap) to proapoptotic phenotype, thereby driving resolution. These compelling data demonstrate the utility of therapeutic strategies based on resolution biologics in effective management of thromboinflammatory complications.
Material and methods
See the supplemental Materials and Methods, available on the Blood Web site, for expanded, in-depth descriptions of the methods.
Drugs, reagents, and antibodies
For in vivo experiments, vehicle (saline), annexin A1 (AnxA1), mimetic peptide Ac2-26 (AnxA1Ac2-26, Ac-AMVSEFLKQAWFIENEEQEYVQTVK, Cambridge Research Biochemicals) 100 µg per mouse,19 Boc2 (N-tert-butoxycarbonyl-L-Phe-D-Leu-L-Phe-D-Leu-L-Phe; MP Biomedicals, Cambridge, United Kingdom) 10 µg per mouse,19 and WRW4 (55 μg per mouse) (EMD Biosciences Inc) were administered (100 µL) IV at the start of cerebral reperfusion.20
For in vitro experiments, 1× phosphate buffered saline vehicle (PBS; Life Technologies), AnxA1Ac2-26 (30 µM), Boc2 (10 µM),WRW4 (10 µM),21 Akti-1/2 (10 µM),18 U0126 (10 µM) (Tocris), and caspase-3 inhibitor Z-DEVD-FMK (20 µM; R&D Systems, Minneapolis, MN) were used as pharmacological tools. NETs were induced by ionomycin (4 µM; Life Technologies). NET-specific stains included neutrophil elastase (NE) rabbit anti-NE (1:200) (Abcam) and histone H3 mouse anti-H3Cit (1:200; Cell Signaling Technology and Sigma-Aldrich).
Animals
Male control and Sickle Cell Transgenic mice (STM; Townes) that were homozygous at the Hba locus for the hα mutation (Hbatm1(HBA)Tow) and homozygous at the Hbb locus for the −383 γ-βA mutation (Hbbtm3(HBG1,HBB)Tow) were purchased from Jackson Laboratory (Bar Harbor, ME). The Animal Care and Use Committee of Louisiana State University Health Sciences Center-Shreveport (LSUHSC-S) approved experimental procedures performed on the mice. All studies were performed blinded and randomized, and all studies complied with ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
Human samples
The study was approved by the institutional review board of the LSUHSC-S (STUDY00000572 and STUDY00000261) and conducted in accordance with the Declaration of Helsinki. After signed consent was obtained, blood was taken from control volunteers (18-52 years old; 26 males, 18 females) and patients with SCD (18-52 years old; 19 males, 28 females). All but one of the patients with SCD were of the HbSS (homozygous hemoglobin S) genotype. One patient was of the HbSC (sickle hemoglobin C disease) genotype. Patients with SCD were recruited upon routine clinical visits to the Feist-Weiller Cancer Center at LSUHSC-S. All patients with SCD were on long-term hydroxyurea therapy, and blood was obtained just before exchange transfusion. Hydroxyurea was started at 15 mg/kg body weight per day and then escalated by 5 mg/kg every 12 weeks until the maximum tolerated dose was achieved on the basis of peripheral blood counts. Patients were on partial exchange transfusion every 2 weeks. Patients with acute infection or other chronic blood-borne diseases (HIV, hepatitis B and C) were excluded from the study. Demographic and clinical characteristics of controls and patients with SCD are included in supplemental Table 3.
Neutrophil depletion and DNase-I treatment
Neutropenia was induced with mouse antineutrophil serum (ANS; 1A8; 150 μg per mouse, intraperitoneally 24 hours before the experiment).22 DNase-I (2000 U) was administered IV for NET degradation.
Thrombosis
Anesthetized mice (ketamine/xylazine, 1:1) were kept under the microscope after jugular vein cannulation and open-window craniotomy. Thrombosis in cerebral vessels was induced using the light/dye thrombosis model.23 After 20 minutes of equilibration, 10 mg/kg of 5% fluorescein isothiocyanate (FITC)-dextran (150 000 molecular weight; Sigma-Aldrich) was injected via the femoral vein and allowed to circulate for 10 minutes. Photoactivation was initiated (excitation, 495 nm; emission, 519 nm) by exposing 100 μm of vessel length to epi-illumination with a 175-W xenon lamp (Lamda LS; Sutter) and a fluorescein filter cube (HQ-FITC; Chroma). Time was recorded when the platelet aggregates first appeared (onset time) and when the flow stopped for 30 seconds (cessation time). Thirty minutes before the onset of thrombosis, the mice were treated with the vehicle, AnxA1Ac2-26 (100 µg per mouse), Boc2 (10 µg per mouse), or WRW4 (55 μg per mouse).
Enzyme-linked immunosorbent assay for NETs
To quantify the NETs in the circulating blood, 96-well immunoassay plates (9018 Costar; Corning) were coated with a NE antibody (1:250; Abcam) in 15 mM Na2CO3, at pH 9.6 (250 μL per well) overnight at 4°C. The following day, the wells were washed 4 times with PBS, followed by blocking with 5% bovine serum albumin for 2 hours at room temperature. After blocking, the wells were washed again (3 times) with PBS. 50 μL of murine plasma was added to the wells and incubated for 2 hours at room temperature on a microplate shaker at 250 rpm. The plates were then washed (3 times) with washing buffer (1% bovine serum albumin and 0.05% Tween 20 in PBS), followed by incubation with the immunoreagent (100 μL in each well). The immunoreagent was prepared by mixing 1:20 volume of anti-DNA peroxidase-conjugated antibody (Anti-DNA-POD) with 19:20 volume incubation buffer (Cell Death Detection ELISAPLUS; Roche) for 2 hours at room temperature on a microplate shaker at 250 rpm. The solution was removed, and each well was rinsed 4 times with 250 μL of incubation buffer. Next, 100 μL of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid [ABTS]; Roche) was added and the plate was incubated for 10 to 20 minutes on a microplate shaker at 250 rpm. The reaction was stopped by pipetting 100 μL of ABST stop solution and the absorbance was measured at 405 nm on a plate reader (Synergy H1, BioTek; reference wavelength, ∼490 nm). Data were analyzed per the manufacturer’s instructions (Roche).
Murine neutrophil isolation and adoptive transfer
Murine neutrophils were isolated, kept at 5 × 106/mL, and treated with AnxA1Ac2-26 (30 µM), WRW4 (10 μM), or AnxA1Ac2-26+WRW4 for 10 minutes before injection (5 × 105 cells per mouse) via the jugular vein of the recipient neutropenic mouse.
Isolation of human neutrophils
Neutrophils were isolated from control donors and patients with SCD by using dextran (spectrum chemical) sedimentation followed by Histopaque 1077.24
Visualization and quantification of NETs
Neutrophils (1 × 105 per well) were seeded on poly-l-lysine–coated coverslips and stimulated for 3 hours at 37°C in 5% CO2. The cells were fixed (10% formalin), permeabilized (0.5% Triton X-100), blocked (10% goat serum), and incubated with NET-specific antibodies (NE rabbit anti-NE [1:200]; histone H3 mouse anti-H3Cit [1:200]) and species-specific secondary antibodies and visualized.
Annexin A1 quantification in plasma
Human and murine AnxA1 enzyme-linked immunosorbent assay (ELISA) kits (MyBioSource) were used to quantify the plasma levels of AnxA1. Plasma preparation and the ELISA assay were performed according to the manufacturer’s instructions. Results were reported in nanograms per milliliter of AnxA1 concentration in plasma of control volunteers and patients with SCD or in plasma of control and STM mice.
Western blot analysis
Samples were immunoblotted with rabbit anti-phospho-Akt1 (1:1000), goat anti-Akt1 (1:500), rabbit anti-phospho-ERK1/2 (1:1000), or rabbit anti-ERK1/2 (1:5000) antibodies overnight followed by species-specific secondary antibodies.25
Cleaved caspase-3 for apoptotic cells
Neutrophils (2 × 105/well) were seeded on poly-l-lysine–coated coverslips and treated with 1× PBS or AnxA1Ac2-26 (30 µM; 3 hours, 37°C, 5% CO2). The cells were incubated with caspase-3 antibody overnight (1:500) followed by species-specific secondary antibody.
Myeloperoxidase release assay
Neutrophils (1 × 105 per well) were left unstimulated, stimulated with ionomycin (4 μM), or pretreated with AnxA1Ac2-26 (30 µM; 15 minutes before any pretreatment with vehicle/ionomycin; 3 hours at 37°C, 5% CO2). After 3 hours, supernatant was collected, added to the myeloperoxidase (MPO) solution, the reaction terminated with H2SO4(2N), and the absorbance read (450 nm).
Chemotaxis assay
Human neutrophils tagged with calcein-AM (Invitrogen) were left unstimulated, stimulated with ionomycin (4 μM), or pretreated with AnxA1Ac2-26 (30 µM; 15 minutes before any pretreatment. Neutrophils were added to the ChemoTx System. After 3 hours, fluorescence intensity was measured (485/530 nm excitation/emission) to determine neutrophil chemotaxis toward leukotriene B4 (10−6 M) or PBS (control).
Quantification of interleukin-1β ELISA
Human neutrophils were left unstimulated, stimulated with ionomycin (4 μM), or pretreated with AnxA1Ac2-26 (30 µM) 15 minutes prior prestimulation. After 5 hours, the supernatant was collected for ELISA.
Statistical analysis
All data were tested for normal distribution, using the Kolmogorov-Smirnov test of normality with the Dallal-Wilkinson-Lillie D’Agostino-Pearson omnibus normality test for corrected P-value. Data that passed the normality assumption were analyzed with the Student t test (2 groups) or by analysis of variance, with the Bonferroni post hoc test (more than 2 groups). Data that failed the normality assumption were analyzed with the nonparametric Mann-Whitney U test (2 groups) or the Kruskal-Wallis test with the Dunn post hoc test (more than 2 groups). Analysis was performed with Prism 6 software (Graph Pad, San Diego, CA). Data are shown as means ± standard error of the mean (SEM). Outliers were defined as ≥2 standard deviations (SD) and have been reported in the figure legends where applicable. Differences were considered statistically significant at P < .05.
Results
AnxA1Ac2-26 ameliorates exacerbated cerebral thrombosis in SCD via Fpr2/ALX
Thromboinflammatory disease states such as SCD are often associated with a microcirculation that assumes a proinflammatory and prothrombotic state.26 To validate our experimental model of thromboinflammation, we performed a light/dye injury model23 (enabling visualization of thrombus formation in real-time [supplemental Videos 1 and 2]) and observed accelerated thrombus formation (decrease in blood flow cessation time) in cerebral arterioles (P < .0001) and venules (P < .01) of STM vs control mice (Figure 1), concurring with our previous findings in a nonhumanized SCD mouse model.23
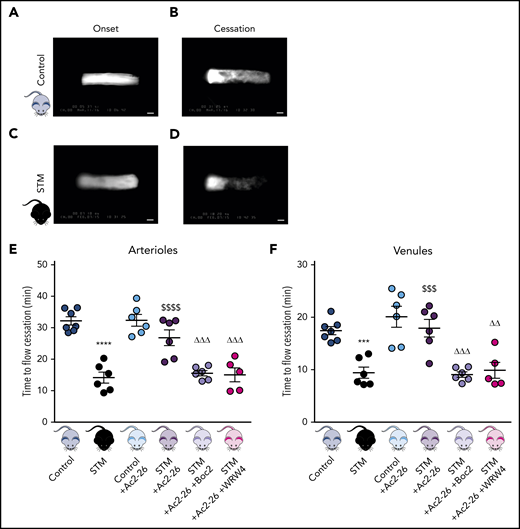
AnxA1Ac2-26 rescues enhanced cerebral thrombus formation. STM and control mice were subjected to light/dye-induced thrombosis with intravenous infusion of 10 mg/kg 5% FITC-dextran followed by photoactivation of cerebral microvessels. (A-D) Images of onset (start of platelet aggregation) and cessation (complete stop of flow for ≥30 seconds) of thrombus formation in control mice and STM. Bars represent 20 μm. The mice were treated with vehicle (saline), AnxA1Ac2-26 (100 μg per mouse), AnxA1Ac2-26+Boc2 (100 μg per mouse + 10 μg per mouse), or AnxA1Ac2-26+WRW4 (100 μg per mouse + 55 μg per mouse), and subjected to light/dye-induced thrombosis, and time of flow cessation (minutes) was quantified in cerebral (E) arterioles and (F) venules. Data are means ± SEM (6-7 mice per group). ***P < .001; ****P < .0001 vs controls. $$$P < .001; $ $ $ $P < .0001 vs STM controls. ΔΔP < .01; ΔΔΔP < .001 vs STM+AnxA1Ac2-26–treated mice.
AnxA1Ac2-26 rescues enhanced cerebral thrombus formation. STM and control mice were subjected to light/dye-induced thrombosis with intravenous infusion of 10 mg/kg 5% FITC-dextran followed by photoactivation of cerebral microvessels. (A-D) Images of onset (start of platelet aggregation) and cessation (complete stop of flow for ≥30 seconds) of thrombus formation in control mice and STM. Bars represent 20 μm. The mice were treated with vehicle (saline), AnxA1Ac2-26 (100 μg per mouse), AnxA1Ac2-26+Boc2 (100 μg per mouse + 10 μg per mouse), or AnxA1Ac2-26+WRW4 (100 μg per mouse + 55 μg per mouse), and subjected to light/dye-induced thrombosis, and time of flow cessation (minutes) was quantified in cerebral (E) arterioles and (F) venules. Data are means ± SEM (6-7 mice per group). ***P < .001; ****P < .0001 vs controls. $$$P < .001; $ $ $ $P < .0001 vs STM controls. ΔΔP < .01; ΔΔΔP < .001 vs STM+AnxA1Ac2-26–treated mice.
AnxA1 and its mimetic peptides (eg, AnxA1Ac2-26) are recognized anti-inflammatory compounds that have shown therapeutic potential in a diverse range of disease models, including ischemia reperfusion–induced lung injury, acute colitis, renal transplantation, diabetic nephropathy, atherosclerosis, acute lung injury, and colitis.27 However, their effects on thrombosis remain somewhat unknown, although we have recently shown that they attenuate platelet responses.16 AnxA1Ac2-26 had no effect on cerebral thrombosis when exogenously administered to control mice, but significantly increased blood flow cessation time in cerebral arterioles (P < .001) and venules (P < .01) of STM mice (Figure 1E-F).
Having established the antithrombotic effect of AnxA1Ac2-26 in STM, we next tested whether the effects are mediated via an interaction with the FPR pathway (especially FPR2/ALX, which is a key receptor involved in resolution and a receptor through which AnxA1 mediates its effects). The FPR pan-antagonist Boc2 blocked the protective actions of AnxA1Ac2-26 (P < .001), suggesting a mechanism of action via the FPR family (Figure 1E-F). To further tease out which receptor, mice were coadministered AnxA1Ac2-26+WRW4 (specific FPR2/ALX antagonist). WRW4-blocked AnxA1Ac2-26 afforded protection in cerebral arterioles (P < .001) and venules (P < .001) thereby confirming an FPR2/ALX mechanism (Figure 1E-F).
Neutrophils contribute to cerebral thrombosis in STM
Compelling evidence has demonstrated a pivotal role of neutrophils in thromboinflammation,28 although the role they play in the context of cerebral thrombosis is less well discerned. To address this knowledge gap we rendered STM mice neutropenic with the well-characterized ANS, 1A8. Using real-time live imaging, we observed a phenotype reversal (ie, no difference in cerebral thrombosis formation between control vs STM/ANS mice; Figure 2B-C), demonstrating that the cerebral microvasculature in STM mice was rendered vulnerable to thrombus formation via a neutrophil-dependent mechanism. Furthermore, transfer of STM, but not control, neutrophils into neutropenic STM mice (Figure 2A) restored the SCD-associated acceleration of thrombus formation in cerebral pial vessels (Figure 2B-C). These results suggest that the STM neutrophil plays a key role in mediating the accelerated cerebral thrombus formation observed in this experimental thromboinflammatory model.
![Neutrophils contribute to cerebral thrombosis in experimental thromboinflammation and exhibit enhanced extracellular DNA activity. Schematic representation of neutrophil transfer from donor control and STM into neutropenic ANS recipient STM followed by light/dye-induced thrombosis (A1-3) time of flow cessation was quantified in cerebral arterioles (B) and venules (C). (D) Cerebral microvessels were analyzed after DNase (2000 U) treatment (n = 4-6 mice/group). (E) Plasma levels of circulating annexin A1 (n = 6 each) were also determined in control mice and STM. (F) NE DNA complex (NE-DNA) levels were determined by ELISA in plasma from saline (vehicle) and AnxA1Ac2-26-treated mice (n = 12 saline-treated and n = 6 AnxA1Ac2-26–treated control and STM). Two values for control-AnxA1Ac2-26 and 1 value for STM-AnxA1Ac2-26 were under detectable levels and are not included. (G) Percentage of histone H3 (H3Cit+)–positive unstimulated (n = 10 [1 outlier removed]; n = 10 STM [1 outlier removed]) and ionomycin-stimulated (n = 10 control; n = 7 STM; 4 µM) neutrophils. Data are as means ± SEM from independent experiments. *P < .05; **P < .01 vs control mice. #P < .05; ###P < .001 vs STM. @@P < .01 vs unstimulated STM neutrophils. $P < .05 vs stimulated STM neutrophils. ØP < .05 vs STM+ANS. ΦP < .05 vs STM+ANS+ control neutrophils.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/11/10.1182_blood.2020009166/5/m_bloodbld2020009166f2.png?Expires=1769129308&Signature=rQvF0Qv4sSZtwAjphqbI~GW-5~dTC3B4wUzjCp9PGqMmhVC-kN-sDjLExmRw2qRNONVqj2IDqIJ3pXvNRhwk-mFwMueOBeYorNedr-vV9IpPlyPh5RiMx4SuI76~e68xbQEOOHGc4Z0TlqdwcDjhFS1Dil9ZdLozHvD-XXjfxjtz4VfUQ4H42qkubInEGXd8lTL5w~6qPBC5TzsmBK3-Sr3mw5pAJgtSPYGSpx3ofpOJDO0VnEuRm6mgbDKuq7vC-tBP-1~Jg9LB8oznaX3H-CxG32tQkjC0p32CcGNhQlStfYUgiyYuUToVNw2D14ulYW5-Wj60gWnfJJg8IvLxNQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Neutrophils contribute to cerebral thrombosis in experimental thromboinflammation and exhibit enhanced extracellular DNA activity. Schematic representation of neutrophil transfer from donor control and STM into neutropenic ANS recipient STM followed by light/dye-induced thrombosis (A1-3) time of flow cessation was quantified in cerebral arterioles (B) and venules (C). (D) Cerebral microvessels were analyzed after DNase (2000 U) treatment (n = 4-6 mice/group). (E) Plasma levels of circulating annexin A1 (n = 6 each) were also determined in control mice and STM. (F) NE DNA complex (NE-DNA) levels were determined by ELISA in plasma from saline (vehicle) and AnxA1Ac2-26-treated mice (n = 12 saline-treated and n = 6 AnxA1Ac2-26–treated control and STM). Two values for control-AnxA1Ac2-26 and 1 value for STM-AnxA1Ac2-26 were under detectable levels and are not included. (G) Percentage of histone H3 (H3Cit+)–positive unstimulated (n = 10 [1 outlier removed]; n = 10 STM [1 outlier removed]) and ionomycin-stimulated (n = 10 control; n = 7 STM; 4 µM) neutrophils. Data are as means ± SEM from independent experiments. *P < .05; **P < .01 vs control mice. #P < .05; ###P < .001 vs STM. @@P < .01 vs unstimulated STM neutrophils. $P < .05 vs stimulated STM neutrophils. ØP < .05 vs STM+ANS. ΦP < .05 vs STM+ANS+ control neutrophils.
Neutrophils contribute to cerebral thrombosis in experimental thromboinflammation and exhibit enhanced extracellular DNA activity. Schematic representation of neutrophil transfer from donor control and STM into neutropenic ANS recipient STM followed by light/dye-induced thrombosis (A1-3) time of flow cessation was quantified in cerebral arterioles (B) and venules (C). (D) Cerebral microvessels were analyzed after DNase (2000 U) treatment (n = 4-6 mice/group). (E) Plasma levels of circulating annexin A1 (n = 6 each) were also determined in control mice and STM. (F) NE DNA complex (NE-DNA) levels were determined by ELISA in plasma from saline (vehicle) and AnxA1Ac2-26-treated mice (n = 12 saline-treated and n = 6 AnxA1Ac2-26–treated control and STM). Two values for control-AnxA1Ac2-26 and 1 value for STM-AnxA1Ac2-26 were under detectable levels and are not included. (G) Percentage of histone H3 (H3Cit+)–positive unstimulated (n = 10 [1 outlier removed]; n = 10 STM [1 outlier removed]) and ionomycin-stimulated (n = 10 control; n = 7 STM; 4 µM) neutrophils. Data are as means ± SEM from independent experiments. *P < .05; **P < .01 vs control mice. #P < .05; ###P < .001 vs STM. @@P < .01 vs unstimulated STM neutrophils. $P < .05 vs stimulated STM neutrophils. ØP < .05 vs STM+ANS. ΦP < .05 vs STM+ANS+ control neutrophils.
AnxA1Ac2-26 inhibits the thrombotic NET phenotype associated with thromboinflammation
Having discovered that neutrophils play a significant role in cerebral thrombosis and exploiting the fact that the AnxA1/FPR2/ALX pathway can mitigate these unwanted responses, we next characterized the mechanisms involved in this process. NET formation is associated with the pathogenesis of several thromboinflammatory diseases (eg, in thrombi associated with deep-vein thrombosis and detected via histology in the lungs of SCD mice29 ). In our study, administration of DNase (which breaks down NETs) delayed cessation of blood flow in STM mice, an effect that was absent in control mice (Figure 2D).
No differences in circulating AnxA1 plasma levels in control vs STM mice were quantified (Figure 2E), but treatment of control and STM mice with AnxA1Ac2-26 resulted in significant reduction of NE (NE-DNA) complexes in STM but not control plasma (P < .05; Figure 2F) suggesting an antithrombotic mechanism and elucidating a drug-sparing effect in normal cohorts.
To test whether AnxA1Ac2-26 antithrombotic effect in vivo occurs by reducing the capability of neutrophils to release NETs (known contributors to thrombosis),30 isolated neutrophils (control and STM mice) were treated with the NET-inducing stimuli ionomycin (a natural calcium ionophore) to stimulate maximal NET production with and without AnxA1Ac2-26. Citrullinated histone-3 (H3Cit) is the most common NET biomarker that has been associated with experimental thrombosis.31 AnxA1Ac2-26 treatment significantly reduced the percentage of ionomycin-stimulated STM neutrophils that were positive for H3Cit+ (P < .05; Figure 2G), an effect not observed in control neutrophils, further emphasizing the specific effect of AnxA1Ac2-26 on neutrophils exposed to a chronic thromboinflammatory milieu associated with SCD.
Targeting Fpr2/ALX reduces human H3Cit+ SCD neutrophils without affecting physiological responses
To translate our findings from mouse models into clinical setting, we isolated neutrophils from control volunteers and patients with SCD (Figure 3A, B). The percentage of unstimulated H3Cit+ SCD neutrophils was increased compared with control neutrophils (Figure 3C), which was further exacerbated with ionomycin (P < .05), suggesting that SCD neutrophils are characterized by extensive histone citrullination potentially contributing to the prothrombosis phenotype. Interestingly, our patients with SCD presented with reduced circulating AnxA1 plasma levels vs those of the control volunteers, highlighting a possible defect in the resolution process, as is often seen in chronic thromboinflammatory states (Figure 3D). Moreover, as observed with murine neutrophils, AnxA1Ac2-26 significantly attenuated ionomycin-induced H3Cit+ SCD neutrophils (P < .0001; Figure 3E) No effect was observed in the control volunteers. Finally, to determine the mechanistic role of the FPR family in these events, the FPR pan-antagonist Boc2 was coadministered with AnxA1Ac2-26. Figure 3E shows that Boc2 significantly abrogated the effects of AnxA1Ac2-26, increasing H3Cit+ SCD neutrophil percentage (P < .05). More specifically, when coadministered with AnxA1Ac2-26, the selective Fpr2/ALX antagonist WRW4 increased the percentage of H3Cit+ SCD neutrophils, thus abrogating the protective actions of AnxA1Ac2-26. These results highlight the protective effects of AnxA1Ac2-26 on NET production were mediated through Fpr2/ALX.
![SCD-associated enhanced H3Cit+ neutrophils can be inhibited by AnxA1Ac2-26. (A) Neutrophil isolation and NET analysis. (B) Images of NETs: H3Cit (green/Alexa Fluor 488), NE (red/Alexa Fluor 568), and nucleus (4′,6-diamidino-2-phenylindole). Bars in main images represent 100 µm; in insets, 10 µm. (C) Percentage of NETs hypercitrullinated at histone H3 (H3Cit+) quantified from unstimulated and ionomycin-stimulated neutrophils from control volunteers (unstimulated [n = 10], 1 outlier removed; and stimulated [n = 10], 1 outlier removed) and from patients with SCD (unstimulated [n = 10]; and stimulated [n = 14]). (D) Plasma levels of circulating annexin A1 (n = 5, 6 respectively) were determined in control volunteers and patients with SCD. Statistical significance was determined by unpaired Student t test and presented as *P < .05 vs control volunteers. (E) Percentage of H3Cit+ ionomycin-stimulated neutrophils from control volunteers (n = 8 vehicle and n = 9 AnxA1Ac2-26 pretreatment) and patients with SCD (n = 14 vehicle, n = 9 AnxA1Ac2-26, n = 10 AnxA1Ac2-26+Boc-2, and n = 9 AnxA1Ac2-26+WRW4). Data are means ± SEM from independent experiments. *P < .05; **P < .01 vs control unstimulated neutrophils. ####P < .0001 vs SCD unstimulated neutrophils. $ $ $ $P < .001 vs ionomycin-stimulated SCD neutrophils. ΔP < .05; ΔΔP < .01 vs SCD+AnxA1Ac2-26–treated SCD neutrophils. φφφP < .001 vs stimulated control neutrophils.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/11/10.1182_blood.2020009166/5/m_bloodbld2020009166f3.png?Expires=1769129308&Signature=1~JsWRkYILEdRbL0wY5oHvEYCrGjonNpgcQjoOKvNTZSke-q3zK-3t0XjDtYy1W5jPPH4T-sYe9Wwuo~HhHii2gniXrJYg2~QvvJwANophIITJvfktwxeqA8lakkIKffm7uFQoTprkSNnUGr8GLaXXV6BeNztUFBk8p3UQeU4CTSv-D06BovUG-AksMTcfk95zyhw71c4-PKDsloiLRXM8Zkp3WGbN29uPNqCgVpCikkjw9GzE0Jb--D~k8y9PBTKQVQTpzZUXOybq6zzUl3RgF8dgbohYi9z4Fum9CQL2AMPB9ocd4nbFYNw5XU4IlQyp-rTKNbqpYzfXTzj-BS-g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SCD-associated enhanced H3Cit+ neutrophils can be inhibited by AnxA1Ac2-26. (A) Neutrophil isolation and NET analysis. (B) Images of NETs: H3Cit (green/Alexa Fluor 488), NE (red/Alexa Fluor 568), and nucleus (4′,6-diamidino-2-phenylindole). Bars in main images represent 100 µm; in insets, 10 µm. (C) Percentage of NETs hypercitrullinated at histone H3 (H3Cit+) quantified from unstimulated and ionomycin-stimulated neutrophils from control volunteers (unstimulated [n = 10], 1 outlier removed; and stimulated [n = 10], 1 outlier removed) and from patients with SCD (unstimulated [n = 10]; and stimulated [n = 14]). (D) Plasma levels of circulating annexin A1 (n = 5, 6 respectively) were determined in control volunteers and patients with SCD. Statistical significance was determined by unpaired Student t test and presented as *P < .05 vs control volunteers. (E) Percentage of H3Cit+ ionomycin-stimulated neutrophils from control volunteers (n = 8 vehicle and n = 9 AnxA1Ac2-26 pretreatment) and patients with SCD (n = 14 vehicle, n = 9 AnxA1Ac2-26, n = 10 AnxA1Ac2-26+Boc-2, and n = 9 AnxA1Ac2-26+WRW4). Data are means ± SEM from independent experiments. *P < .05; **P < .01 vs control unstimulated neutrophils. ####P < .0001 vs SCD unstimulated neutrophils. $ $ $ $P < .001 vs ionomycin-stimulated SCD neutrophils. ΔP < .05; ΔΔP < .01 vs SCD+AnxA1Ac2-26–treated SCD neutrophils. φφφP < .001 vs stimulated control neutrophils.
SCD-associated enhanced H3Cit+ neutrophils can be inhibited by AnxA1Ac2-26. (A) Neutrophil isolation and NET analysis. (B) Images of NETs: H3Cit (green/Alexa Fluor 488), NE (red/Alexa Fluor 568), and nucleus (4′,6-diamidino-2-phenylindole). Bars in main images represent 100 µm; in insets, 10 µm. (C) Percentage of NETs hypercitrullinated at histone H3 (H3Cit+) quantified from unstimulated and ionomycin-stimulated neutrophils from control volunteers (unstimulated [n = 10], 1 outlier removed; and stimulated [n = 10], 1 outlier removed) and from patients with SCD (unstimulated [n = 10]; and stimulated [n = 14]). (D) Plasma levels of circulating annexin A1 (n = 5, 6 respectively) were determined in control volunteers and patients with SCD. Statistical significance was determined by unpaired Student t test and presented as *P < .05 vs control volunteers. (E) Percentage of H3Cit+ ionomycin-stimulated neutrophils from control volunteers (n = 8 vehicle and n = 9 AnxA1Ac2-26 pretreatment) and patients with SCD (n = 14 vehicle, n = 9 AnxA1Ac2-26, n = 10 AnxA1Ac2-26+Boc-2, and n = 9 AnxA1Ac2-26+WRW4). Data are means ± SEM from independent experiments. *P < .05; **P < .01 vs control unstimulated neutrophils. ####P < .0001 vs SCD unstimulated neutrophils. $ $ $ $P < .001 vs ionomycin-stimulated SCD neutrophils. ΔP < .05; ΔΔP < .01 vs SCD+AnxA1Ac2-26–treated SCD neutrophils. φφφP < .001 vs stimulated control neutrophils.
Interestingly, although AnxA1Ac2-26 modulated histone citrullination under pro-NETotic conditions, the resolving peptide did not affect other neutrophil physiological responses, such as MPO release and chemotaxis (supplemental Figure 2A-C). This lack of effect could be related to the fact that under pro-NETotic conditions, neutrophils are at the point of no return when releasing their MPO after membrane disruption. Furthermore, although interleukin-1β (IL-1β) production was increased in unstimulated SCD neutrophils compared with the control, it was dramatically reduced when neutrophils were stimulated with ionomycin, with AnxA1Ac2-26 having no effect (supplemental Figure 2D). These effects may be related to the ability of ionomycin-induced NETs to produce IL-1β–degrading serine proteases.32
AnxA1Ac2-26 regulates the NETosis-apoptosis axis in human SCD neutrophils: impact for therapeutic strategy against thrombosis
Because of the importance of ERK and Akt kinases in NET release,17,18,33 we examined the expression of these kinases in control and SCD neutrophils (Figures 4A). A significant increase in ERK phosphorylation (at 15 minutes, P < .001) and Akt phosphorylation (30 and 60 minutes; P < .01 each) was observed in ionomycin-stimulated SCD neutrophils vs SCD neutrophils at baseline (Figures 4B-E) with no differences observed in baseline ERK and Akt phosphorylation in patients with SCD and control volunteers (Figure 4B-E). Treatment of SCD neutrophils with AnxA1Ac2-26 showed an 85% reduction in ionomycin-induced ERK phosphorylation at 15 minutes (P < .001 vs ionomycin-stimulated control neutrophils at 15 minutes) and ∼75% reduction in ionomycin-induced Akt phosphorylation (30 and 60 minutes; P < .01 at each time point) compared with ionomycin-stimulated control neutrophils (Figures 4B-E). Furthermore, as with AnxA1Ac2-26 treatment, administration of Akt or Erk inhibitors reduced ionomycin-induced H3Cit+SCD neutrophils (Figure 4F). Interestingly, in the presence of Z-DEVD-FMK (caspase-3 inhibitor), AnxA1Ac2-26 no longer reduced the percentage of H3Cit+ neutrophils (Figure 4G), but treatment with AnxA1Ac2-26 alone increased cleaved caspase-3 (Figures 4H-I). Collectively, these data suggest a potential molecular mechanism by which AnxA1Ac2-26 can act as a pharmacological switch between the NETosis-apoptosis axis in SCD.
![AnxA1Ac2-26 dampens ERK and Akt activation in neutrophils isolated from patients with SCD and activates cleaved caspase-3. (A) Procedure for sample preparation for western blot analysis. Representative western blots of neutrophils from control volunteers and patients with SCD for ERK (B) and Akt (C) activation after AnxA1Ac2-26 treatment and ionomycin stimulation. Densitometric analysis of p-ERK/total ERK (D; n = 5) and p-Akt/total Akt (E; [n = 4]. One outlier [defined as ≥2 standard deviations] removed from control ionomycin+AnxA1Ac2-26 [30 minutes]). (F-G) Percentage of H3Cit+ SCD neutrophils after stimulation with ionomycin (4 µM, 3 hours), with/without pretreatment with AnxA1Ac2-26 (30 µM, 15 minutes) and Akt (10 µM, 30 minutes), ERK (10 µM, 60 minutes), or caspase-3 (Z-DEVD-FMK, 20 µM, 45 minutes) inhibitors (n = 5 each group). (H) Representative immunofluorescence images of cleaved caspase-3 staining from SCD neutrophils with/without AnxA1Ac2-26 (30 μM) treatment. Scale bar, 100 µm. (I) Percentage of cleaved caspase-3+ neutrophils from control and patients with SCD with/without AnxA1Ac2-26 treatment (n = 5). Data expressed as mean ± SEM from independent experiments. *P < .05; **P < .01; ***P < .001 vs unstimulated control neutrophils. #P < .05; ##P < .01 vs unstimulated SCD neutrophils. $$P < .01; $$$P < .001; $ $ $ $P < .0001 vs ionomycin-stimulated SCD neutrophils at the corresponding time points. ΔΔP < .01 vs AnxA1Ac2-26-pretreated, ionomycin-stimulated SCD neutrophils.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/11/10.1182_blood.2020009166/5/m_bloodbld2020009166f4.png?Expires=1769129308&Signature=THwsZics1LZpwoHoX1uBzDjbwdvDyB1~dyJYiQS~eXA5unsG0TgkgAW5b8LKpiLgK52zU8BBX6zZo75Lz5LIjALEJKog-AZQAxd5WzBIV7st-KX79zIpRC8RM0QGgzZn0pa-52xLLmflwQD~sRHhHQ9wS9b-mFkdyGKZDWLFdzGvxYl6iD4D698eYUjUMaYE4LeVfNDQ8b~HH~kQNnDoCWKeq2KBeZ0vKhdxDPDu4w0ySmEYZJsVNcDGKkqjcugEs3chPS-mEOQDQTHKN8Fyq4jg0X9wstiHcpm0~54yHe9a1of4BF42kGw~DVT-pcqJt08R5joH2bDZPVWLW14zSA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AnxA1Ac2-26 dampens ERK and Akt activation in neutrophils isolated from patients with SCD and activates cleaved caspase-3. (A) Procedure for sample preparation for western blot analysis. Representative western blots of neutrophils from control volunteers and patients with SCD for ERK (B) and Akt (C) activation after AnxA1Ac2-26 treatment and ionomycin stimulation. Densitometric analysis of p-ERK/total ERK (D; n = 5) and p-Akt/total Akt (E; [n = 4]. One outlier [defined as ≥2 standard deviations] removed from control ionomycin+AnxA1Ac2-26 [30 minutes]). (F-G) Percentage of H3Cit+ SCD neutrophils after stimulation with ionomycin (4 µM, 3 hours), with/without pretreatment with AnxA1Ac2-26 (30 µM, 15 minutes) and Akt (10 µM, 30 minutes), ERK (10 µM, 60 minutes), or caspase-3 (Z-DEVD-FMK, 20 µM, 45 minutes) inhibitors (n = 5 each group). (H) Representative immunofluorescence images of cleaved caspase-3 staining from SCD neutrophils with/without AnxA1Ac2-26 (30 μM) treatment. Scale bar, 100 µm. (I) Percentage of cleaved caspase-3+ neutrophils from control and patients with SCD with/without AnxA1Ac2-26 treatment (n = 5). Data expressed as mean ± SEM from independent experiments. *P < .05; **P < .01; ***P < .001 vs unstimulated control neutrophils. #P < .05; ##P < .01 vs unstimulated SCD neutrophils. $$P < .01; $$$P < .001; $ $ $ $P < .0001 vs ionomycin-stimulated SCD neutrophils at the corresponding time points. ΔΔP < .01 vs AnxA1Ac2-26-pretreated, ionomycin-stimulated SCD neutrophils.
AnxA1Ac2-26 dampens ERK and Akt activation in neutrophils isolated from patients with SCD and activates cleaved caspase-3. (A) Procedure for sample preparation for western blot analysis. Representative western blots of neutrophils from control volunteers and patients with SCD for ERK (B) and Akt (C) activation after AnxA1Ac2-26 treatment and ionomycin stimulation. Densitometric analysis of p-ERK/total ERK (D; n = 5) and p-Akt/total Akt (E; [n = 4]. One outlier [defined as ≥2 standard deviations] removed from control ionomycin+AnxA1Ac2-26 [30 minutes]). (F-G) Percentage of H3Cit+ SCD neutrophils after stimulation with ionomycin (4 µM, 3 hours), with/without pretreatment with AnxA1Ac2-26 (30 µM, 15 minutes) and Akt (10 µM, 30 minutes), ERK (10 µM, 60 minutes), or caspase-3 (Z-DEVD-FMK, 20 µM, 45 minutes) inhibitors (n = 5 each group). (H) Representative immunofluorescence images of cleaved caspase-3 staining from SCD neutrophils with/without AnxA1Ac2-26 (30 μM) treatment. Scale bar, 100 µm. (I) Percentage of cleaved caspase-3+ neutrophils from control and patients with SCD with/without AnxA1Ac2-26 treatment (n = 5). Data expressed as mean ± SEM from independent experiments. *P < .05; **P < .01; ***P < .001 vs unstimulated control neutrophils. #P < .05; ##P < .01 vs unstimulated SCD neutrophils. $$P < .01; $$$P < .001; $ $ $ $P < .0001 vs ionomycin-stimulated SCD neutrophils at the corresponding time points. ΔΔP < .01 vs AnxA1Ac2-26-pretreated, ionomycin-stimulated SCD neutrophils.
Discussion
Using pharmacological and genetic approaches, coupled with murine and clinical samples, we discovered several key conceptual and novel findings that we believe advance knowledge and understanding in the field of SCD, thromboinflammation, and resolution biology. Specifically, we found that (1) neutrophils from SCD, a known model of thromboinflammation, play a major role in cerebral thrombosis; (2) targeting Fpr2/ALX (a key receptor of inflammation resolution) mitigates these effects; and (3) the proresolution, anti-inflammatory mediator AnxA1Ac2-26 reduces H3Cit+ rich NET production, transforming neutrophil phenotype from pro-NETotic to proapoptotic thereby driving thromboinflammation resolution in SCD (Figure 5).
![Schematic of proposed mechanisms. (A) Our data show that SCD neutrophils produce increased NETs, which are exacerbated upon stimulation (eg, ionomycin), leading to increased phosphorylation of NET specific kinases (ERK and Akt), which has been shown to result in histone citrullination and inhibition of apoptosis via upregulation of antiapoptotic proteins (eg, Mcl-133 [prothrombotic state]). Peptidylarginine deiminase 4 (PAD4) forms a complex with intracellular calcium to catalyze histone citrullination.31 NET stimuli activate PKC, PLC, and PI3K,33 which in turn activate ERK and AKT, resulting in calcium-PAD4 complex formation, which catalyzes histone citrullination. (B) We found that AnxA1Ac2-26 interacts with Fpr2/ALX, suppressing ERK and Akt phosphorylation, preventing histone citrullination, and enabling apoptosis by activating caspase-3 (proresolving state). Mcl-1, myeloid cell leukemia protein-1; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PKC, protein kinase C; PLC, phospholipase C.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/11/10.1182_blood.2020009166/5/m_bloodbld2020009166f5.png?Expires=1769129308&Signature=KU-Hcsvgy4~Pns4ngNpffwUmPxt5Y0I0UxmIs11Vr2Wet2Xy5EHFTSGfXvpS9a7ZWyJFuevoT6MsQvcO0XWr0BRAOZGG2yfOD8oBZTq~d90uL3IUBWvCpWBoozFe12gNjGe25PqqV66tSGjJ5KeeFJWADf97qWteDDuMWoQWKu-8TuyLQRu6VLxtVUQzfn6CgP2gWiBojycw78zghSDcAm2i8NWNlNKZ80BW4T649eHK7u-qEHilbqkxP31eyqAqQ76-Tg6mxGhogAxAmp4urrDJEWGa2YNCsUWHCmVinxvkDj3VdGoAR~JikV0nQzJpXhLza9f6YORMYt9-0c-mFA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematic of proposed mechanisms. (A) Our data show that SCD neutrophils produce increased NETs, which are exacerbated upon stimulation (eg, ionomycin), leading to increased phosphorylation of NET specific kinases (ERK and Akt), which has been shown to result in histone citrullination and inhibition of apoptosis via upregulation of antiapoptotic proteins (eg, Mcl-133 [prothrombotic state]). Peptidylarginine deiminase 4 (PAD4) forms a complex with intracellular calcium to catalyze histone citrullination.31 NET stimuli activate PKC, PLC, and PI3K,33 which in turn activate ERK and AKT, resulting in calcium-PAD4 complex formation, which catalyzes histone citrullination. (B) We found that AnxA1Ac2-26 interacts with Fpr2/ALX, suppressing ERK and Akt phosphorylation, preventing histone citrullination, and enabling apoptosis by activating caspase-3 (proresolving state). Mcl-1, myeloid cell leukemia protein-1; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PKC, protein kinase C; PLC, phospholipase C.
Schematic of proposed mechanisms. (A) Our data show that SCD neutrophils produce increased NETs, which are exacerbated upon stimulation (eg, ionomycin), leading to increased phosphorylation of NET specific kinases (ERK and Akt), which has been shown to result in histone citrullination and inhibition of apoptosis via upregulation of antiapoptotic proteins (eg, Mcl-133 [prothrombotic state]). Peptidylarginine deiminase 4 (PAD4) forms a complex with intracellular calcium to catalyze histone citrullination.31 NET stimuli activate PKC, PLC, and PI3K,33 which in turn activate ERK and AKT, resulting in calcium-PAD4 complex formation, which catalyzes histone citrullination. (B) We found that AnxA1Ac2-26 interacts with Fpr2/ALX, suppressing ERK and Akt phosphorylation, preventing histone citrullination, and enabling apoptosis by activating caspase-3 (proresolving state). Mcl-1, myeloid cell leukemia protein-1; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PKC, protein kinase C; PLC, phospholipase C.
Using an experimental model of thromboinflammation (ie, the Townes STM mouse, which recapitulates many clinical manifestations of SCD), we revealed heightened cerebral thrombotic responses. By exploiting the Fpr pathway as a therapeutic target, administration of AnxA1Ac2-26 resulted in blood flow prolongation in the cerebral microcirculation of STM (no change was observed in circulating neutrophil counts; supplemental Table 1), with effects being equally robust in cerebral venules and arterioles (despite clear differences between vessel types; eg, shear rates and leukocyte-endothelial interaction).34 These results demonstrate the potent activity and versatility of the AnxA1Ac2-26 to mitigate SCD-associated thromboinflammation.
There are 3 FPRs in humans: FPR1, FPR2/lipoxin A4 (Fpr2/ALX, orthologue in the mouse), and FPR3.13 Coadministration of AnxA1Ac2-26 with the Fpr pan-antagonist Boc2 elicited an abrogation of the peptide’s protective responses. Moreover, this effect was consistent when the Fpr2/ALX-specific antagonist WRW4 was used, confirming not only the involvement of the Fpr-family in mediating the effects of AnxA1Ac2-26, but more specifically Fpr2/ALX. These findings are the first to show the protective effects of AnxA1Ac2-26 in cerebral thrombosis, demonstrating that AnxA1Ac2-26 not only possesses anti-inflammatory capabilities (eg, attenuation of leukocyte-platelet responses after stroke, reduction of lipopolysaccharide-induced leukocyte adhesion and migration), but has antithrombotic capabilities, making it a promising therapeutic candidate for promoting resolution in the context of thromboinflammatory diseases such as SCD.
Under normal physiological conditions, the host produces an adequate resolution response to inflammation and coagulation, characterized by specific immunoresolvents that induce clearance mechanisms (eg, apoptosis and efferocytosis).10,16,35 However, if the resolution process is defective, as observed in chronic thromboinflammatory states,11,36 then it results in the reduction or altered production of proresolving mediators, as demonstrated by our patients with SCD having reduced circulating AnxA1 plasma levels vs the control volunteers. We speculate that this chronic deficit contributes to a nonresolving state of inflammation, as previously observed.16 Interestingly, AnxA1 plasma levels in STM mice were similar to those in control mice.11 Although AnxA1 distribution is similar between human and murine neutrophils,37 differences in AnxA1 levels between species could be related to variances in neutrophil populations (eg, human neutrophils constitute 65%-75% of all peripheral blood leukocytes, unlike in the mouse, where ∼10%-25% of all leukocytes are neutrophils). In addition, it was recently shown that resolvin (Rv)D1 (another endogenous proresolving mediator) levels were similar in SCD mice and controls. However, lower levels of RvD1 were detected in spleens (a known target of acute vasoocclusive crises) of SCD mice vs controls under normoxia or when exposed to hypoxia/reoxygenation,11 suggesting like AnxA1, that other endogenous mediators may play important roles in resolution of thromboinflammation. As observed in the clinic, STM mice presented with neutrophilia (supplemental Table 2) and exhibited profound protection against microvascular thrombosis when made neutropenic. Furthermore, upon transfer of donor STM neutrophils into recipient controls, the animals displayed a phenotype similar to that of the full STM mouse, highlighting a key role that circulating STM neutrophils play in mediating cerebral thrombosis, which may translate to the clinical setting.
NET release results in production of circulating free DNA (cfDNA), which is significantly increased in thromboinflammatory diseases, with detrimental effects.38 In this study, we discovered that increased cfDNA in STM plasma was complexed with NE, which AnxA1Ac2-26 reduced, possibly by blocking the attachment of elastase to chromatin in the neutrophil. In vivo, DNase I (main factor regulating elimination of the cfDNA) heightened blood flow cessation times in STM mice, supporting the important role that NETs play in a thromboinflammatory environment. Our results suggest that a distinctive NETotic phenotype exists in STM, and AnxA1Ac2-26 specifically targets and reduces excessive NETosis in this thromboinflammatory model.
Different studies of thrombosis, as well as other inflammatory models, including SCD, have shown the importance of NET-associated citrullinated histone H3.29,30 In addition, histones comprise 65% of the total protein content in neutrophils, and extracellular histones (which are the backbone of NETs) are known to participate in immunothrombosis because of their prothrombotic,39 proinflammatory, and cytotoxic effects.40 In the present study, we demonstrated that AnxA1Ac2-26 significantly modified both the prothrombotic STM-NET and SCD-NET phenotypes by suppressing the production of citrullinated histone-rich NETs in an AnxA1/FPR2/ALX-dependent mechanism. In addition, extracellular DNA production was not affected by AnxA1Ac2-26, suggesting that the peptide does not reduce physiological netosis.17,41 These results are in accordance with our in vivo data and may help to further explain the antithrombotic action of AnxA1Ac2-26. Although we observed some differences in findings from murine neutrophils vs those with human neutrophils (eg, NETs released from mice are more compact than those observed from humans42 ), AnxA1Ac2-26 was still effective at specifically reducing histone-rich NET production. Interestingly, the effects of AnxA1Ac2-26 on H3cit+ neutrophils were observed only in STM and SCD neutrophils and not control neutrophils, suggesting that AnxA1Ac2-26 spares physiological NETosis. Furthermore, our data show that once AnxA1Ac2-26 was involved in regulating NET formation, it did not affect other neutrophil functions, such as MPO release, chemotaxis, or cytokine (IL-1β) production but promoted resolution by changing the SCD neutrophil phenotype from a pro-NETotic to a proapoptotic phenotype.
There is paucity in studies on the cellular mechanisms by which AnxA1/FPR2/ALX can be channelled as an anti-inflammatory or proresolving pathway. We further discovered that SCD neutrophils displayed increased ERK and Akt (the main driver of NADPH-independent NET production)41,43 activation, ensuing in extensive histone citrullination, as indicated by NET-based assays.41 AnxA1Ac2-26 inhibited ERK and Akt activity in SCD neutrophils, with inhibitors of ERK and Akt suppressing H3Cit+ neutrophil production to levels similar to those with AnxA1Ac2-26 treatment. These data demonstrate a molecular mechanism by which AnxA1Ac2-26 switches off the prothrombotic H3Cit+ NETotic drive and enhances the resolution.35 More important, AnxA1Ac2-26 suppression of ERK and Akt phosphorylation was absent in control neutrophils. These disparate effects of AnxA1Ac2-26 on SCD vs control phenotype may point toward the configurational plasticity of the FPR signaling axis in response to biased agonism.44 We also discovered AnxA1Ac2-26 to activate cleaved caspase-3 in SCD neutrophils, an effect that was abrogated in the presence of the specific caspase-3 inhibitor (Z-DEVD-FMK). Interestingly, these effects of AnxA1Ac2-26 on neutrophil apoptosis are also observed in an acute inflammation model of lipopolysaccharide-induced pleurisy.45 Taken together, our data provide a previously unknown phenomenon regarding the ability of AnxA1Ac2-26 to act as a natural homeostatic clearance ligand during an ensuing thrombotic process.
In summary, our study provides substantial evidence that targeting the AnxA1-FPR2/ALX pathway may provide a viable strategy for the management of thrombotic complications associated with SCD. More specifically, from an intravascular perspective targeting SCD neutrophils via the AnxA1-FPR2/ALX pathway reduces H3Cit+ NETotic drive, which plays a key role (directly or indirectly) in preventing the activation of various downstream processes including platelet aggregation, thereby enabling and promoting resolution. These unique findings may provide impetus to drug discovery programs based on resolution biologics in the management of not only SCD, but also other disease states with an underlying thromboinflammatory phenotype.
For original data, please contact Felicity N. E. Gavins (felicity.gavins@brunel.ac.uk).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank R. Mansour, S. Master, and V. Vanchiere (LSUHSC-S) for providing clinical input, and J. Walton, L. Latiolais, M. Linville, and L. Pipkin (LSUHSC-S) for performing the phlebotomies.
This work was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) grants HL134959-01A1 (F.N.E.G.); grants HL098435, HL133497, and HL141155 and National Institute of General Medical Sciences grant GM12130 (A.W.O.); and grant HL142604 (R.P.); American Heart Association grant 19PRE34380751 (Z.A.-Y.); and Royal Society Wolfson Foundation grant RSWF\R3\183001 (F.N.E.G.).
Authorship
Contribution: J.A. performed experiments, analyzed the data and wrote the manuscript; J.A., E.Y.S., S.A.V., Z.A.-Y., E.M.S., and G.K. performed experiments and analyzed the data; J.A., E.Y.S., S.A.V., A.W.O., R.P., R.P.H., D.N.G., P.K., and F.N.E.G. wrote the manuscript; and A.W.O. and F.N.E.G. provided reagents, designed experiments, analyzed the data, and supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Felicity N. E. Gavins, The Centre for Inflammation Research and Translational Medicine (CIRTM), Brunel University London, Kingston Lane, London UB8 3PH, United Kingdom; e-mail: felicity.gavins@brunel.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal