

Key Points
LUBAC accelerates B-cell lymphomagenesis through protection of DNA damage–induced apoptosis, thereby promoting AID-mediated mutations.
Inhibition of LUBAC by small molecules is a promising therapeutic strategy for B-cell lymphomas with NF-κB activation.

Abstract
The linear ubiquitin chain assembly complex (LUBAC) is a key regulator of NF-κB signaling. Activating single-nucleotide polymorphisms of HOIP, the catalytic subunit of LUBAC, are enriched in patients with activated B-cell–like (ABC) diffuse large B-cell lymphoma (DLBCL), and expression of HOIP, which parallels LUBAC activity, is elevated in ABC-DLBCL samples. Thus, to clarify the precise roles of LUBAC in lymphomagenesis, we generated a mouse model with augmented expression of HOIP in B cells. Interestingly, augmented HOIP expression facilitated DLBCL-like B-cell lymphomagenesis driven by MYD88-activating mutation. The developed lymphoma cells partly shared somatic gene mutations with human DLBCLs, with increased frequency of a typical AID mutation pattern. In vitro analysis revealed that HOIP overexpression protected B cells from DNA damage-induced cell death through NF-κB activation, and analysis of the human DLBCL database showed that expression of HOIP positively correlated with gene signatures representing regulation of apoptosis signaling, as well as NF-κB signaling. These results indicate that HOIP facilitates lymphomagenesis by preventing cell death and augmenting NF-κB signaling, leading to accumulation of AID-mediated mutations. Furthermore, a natural compound that specifically inhibits LUBAC was shown to suppress the tumor growth in a mouse transplantation model. Collectively, our data indicate that LUBAC is crucially involved in B-cell lymphomagenesis through protection against DNA damage–induced cell death and is a suitable therapeutic target for B-cell lymphomas.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most frequent lymphoma subtype in adults,1,2 and it is classified into 2 major categories, germinal center B-cell–like (GCB) DLBCL and activated B-cell–like (ABC)-DLBCL, based on the gene expression profiling.3-6 Since ABC-DLBCL has been shown to have a worse prognosis than GCB-DLBCL, new therapeutic strategies against ABC-DLBCL are warranted.7-9
ABC-DLBCL is characterized by constitutive NF-κB activation mediated by the B-cell receptor and Toll-like receptor (TLR) signaling pathways, and many oncogenic mutations within these pathways have been identified. Among them, activating mutations of MYD88, a signaling molecule in the TLR pathway, including L265P, are present in ∼30% of ABC-DLBCL cases10 and constitute the most frequent genetic abnormalities leading to aberrant NF-κB activation.
Protein ubiquitination is involved in multiple steps of the NF-κB pathway.11 The linear ubiquitin chain assembly complex (LUBAC), which consists of the catalytic subunit HOIP (RNF31) and 2 accessory subunits, HOIL-1L and SHARPIN, promotes NF-κB activation and protects against cell death by synthesizing unique N-terminally linked linear polyubiquitin chains.12-19 We previously reported that rare germline single-nucleotide polymorphisms (SNPs) in HOIP that increase LUBAC ligase activity are significantly enriched in ABC-DLBCL patients, suggesting that augmentation of LUBAC activity contributes to ABC-DLBCL pathogenesis.20 The majority of ABC-DLBCLs in patients with these HOIP SNPs also harbor the MYD88 L265P mutation. Given that LUBAC plays a pivotal role in NF-κB activation by linearly polyubiquitinating substrates, including the key NF-κB regulator NEMO,18,19,21-24 it is speculated that LUBAC collaborates with MYD88 signaling in B-cell lymphomagenesis by further amplifying NF-κB activation.
By analyzing published clinical RNA sequencing (RNA-seq) gene expression data,25 we found that expression of HOIP is elevated in human ABC-DLBCL (Figure 1A). As we previously reported that enforced expression of the catalytic subunit HOIP augments LUBAC functions,17 we assumed that LUBAC activation is frequently involved in the pathogenesis of ABC-DLBCL, independent of SNPs in HOIP. To clarify the roles of LUBAC played in the pathogenesis of B-cell lymphoma, we established a gene-engineered mouse with enforced expression of HOIP in B cells. We found that increased expression of HOIP enhanced LUBAC activity, and it facilitated generation of MYD88-mediated DLBCL, whereas it could not lead to B-cell lymphoma development per se. Elevated expression of LUBAC was suggested to accelerate B-cell lymphomagenesis, not only by activating NF-κB in concert with MYD88-mediated signals but also by protecting cells from DNA-damage–induced apoptosis. Importantly, the mutations in B-cell lymphomas that arose in mice expressing an oncogenic MYD88 mutant and high levels of HOIP partially overlap with those reported in human DLBCLs, indicating the biological similarity between these tumors. Finally, by using a mouse lymphoma model with secondary transplantation of a newly established lymphoma cell line, we demonstrated that LUBAC inhibition represents a novel and promising therapeutic strategy against B-cell lymphomas.
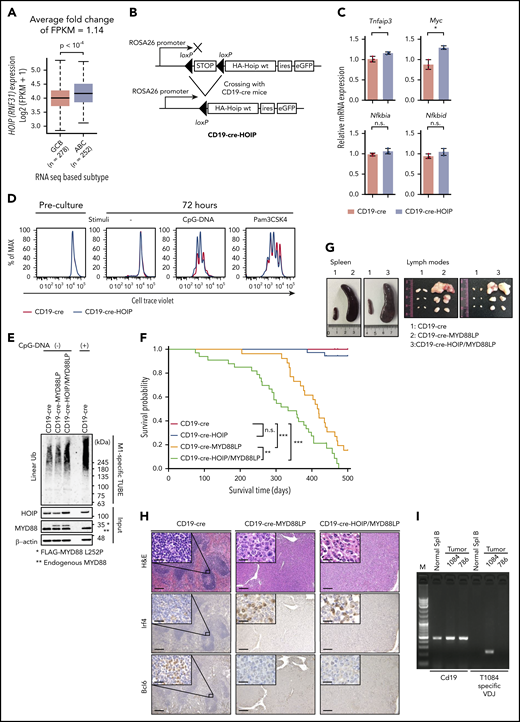
Augmented LUBAC expression accelerates oncogenic MYD88-mediated B-cell lymphomagenesis in mice. (A) Association of HOIP (RNF31) expression with cell of origin in human DLBCL. Boxes represent the median and the first and third quartiles, and whiskers represent the minimum and maximum of all data points. (B) Schematic representation of conditional expression of HOIP in mice. (C) Transcript levels of NF-κB target genes in unstimulated splenic B cells from mice (10 weeks old), normalized against Actb messenger RNA; n = 3 per genotype. Data are presented as means ± standard deviation (SD). (D) Cell Trace Violet–labeled splenic B cells were cultured with or without stimuli. (E) Cell lysates of splenic B cells derived from CD19-cre, CD19-cre-MYD88LP, and CD19-cre-HOIP/MYD88LP mice were subjected to Halo-tagged linear ubiquitin-specific tandem ubiquitin binding entity (M1-specific TUBE) binding and Halo tag–based purification and analyzed by immunoblotting. (F) Kaplan-Meier plots of survival of transgenic mice (n = 18, CD19-cre; n = 36, CD19-cre-HOIP; n = 26, CD19-cre-MYD88LP; and n = 33, CD19-cre-HOIP/MYD88LP). (G-I) Representative tumor involvement of lymphoid organs isolated from 9-month-old CD19-Cre-MYD88LP and CD19-cre-HOIP/MYD88LP mice. (G) Macroscopic appearance of spleens (left) and lymph nodes (right). (H) Representative H&E and immunohistochemical staining for Irf4 and Bcl6 of spleens (CD19-cre mice) or tumors (CD19 cre-MYD88LP and CD19-cre-HOIP/MYD88LP mice). Scale bars represent 200 μm (inset 20 μm). (I) Representative analyses of clonality. Tumor 1084–specific primers specifically amplified tumor 1084–specific V(D)J but did not amplify V(D)J from genomic DNA of normal splenic B cells or tumor 786. *P < .05, **P < .01, and ***P < .001, 2-tailed unpaired Student t test (A and C) or log-rank test (F). See also supplemental Figures 1 and 2 and supplemental Table 1. CpG, cytosine guanine dinucleotide; FPKM, fragments per kilobase million; HA, hemagglutinin; n.s., not significant; Spl, splenic; wt, wild-type.
Augmented LUBAC expression accelerates oncogenic MYD88-mediated B-cell lymphomagenesis in mice. (A) Association of HOIP (RNF31) expression with cell of origin in human DLBCL. Boxes represent the median and the first and third quartiles, and whiskers represent the minimum and maximum of all data points. (B) Schematic representation of conditional expression of HOIP in mice. (C) Transcript levels of NF-κB target genes in unstimulated splenic B cells from mice (10 weeks old), normalized against Actb messenger RNA; n = 3 per genotype. Data are presented as means ± standard deviation (SD). (D) Cell Trace Violet–labeled splenic B cells were cultured with or without stimuli. (E) Cell lysates of splenic B cells derived from CD19-cre, CD19-cre-MYD88LP, and CD19-cre-HOIP/MYD88LP mice were subjected to Halo-tagged linear ubiquitin-specific tandem ubiquitin binding entity (M1-specific TUBE) binding and Halo tag–based purification and analyzed by immunoblotting. (F) Kaplan-Meier plots of survival of transgenic mice (n = 18, CD19-cre; n = 36, CD19-cre-HOIP; n = 26, CD19-cre-MYD88LP; and n = 33, CD19-cre-HOIP/MYD88LP). (G-I) Representative tumor involvement of lymphoid organs isolated from 9-month-old CD19-Cre-MYD88LP and CD19-cre-HOIP/MYD88LP mice. (G) Macroscopic appearance of spleens (left) and lymph nodes (right). (H) Representative H&E and immunohistochemical staining for Irf4 and Bcl6 of spleens (CD19-cre mice) or tumors (CD19 cre-MYD88LP and CD19-cre-HOIP/MYD88LP mice). Scale bars represent 200 μm (inset 20 μm). (I) Representative analyses of clonality. Tumor 1084–specific primers specifically amplified tumor 1084–specific V(D)J but did not amplify V(D)J from genomic DNA of normal splenic B cells or tumor 786. *P < .05, **P < .01, and ***P < .001, 2-tailed unpaired Student t test (A and C) or log-rank test (F). See also supplemental Figures 1 and 2 and supplemental Table 1. CpG, cytosine guanine dinucleotide; FPKM, fragments per kilobase million; HA, hemagglutinin; n.s., not significant; Spl, splenic; wt, wild-type.
Methods
Mice
Tissue-specific HOIP transgenic mice (ROSA26-STOP-Hoip-ires-eGFP-pA) and MYD88 L252P transgenic mice (ROSA26-STOP-Myd88_L252P-ires-eGFP-pA) (accession No. CDB 1320K; http://www2.clst.riken.jp/arg/mutant%20mice%20list.html) were established as described in supplemental Methods (available on the Blood Web site). ROSA26-STOP-Hoip-ires-eGFP-pA or ROSA26-STOP-Myd88_L252P-ires-eGFP-pA transgenic mice were crossed with CD19-cre mice to express transgenic HOIP or MYD88 protein specifically in B cells from the pre–B-cell stage.26 All mice were maintained under specific-pathogen–free conditions. All animal protocols were approved by Kyoto University and RIKEN Center for Biosystems Dynamics Research.
Analysis of EGA and TCGA datasets
Clinical and RNA-seq gene expression data derived from the core set of 624 human DLBCL samples were obtained from the European Genome–phenome Archive (EGA) (dataset identifier [ID]: EGAD00001003600)25 and the Cancer Genome Atlas (TCGA). Whole-exome sequencing and RNA-seq data of 48 DLBCL samples (project ID: TCGA-DLBC) were obtained from the Broad Institute Firehose (http://gdac.broadinstitute.org)27,28 and analyzed as described in supplemental Methods and supplemental Tables 1 to 4.
Whole-exome sequencing
Lymphoma tissues obtained from the transgenic mouse model were analyzed by whole-exome sequencing using the SureSelect XT Mouse All Exon V2 kit (Agilent). Mouse tail DNA was used as a germline control. Sequence alignment to GRCm38/mm10 and mutation calling were performed using the Genomon pipeline (https://github.com/Genomon-Project) as previously described,29 with minor modifications. Candidate mutations with (1) P < .01 (Fisher’s exact test), (2) >4 variant reads in tumor samples, and (3) variant allele frequency (VAF) in tumor samples >0.05 or >0.2 were selected and manually reviewed. Human orthologs of mouse genes were assigned with the Ensembl 92 database. For each sample, the number of mutations, single-nucleotide variations (SNVs) at C:G base pairs, transitions, and SNVs within the WRCY/RGYW motifs were calculated and compared using the Brunner-Munzel test. Enrichment of SNVs at C/G within the WRCY/RGYW motifs in genes were performed by binomial test. Gene enrichment analyses were performed with Fisher’s exact test using the gene sets derived from supplemental Tables 5 to 8.30,31
AlphaScreen binding assay for LUBAC inhibitors
To search for inhibitors of linear polyubiquitination, an AlphaScreen-based high-throughput screening (HTS) system was established using N-terminally FLAG-His–tagged ubiquitin, C-terminally glutathione S-transferase–tagged ubiquitin, ubiquitin-activating enzyme E1, UbcH7 as the E2 ubiquitin–conjugating enzyme, and Petit-LUBAC or Petit-SHARPIN as the E3 ubiquitin ligase, as described in supplemental Methods.
Generation of a preclinical model for validation of the LUBAC inhibitor
The cell line HM876 was established as described in supplemental Methods. Transplantation of HM876 tumor cells was performed by subcutaneously injecting 5 × 106 cells into 6-week-old C57BL/6 females that were sublethally irradiated (4.5 Gy) 6 hours before transplantation. The animals were divided into 3 groups; the control group (n = 7) received intraperitoneal injection of dimethyl sulfoxide (DMSO) diluted in 5% glucose, and the other 2 groups were injected intraperitoneally with thiolutin diluted in 5% glucose at 2.5 or 5.0 mg/kg per day (n = 7). Thiolutin was administered from days 2 to 6 and days 9 to 13. On day 14, the animals were killed, and tumor weight was assessed.
Results
Augmented HOIP expression accelerates MYD88-mediated B-cell lymphomagenesis in mice
Based on the analysis of a publicly available database of gene expression in human B cells,32 HOIP is physiologically expressed throughout B-cell development (supplemental Figure 1A). However, we previously reported that 2 rare SNPs of HOIP that augment LUBAC activity were enriched specifically in patients with ABC-DLBCL.20 Because the protein expression level of HOIP determines the amount of other LUBAC subunits and activity of LUBAC,17 we hypothesized that HOIP plays a key role in the activation of the NF-κB pathway in ABC-DLBCL,21,33,34 irrespective of the SNP status. We examined expression of HOIP in RNA-seq data from 624 DLBCL samples in the EGA (dataset ID: EGAD00001003600)25 and found that its expression level was significantly higher in ABC-DLBCL than in GCB-DLBCL, as well as HOIL-1L and SHARPIN, which encode other subunits of LUBAC, although statistical significance was not observed for SHARPIN (Figure 1A; supplemental Figure 1B; supplemental Table 1). On the other hand, the expression level of OTULIN, encoding a linear ubiquitin-specific deubiquitinase that negatively regulates LUBAC signaling,35,36 was lower in ABC-DLBCL (supplemental Figure 1B; supplemental Table 1). These results are compatible with the increased LUBAC activity in ABC-DLBCL.
Therefore, to investigate the role of LUBAC in B-cell lymphomagenesis, we generated mice expressing high levels of HOIP specifically in B cells from the pre–B-cell stage (CD19-cre-HOIP) (Figure 1B). Bicistronic expression of eGFP allowed us to confirm that transgenic HOIP was specifically expressed in CD19+ B cells (supplemental Figure 1C). In CD19-cre-HOIP mice, elevated expression of HOIP increased expression of the other LUBAC subunits, thereby increasing the amount of trimeric LUBAC in B cells (supplemental Figure 1D). As expected, high levels of LUBAC increased expression of NF-κB target genes in splenic B cells, albeit mildly (Figure 1C; supplemental Figure 1E). Although some of the CD19-cre-HOIP mice aged >14 months showed splenomegaly, no lymphoma development was observed (supplemental Figure 1F).
The majority of ABC-DLBCLs with LUBAC-activating HOIP SNPs also carry the oncogenic MYD88 L265P mutation.20 Consistent with this, we found that B cells with enforced HOIP expression proliferated more efficiently by TLR stimulation (CpG-DNA and Pam3CSK4) (Figure 1D), which suggested the synergistic effect of LUBAC and MYD88 signaling. To evaluate the combinatorial effect of LUBAC and MYD88 L265P, we generated mice in which Myd88 L252P, the equivalent of human L265P, was expressed specifically in B cells from the pre–B-cell stage (CD19-cre-MYD88LP) (supplemental Figure 1G-I). MYD88 L252P increased proliferation and NF-κB activity of splenic B cells (supplemental Figure 1J-K). Hence, we assessed the synergistic effects of HOIP and MYD88 L252P on B-cell tumorigenesis in these mice. We evaluated the linear ubiquitin chains in B cells by using linear ubiquitin-specific tandem ubiquitin binding entity (supplemental Figure 1L)37,38 and found that the amount of linear ubiquitin chains was higher in splenic B cells of CD19-cre-HOIP/MYD88LP mice than in those of CD19-cre-MYD88LP mice (Figure 1E). As reported previously, B-cell–specific expression of MYD88 L252P led to decreased survival.39 We found that introduction of a HOIP transgenic allele significantly shortened the survival of CD19-cre-MYD88LP mice (Figure 1F).
Next, we examined pathological changes in CD19-cre-HOIP/MYD88LP and CD19-cre-MYD88LP mice. Mice with both genotypes developed marked lymphosplenomegaly, and histological examination of spleens and lymph nodes revealed infiltrates of lymphoid cells in these organs (Figure 1G-H). In addition, human DLBCL-like eGFP and CD19-positive large abnormal B cells diffusely infiltrated into the affected organs in mice with both genotypes (Figure 1H; supplemental Figure 2A). Assessment of V(D)J recombination of immunoglobulin heavy chain loci using a polymerase chain reaction–based method confirmed the presence of monoclonal B-cell populations in all involved tissues derived from 14 mice (4 CD19-cre-MYD88LP and 10 CD19-cre-HOIP/MYD88LP) (Figure 1I; supplemental Figure 2B; Table 1). Lymphomas developed in 4 CD19-cre-MYD88LP mice, and those in 8 of the 10 CD19-cre-HOIP/MYD88LP mice were positive for CD19, B220, and immunoglobulin M (IgM) and negative for CD138 by flow cytometric analysis (DLBCL-like lymphomas). These tumors were Irf4 positive and Bcl6 negative by immunohistochemical staining (Figure 1H). Moreover, sequence analysis of the variable regions of the clonally rearranged IgH gene revealed the presence of somatic hypermutations in most of the DLBCL-like lymphomas (supplemental Table 9). These results suggested that these tumors are mostly derived from post–germinal center B cells and are compatible with human ABC-DLBCL.40 Tumor cells of the remaining 2 CD19-cre-HOIP/MYD88LP mice exhibited a plasma-cell–like phenotype of CD19- and B220-negative and CD138-positive expression (Table 1). These results indicated that elevated expression of LUBAC potentially has a function to facilitate MYD88-mediated B-cell tumorigenesis.
Surface phenotypes of lymphomas in transgenic mice
| Tumor ID | Genotype | Surface phenotypes | Major site of involvement | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD19 | B220 | IgM | IgD | CD5 | CD21 | CD23 | CD38 | CD138 | Igκ | Igλ | |||
| 786 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Spleen |
| 950 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Spleen |
| 1032 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Extranodal (subcutaneous) |
| 1074 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Mesenteric lymph nodes |
| 1078 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | − | + | Peripheral lymph nodes |
| 1083 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peripheral lymph nodes |
| 1084 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peripheral lymph nodes |
| 1182 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | NA | NA | NA | − | + | − | Mesenteric lymph nodes |
| 1236 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Mesenteric lymph nodes |
| 1237 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Extranodal (subcutaneous) |
| 1289 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peritoneal |
| 1385 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peripheral lymph nodes |
| 876 | CD19-cre-HOIP/MYD88LP | − | − | + | − | − | − | − | + | + | + | − | Peritoneal |
| 1027 | CD19-cre-HOIP/MYD88LP | − | − | + | − | − | − | − | − | + | + | − | Peritoneal |
| Tumor ID | Genotype | Surface phenotypes | Major site of involvement | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD19 | B220 | IgM | IgD | CD5 | CD21 | CD23 | CD38 | CD138 | Igκ | Igλ | |||
| 786 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Spleen |
| 950 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Spleen |
| 1032 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Extranodal (subcutaneous) |
| 1074 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Mesenteric lymph nodes |
| 1078 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | − | + | Peripheral lymph nodes |
| 1083 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peripheral lymph nodes |
| 1084 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peripheral lymph nodes |
| 1182 | CD19-cre-HOIP/MYD88LP | + | + | + | − | − | NA | NA | NA | − | + | − | Mesenteric lymph nodes |
| 1236 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Mesenteric lymph nodes |
| 1237 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Extranodal (subcutaneous) |
| 1289 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peritoneal |
| 1385 | CD19-cre-MYD88LP | + | + | + | − | − | − | − | + | − | + | − | Peripheral lymph nodes |
| 876 | CD19-cre-HOIP/MYD88LP | − | − | + | − | − | − | − | + | + | + | − | Peritoneal |
| 1027 | CD19-cre-HOIP/MYD88LP | − | − | + | − | − | − | − | − | + | + | − | Peritoneal |
NA, not available.
High LUBAC expression is associated with increased accumulation of activation-induced cytidine deaminase (AID)–mediated somatic mutations
We did not find any significant macroscopic, histological, and immunophenotypic differences between DLBCL-like lymphomas that developed in CD19-cre-MYD88LP and CD19-cre-HOIP/MYD88LP mice (Figure 1G-H; Table 1). To understand the biological background of the accelerated MYD88-mediated lymphomagenesis in the condition of augmented LUBAC activity, we performed whole-exome sequencing analyses of genomic DNA isolated from 12 lymphomas derived from 12 different mice (8 from CD19-cre-HOIP/MYD88LP and 4 from CD19-cre-MYD88LP mice). Significantly more mutations were detected in the whole exons of lymphoma cells derived from CD19-cre-HOIP/MYD88LP mice than in those from CD19-cre-MYD88LP mice (Figure 2A; supplemental Figure 3A), indicating that elevated LUBAC expression increased the number of somatic mutations. Twenty-six genes were found to be recurrently mutated nonsynonymously in ≥2 samples among 12 mice. Twenty-three of them were recurrently mutated among those from 8 CD19-cre-HOIP/MYD88LP mice. Moreover, 6 of them, including Irf2bp2 and Pim1, were reported to be frequently mutated in human DLBCLs, especially in ABC-DLBCL (Figure 2B-C; supplemental Table 5).30 These results suggested that B-cell lymphomas generated in mice expressing HOIP with MYD88 mutant transgene share some genome mutations with human DLBCLs.25,30
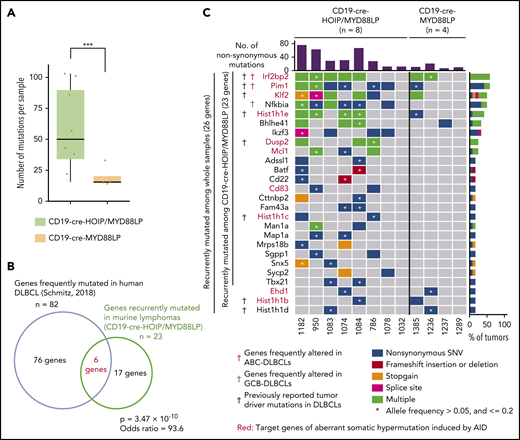
LUBAC facilitates somatic mutations in genes frequently mutated in human DLBCL. (A-C) Mutations with VAF >0.05 in tumor samples were selected and analyzed. (A) Numbers of total mutations including both synonymous and nonsynonymous mutations in each tumor sample. Boxes represent the median and the first and third quartiles, and whiskers represent the minimum and maximum of all data points. (B) Venn diagram depicting the overlap between genes recurrently mutated nonsynonymously in lymphoma cells derived from CD19-cre-HOIP/MYD88LP mice and those frequently detected in human DLBCL.30 (C) Mutational heatmap showing recurrently mutated genes across sequenced samples, color-coded according to 5 types of genetic alteration. Above the mutational heatmap, the bar graph indicates the number of nonsynonymous mutations in each sample. To the right of the mutational heatmap, the stacked bar graph indicates the percentage of tumors that have each mutations, using the same 5-color scheme. Target genes of aberrant somatic hypermutation induced by AID in human DLBCL are labeled in red. Black daggers indicates murine homologous genes frequently mutated in human DLBCL.30 Red and blue daggers indicate murine homolog of previously reported altered genes significantly enriched in human ABC-DLBCL and GCB-DLBCL, respectively.30 ***P < .001, Brunner-Munzel test (A). (B) Fisher’s exact test. See also supplemental Figure 3 and supplemental Table 5.
LUBAC facilitates somatic mutations in genes frequently mutated in human DLBCL. (A-C) Mutations with VAF >0.05 in tumor samples were selected and analyzed. (A) Numbers of total mutations including both synonymous and nonsynonymous mutations in each tumor sample. Boxes represent the median and the first and third quartiles, and whiskers represent the minimum and maximum of all data points. (B) Venn diagram depicting the overlap between genes recurrently mutated nonsynonymously in lymphoma cells derived from CD19-cre-HOIP/MYD88LP mice and those frequently detected in human DLBCL.30 (C) Mutational heatmap showing recurrently mutated genes across sequenced samples, color-coded according to 5 types of genetic alteration. Above the mutational heatmap, the bar graph indicates the number of nonsynonymous mutations in each sample. To the right of the mutational heatmap, the stacked bar graph indicates the percentage of tumors that have each mutations, using the same 5-color scheme. Target genes of aberrant somatic hypermutation induced by AID in human DLBCL are labeled in red. Black daggers indicates murine homologous genes frequently mutated in human DLBCL.30 Red and blue daggers indicate murine homolog of previously reported altered genes significantly enriched in human ABC-DLBCL and GCB-DLBCL, respectively.30 ***P < .001, Brunner-Munzel test (A). (B) Fisher’s exact test. See also supplemental Figure 3 and supplemental Table 5.
Notably, a significant proportion of recurrently mutated genes in lymphomas from CD19-cre-HOIP/MYD88LP mice were identified as known or predicted targets of aberrant somatic hypermutation induced by AID (Figures 2C and 3A; supplemental Table 6).25,30,31,41,42 AID plays essential roles in class-switch recombination and somatic hypermutation of the immunoglobulin genes during physiological B-cell maturation,43 and it is also involved in the pathogenesis of human DLBCL by introducing aberrant somatic hypermutations in nonimmunoglobulin genes.34,44-46 We found that somatic SNVs within WRCY/RGYW motifs or at C:G sites and transition mutations accumulated at higher levels in tumors derived from CD19-cre-HOIP/MYD88LP mice than in hose from CD19-cre-MYD88LP mice (Figure 3B; supplemental Figure 3B-D; supplemental Tables 7 and 8). Additionally, most of these mutations were located within 2 kb downstream of the transcription start site of each target gene (Figure 3C; supplemental Figure 3E). Since all of these characteristics are known as hallmarks of AID-mediated somatic mutations, it is indicated that AID-mediated mutagenesis is involved in B-cell lymphoma development in CD19-cre-HOIP/MYD88LP mice.47-49 Meanwhile, in the analyses of the whole-exome sequencing and RNA-seq data from 48 human DLBCL samples (project ID: TCGA-DLBC),27,28 we found that the frequency of AID-induced mutations positively correlated with the expression level of HOIP (Figure 3D-E; supplemental Table 4). Taken together, these results indicated that elevated expression of HOIP is associated with increased accumulation of somatic mutations of AID pattern, and augmented LUBAC activity is suspected to explain the facilitation of MYD88-mediated B-cell lymphomagenesis.
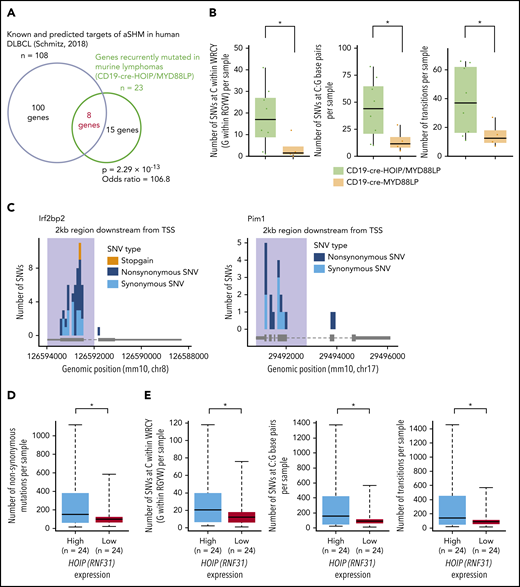
LUBAC facilitates aberrant somatic hypermutations mediated by AID. (A-C) Mutations with VAF >0.05 in tumor samples were selected and analyzed. (A) Venn diagram depicting the overlap between genes recurrently mutated in lymphoma cells derived from CD19-cre-HOIP/MYD88LP mice and murine homolog of known or predicted targets of aberrant somatic hypermutation mediated by AID.30 (B) Numbers of SNVs at C/G within the WRCY/RGYW motifs (left), and numbers of C:G (center) and transition mutations (right) in each tumor sample. (C) Mutation distribution in targeted genes observed in lymphoma cells derived from 8 CD19-cre-HOIP/MYD88LP mice. Shadows indicate the 2-kb region downstream of the transcription start site (TSS). (D) Numbers of nonsynonymous mutations in each human DLBCL sample. (E) Numbers of SNVs at C/G within the WRCY/RGYW motifs (left), and numbers of C:G (center) and transition mutations (right) in each human DLBCL sample. (D and E) Average fold change of FPKM (high vs low) = 1.36. Boxes represent the median and the first and third quartiles, and whiskers represent the minimum and maximum of all data points. *P < .05, **P < .01, and ***P < .001, Fisher’s exact test (A), Brunner-Munzel test (B), or 2-tailed unpaired Student t test (D and E). See also supplemental Figure 3 and supplemental Tables 6-8.
LUBAC facilitates aberrant somatic hypermutations mediated by AID. (A-C) Mutations with VAF >0.05 in tumor samples were selected and analyzed. (A) Venn diagram depicting the overlap between genes recurrently mutated in lymphoma cells derived from CD19-cre-HOIP/MYD88LP mice and murine homolog of known or predicted targets of aberrant somatic hypermutation mediated by AID.30 (B) Numbers of SNVs at C/G within the WRCY/RGYW motifs (left), and numbers of C:G (center) and transition mutations (right) in each tumor sample. (C) Mutation distribution in targeted genes observed in lymphoma cells derived from 8 CD19-cre-HOIP/MYD88LP mice. Shadows indicate the 2-kb region downstream of the transcription start site (TSS). (D) Numbers of nonsynonymous mutations in each human DLBCL sample. (E) Numbers of SNVs at C/G within the WRCY/RGYW motifs (left), and numbers of C:G (center) and transition mutations (right) in each human DLBCL sample. (D and E) Average fold change of FPKM (high vs low) = 1.36. Boxes represent the median and the first and third quartiles, and whiskers represent the minimum and maximum of all data points. *P < .05, **P < .01, and ***P < .001, Fisher’s exact test (A), Brunner-Munzel test (B), or 2-tailed unpaired Student t test (D and E). See also supplemental Figure 3 and supplemental Tables 6-8.
Augmented LUBAC activity overcomes cell death induced by DNA damage, thereby accelerating accumulation of somatic mutations
Although AID has a strong preference for immunoglobulin genes, it produces off-target DNA damage as well, resulting in aberrant somatic mutations.34,44,46,50 As shown above, AID-mediated mutations accumulated more prominently in CD19-cre-HOIP/MYD88LP mice compared with CD19-cre-MYD88LP mice. However, the expression levels of AID and the percentages of germinal center B cells in mesenteric lymph nodes were comparable between CD19-cre-HOIP/MYD88LP and CD19-cre-MYD88LP mice (Figure 4A). In addition, no correlation was found in the expression level of AID and HOIP in human DLBCLs (data not shown). Therefore, the altered expression level of AID did not seem to be the main reason for increased somatic mutations in lymphomas derived from CD19-cre-HOIP/MYD88LP mice.
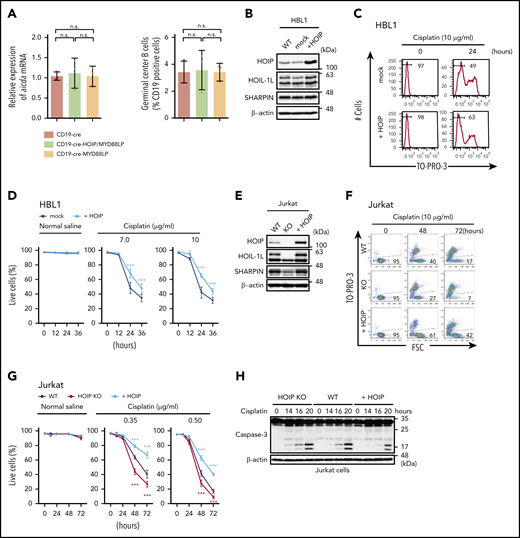
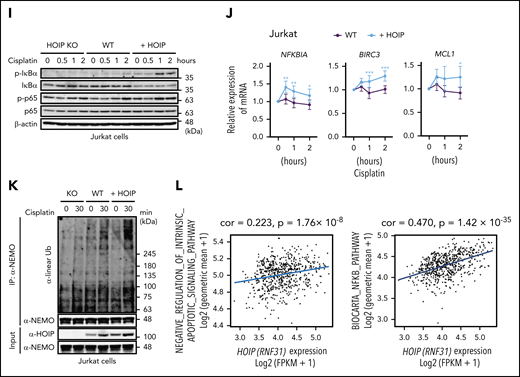
Augmented LUBAC activity overcomes cell death induced by DNA damage thereby accelerating accumulation of somatic mutations. (A) Transcript levels of Aicda, normalized to Actb (left panel), and percentages of germinal center B cells (right panel) in mesenteric lymph nodes from 10-week-old mice; n = 3 per genotype. Data represent means ± SD. (B) HOIP-overexpressing HBL1 cells were established, and immunoblot analyses were performed using lysates from wild-type (WT), mock-transfected, or HOIP-overexpressing HBL1 cells. (C) Live cells were analyzed by fluorescence-activated cell sorting using TO-PRO-3 staining. HBL1 cells were treated with or without 10 μg/mL cisplatin for 0 to 24 hours. (D) Percentage of live cells (±SD); n = 6 per group in 3 independent experiments. (E) Immunoblot analyses were performed using lysates from wild-type, HOIP-knockout (KO), or HOIP-overexpressing Jurkat cells. (F) Live cells were analyzed by fluorescence-activated cell sorting using FSC and TO-PRO-3 staining. Jurkat cells were treated with or without 0.5 μg/mL cisplatin for 0 to 72 hours. (G) Percentage of live cells (±SD) in 3 independent experiments. (H-J) Jurkat cells were treated with 3 μg/mL cisplatin for the indicated periods, followed by immunoblotting (H-I) or quantitative reverse-transcription polymerase chain reaction, normalized against Actb messenger RNA (mRNA) (J). (K) Jurkat cells were treated with 5 μg/mL cisplatin for the indicated periods. Whole-cell lysates were analyzed by anti-NEMO immunoprecipitation (IP), followed by immunoblotting using antibodies against linear polyubiquitin (Ub) and NEMO. (L) Correlation of expression of HOIP (RNF31) and negative regulation of intrinsic apoptotic signaling signature (left) and NF-κB signaling signature (right). *P < .05, **P < .01, and ***P < .001, 1-way analysis of variance (ANOVA) with Turkey’s post hoc test (A), 2-way ANOVA with Bonferroni post hoc test (D, G, and J), or Pearson’s correlation (L). See also supplemental Figures 4 and 5 and supplemental Tables 1 to 4.
Augmented LUBAC activity overcomes cell death induced by DNA damage thereby accelerating accumulation of somatic mutations. (A) Transcript levels of Aicda, normalized to Actb (left panel), and percentages of germinal center B cells (right panel) in mesenteric lymph nodes from 10-week-old mice; n = 3 per genotype. Data represent means ± SD. (B) HOIP-overexpressing HBL1 cells were established, and immunoblot analyses were performed using lysates from wild-type (WT), mock-transfected, or HOIP-overexpressing HBL1 cells. (C) Live cells were analyzed by fluorescence-activated cell sorting using TO-PRO-3 staining. HBL1 cells were treated with or without 10 μg/mL cisplatin for 0 to 24 hours. (D) Percentage of live cells (±SD); n = 6 per group in 3 independent experiments. (E) Immunoblot analyses were performed using lysates from wild-type, HOIP-knockout (KO), or HOIP-overexpressing Jurkat cells. (F) Live cells were analyzed by fluorescence-activated cell sorting using FSC and TO-PRO-3 staining. Jurkat cells were treated with or without 0.5 μg/mL cisplatin for 0 to 72 hours. (G) Percentage of live cells (±SD) in 3 independent experiments. (H-J) Jurkat cells were treated with 3 μg/mL cisplatin for the indicated periods, followed by immunoblotting (H-I) or quantitative reverse-transcription polymerase chain reaction, normalized against Actb messenger RNA (mRNA) (J). (K) Jurkat cells were treated with 5 μg/mL cisplatin for the indicated periods. Whole-cell lysates were analyzed by anti-NEMO immunoprecipitation (IP), followed by immunoblotting using antibodies against linear polyubiquitin (Ub) and NEMO. (L) Correlation of expression of HOIP (RNF31) and negative regulation of intrinsic apoptotic signaling signature (left) and NF-κB signaling signature (right). *P < .05, **P < .01, and ***P < .001, 1-way analysis of variance (ANOVA) with Turkey’s post hoc test (A), 2-way ANOVA with Bonferroni post hoc test (D, G, and J), or Pearson’s correlation (L). See also supplemental Figures 4 and 5 and supplemental Tables 1 to 4.
Previous studies showed that LUBAC has functions in protecting cells from genotoxic-damage–induced apoptosis and mediating NF-κB activation via plasma membrane receptors.51,52 Therefore, we examined the cell protective effect of LUBAC against genotoxic stress. Enforced expression of HOIP protected HBL1, a human ABC-DLBCL–derived cell line,7 and murine splenic B cells from cisplatin-induced cell death (Figure 4B-D; supplemental Figure 4A-B). We also found that enforced expression of LUBAC protected Jurkat cells from cisplatin-induced cell death by suppressing apoptosis (Figure 4E-H). It has been reported that LUBAC-mediated linear ubiquitination of NEMO is involved in genotoxic NF-κB activation and protects cells from DNA-damage–induced cell death.52-55 Indeed, expression of NF-κB target genes, including antiapoptotic genes, was modestly but significantly higher in splenic B cells of the CD19-cre-HOIP/MYD88LP mice than in those of CD19-cre-MYD88LP mice (supplemental Figure 5). Elevated levels of HOIP not only augmented activation of NF-κB and expression of several antiapoptotic genes but also enhanced linear ubiquitination of NEMO induced by cisplatin (Figure 4I-K; supplemental Figure 4C). In accordance with these results, RNA-seq analyses of human DLBCLs revealed that expression of HOIP positively correlated with expression of the genes involved in negative regulation of intrinsic apoptotic signaling, as well as those involved in the NF-κB pathway (Figure 4L). These results suggested that enhanced HOIP expression increases LUBAC activity and confers tumor cell resistance to cisplatin-induced DNA damage by modulating expression of genes associated with cell death.
On the other hand, AID-induced DNA alterations are repaired by the DNA double-strand break repair machinery, which also functions in repairing cisplatin-induced DNA damage.41,56 Based on the observation that somatic mutations of AID signature are increased in mouse lymphoma cells, it can be speculated that increased LUBAC activity would also promote the accumulation of oncogenic somatic mutations caused by AID and, in turn, facilitate the development of MYD88-mediated B-cell lymphoma.
LUBAC is an effective target for the treatment of DLBCL
Analysis of publicly available RNA-seq gene expression data25,30 suggested that the prognosis in primary refractory or relapsed DLBCL is worse in patients with high HOIP expression than in those with low HOIP expression (supplemental Figure 6A). Indeed, we showed that LUBAC is involved in B-cell lymphomagenesis by protecting cells from DNA-damage–induced apoptosis (Figure 4D,G; supplemental Figure 4B), which may lead to resistance to cytotoxic chemotherapies.51 We have previously described that LUBAC represents a novel therapeutic target against this cancer because reduction of LUBAC suppresses NF-κB activation and proliferation of ABC-DLBCL cells in vitro cell culture.20,57 We then tried to establish a preclinical model for B-cell lymphomas using CD19-cre-HOIP/MYD88LP mice to evaluate whether LUBAC is a promising drug target for B-cell lymphomas in vivo.
The cell line HM876, derived from a B-cell lymphoma with plasma-cell–like surface phenotype in a CD19-cre-HOIP/MYD88LP mouse, exhibited elevated expression of trimeric LUBAC and constitutive activation of NF-κB, manifested by phosphorylation and reduced expression of IκBα (Figure 5A; supplemental Figure 6B; Table 1). Using HM876 cells, we established a mouse lymphoma model by secondary transplantation of HM876 cells for in vivo drug evaluation (Figure 5B).
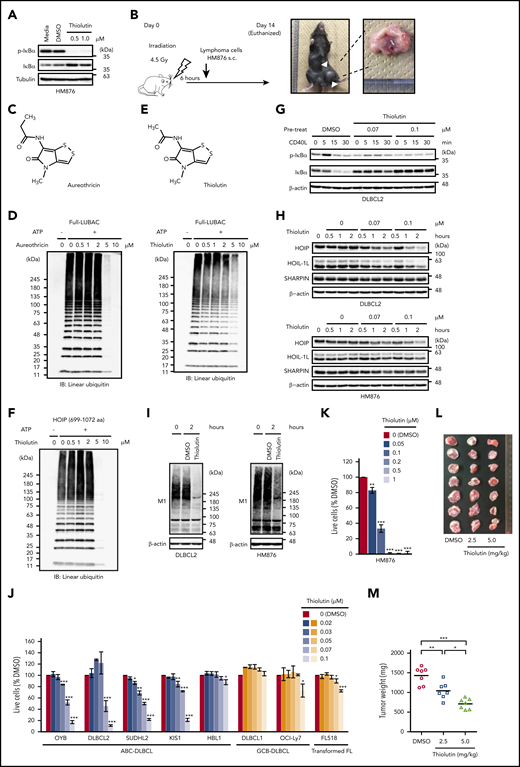
LUBAC is an effective target for the treatment of DLBCL. (A) Elevated phosphorylation and degradation of IκBα in unstimulated HM876 cells were suppressed by thiolutin. (B) Diagram of allogeneic transplantation model. s.c., subcutaneously. (C) Chemical structure of aureothricin. (D) Inhibition of LUBAC ligase activity by aureothricin and thiolutin in vitro. ATP, adenosine triphosphate; IB, immunoblotting. (E) Chemical structure of thiolutin. (F) Thiolutin inhibited linear polyubiquitination mediated by HOIP (amino acids 699-1072). (G) Upon stimulation of DLBCL2 cells with CD40 ligand, thiolutin suppressed phosphorylation and degradation of IκBα in a dose-dependent manner. DLBCL2 cells were exposed to thiolutin or DMSO for 2 hours and then stimulated with CD40 ligand (30 ng/mL) for the indicated times. (H) Levels of LUBAC components in DLBCL2 (upper panel) and HM876 (lower panel) cells treated with thiolutin were reduced in a dose-dependent manner. (I) Cell lysates of DLBCL2 (left panel) and HM876 (right panel) cells treated with or without thiolutin (0.1 μM) for 2 hours were analyzed by immunoblotting. Samples were probed with anti-linear ubiquitin specific antibody (LUB9). (J) Viability of DLBCL lines after 48 hours treatment with the indicated concentrations of thiolutin, normalized against that of control (DMSO-treated) cells. Data are means ± SD from three experiments. (K) Viability of HM876 cells after 48 hours treatment with the indicated concentrations of thiolutin, normalized against that of control (DMSO-treated) cells. Data are means ± SD from three experiments. (L and M) Thiolutin suppressed growth of lymphomas in vivo. (L) Gross appearance of engrafted tumors. (M) Tumor weight. Data represent means ± SD. *P < .05, **P < .01, and *** P < .001, 2-tailed unpaired Student t test (J and K) or one-way ANOVA with Turkey’s post hoc tests (M). See also supplemental Figure 6.
LUBAC is an effective target for the treatment of DLBCL. (A) Elevated phosphorylation and degradation of IκBα in unstimulated HM876 cells were suppressed by thiolutin. (B) Diagram of allogeneic transplantation model. s.c., subcutaneously. (C) Chemical structure of aureothricin. (D) Inhibition of LUBAC ligase activity by aureothricin and thiolutin in vitro. ATP, adenosine triphosphate; IB, immunoblotting. (E) Chemical structure of thiolutin. (F) Thiolutin inhibited linear polyubiquitination mediated by HOIP (amino acids 699-1072). (G) Upon stimulation of DLBCL2 cells with CD40 ligand, thiolutin suppressed phosphorylation and degradation of IκBα in a dose-dependent manner. DLBCL2 cells were exposed to thiolutin or DMSO for 2 hours and then stimulated with CD40 ligand (30 ng/mL) for the indicated times. (H) Levels of LUBAC components in DLBCL2 (upper panel) and HM876 (lower panel) cells treated with thiolutin were reduced in a dose-dependent manner. (I) Cell lysates of DLBCL2 (left panel) and HM876 (right panel) cells treated with or without thiolutin (0.1 μM) for 2 hours were analyzed by immunoblotting. Samples were probed with anti-linear ubiquitin specific antibody (LUB9). (J) Viability of DLBCL lines after 48 hours treatment with the indicated concentrations of thiolutin, normalized against that of control (DMSO-treated) cells. Data are means ± SD from three experiments. (K) Viability of HM876 cells after 48 hours treatment with the indicated concentrations of thiolutin, normalized against that of control (DMSO-treated) cells. Data are means ± SD from three experiments. (L and M) Thiolutin suppressed growth of lymphomas in vivo. (L) Gross appearance of engrafted tumors. (M) Tumor weight. Data represent means ± SD. *P < .05, **P < .01, and *** P < .001, 2-tailed unpaired Student t test (J and K) or one-way ANOVA with Turkey’s post hoc tests (M). See also supplemental Figure 6.
We next looked for small compounds that can inhibit the activity of LUBAC. HTS of 41 760 compounds in total using an AlphaScreen-based method (supplemental Figure 6C) identified aureothricin as a candidate LUBAC inhibitor (Figure 5C-D; supplemental Figure 6D-E). Because thiolutin is a molecular derivative of aureothricin, we examined both compounds in subsequent experiments (Figure 5C-E; supplemental Figure 6E).
Ubiquitin ligases are classified into 3 groups: RING, HECT, and RING-IBR-RING (RBR). LUBAC is an RBR ligase,58 and thiolutin inhibited catalytic RBR domain of HOIP in LUBAC (Figure 5F). Thiolutin did not noticeably inhibit the activities of a HECT ligase (Nedd4) or a RING ligase (cIAP2) in vitro and only slightly inhibited another RBR ligase (Parkin) when used in higher concentrations (supplemental Figure 6F), suggesting that its inhibitory function is specific for LUBAC. Thiolutin effectively suppressed CD40 ligand–mediated NF-κB activation and decreased the protein expression levels of LUBAC subunits at concentrations as low as 0.07 μM in ABC-DLBCL-derived DLBCL2 cells and HM876 cells (Figure 5G-H). Thiolutin did not decrease the amount of LUBAC components in the in vitro ubiquitination assay, nor did it obviously alter the gene expression of LUBAC subunits in DLBCL cells (supplemental Figure 6G-H). Thiolutin decreased the amount of linear ubiquitin chains without affecting the amount of K48- or K63-ubiquitin chains in cells (Figure 5I; supplemental Figure 6I) and appeared to decrease survival of ABC-DLBCL–derived cell lines more effectively than GCB-DLBCL–derived cell lines (Figure 5J). Likewise, thiolutin suppressed NF-κB activation and exerted significant cytotoxicity in HM876 cells in vitro (Figure 5A,K). These results suggested that the cytotoxic effect of thiolutin is mainly caused by the inhibition of LUBAC and the blockade of NF-κB signaling. To validate whether LUBAC is an effective therapeutic target for B-cell lymphomas, we intraperitoneally administered thiolutin to mice inoculated with HM876 cells and found that thiolutin significantly decreased the tumor burden (Figure 5L-M), indicating that inhibition of LUBAC represents an effective treatment of B-cell lymphomas with NF-κB activation. Moreover, our results demonstrate that our mouse model provides a valuable preclinical platform for the development of novel therapeutic approaches for B-cell lymphomas.
Discussion
Constitutive activation of NF-κB signaling is required for survival and proliferation of B cells and plays a crucial role in pathogenesis of ABC-DLBCL. Previously, we reported that rare germline SNPs in the gene encoding HOIP, which activates LUBAC ligase activity, are accumulated in individuals with ABC-DLBCL.20 We also found that expression of HOIP is elevated in ABC-DLBCL compared with GCB-DLBCL (Figure 1A). According to these observations, it is suspected that HOIP plays important roles broadly in ABC-DLBCL, whereas the precise contribution of HOIP and its functional protein complex LUBAC to lymphomagenesis has been poorly understood. Hence, we established mice models that allow enhanced expression of HOIP in B cells and assessed the roles of LUBAC in the pathogenesis of ABC-DLBCL. Because the LUBAC-activating SNPs of HOIP accumulate in patients with ABC-DLBCL with oncogenic MYD88 L265P mutation,20 we investigated the functional synergism of LUBAC and MYD88 in B-cell lymphomagenesis in mice. The results revealed that elevated expression of LUBAC accelerates MYD88-driven lymphomagenesis.
Our data showed that overexpression of HOIP increased NF-κB activation and enhanced proliferation of B cells upon MYD88-dependent signal activation (Figure 1C-D; supplemental Figure 1E), although it could not induce lymphomas in mice by itself (Figure 1F). However, enforced HOIP expression with oncogenic MYD88 L252P signaling facilitates tumor formation in mice, of which phenotype is DLBCL-like. More importantly, whole-exome sequence analysis of lymphomas developed in mice revealed higher somatic mutations in lymphomas with coexpression of HOIP, many of which are of AID signature and partially resemble those often seen in DLBCL patient samples (Figures 2B-C and 3A-C; supplemental Figure 3A-E). This suggests that the mouse model expressing HOIP and MYD88 L252P shares biological features with human DLBCL.
NF-κB is known to be activated by genotoxic damage, including that triggered by AID, and it aids in cell survival by inducing a variety of antiapoptotic genes.53 We previously reported that LUBAC-mediated linear ubiquitination of NEMO plays a key role in transducing nuclear genotoxic signals to the cytoplasm, in turn inducing genotoxic stress–induced NF-κB activation.52 As no significant difference in the expression levels of AID could be observed in B cells between CD19-cre-HOIP/MYD88LP and CD19-cre-MYD88LP mice (Figure 4A), we assume that the increased mutation burden in the tumors of CD19-cre-HOIP/MYD88LP mice is rather a result of a higher tolerability to genotoxic stress in the condition of higher catalytic activity of LUBAC (Figure 4D,G,J; supplemental Figure 4B). Therefore, elevated expression of LUBAC is considered to facilitate MYD88-mediated B-cell lymphomagenesis by conferring resistance to genotoxic stress in B cells and, in turn, augmenting the accumulation of oncogenic mutations. Our findings are compatible with previous reports that the apoptotic pathway counters MYD88-driven B-cell proliferation59 and that aberrant expression of the antiapoptotic protein Bcl-2 facilitates the generation of oncogenic MYD88-driven DLBCL.39,60,61
In accordance with our mouse experiment, the expression level of HOIP appears to positively correlate with the number of somatic mutations of AID signature in human DLBCL (Figure 3E). Since no correlation was found in the expression level of AID and HOIP in human DLBCLs (data not shown), augmented protection of DNA-damage–induced cell death by enhanced LUBAC expression might rather be a main cause of the high mutation rates in human DLBCLs with high HOIP expression. Gene mutations recurrently found in human DLBCLs could barely be detected in lymphomas generated in mice with oncogenic MYD88 transgene alone. This could be simply due to the limited number of tumors that could be analyzed in CD19-cre-MYD88LP mice or the potential differences in B-cell developmental stage in which oncogenic MYD88 transgenes are acquired between our model and human DLBCL.
We observed a population of mice with tumors of a more differentiated phenotype with CD138 and IgM expression (Table 1) and the presence of serum M proteins (data not shown). These tumors may possibly be the equivalent of lymphoplasmacytic lymphoma in humans, another B-cell malignancy in which MYD88 L265P is closely involved.62,63 Considering that these tumors were observed only in CD19-cre-HOIP/MYD88LP mice (Table 1), augmented LUBAC activity may have played some role in their development, whereas there is presently no data for the involvement of LUBAC in the pathogenesis of lymphoplasmacytic lymphoma.
Finally, we established a preclinical tumor transplantation model for human B-cell lymphomas using a cell line derived from a CD19-cre-HOIP/MYD88LP mouse. In this model, we showed that thiolutin, a specific inhibitor of LUBAC, suppressed the growth of lymphoma cells. Reduction or deletion of LUBAC counteracts resistance to cytotoxic chemotherapy, possibly by decreasing the expression of antiapoptotic genes that are induced by NF-κB activation.51
In summary, our results suggest that LUBAC has a function to accelerate B-cell lymphomagenesis by conferring resistance to genotoxic stress in B cells. We have also shown that, as a direct regulator of NF-κB pathway, LUBAC is an effective treatment target for lymphoma. Considering that resistance to genotoxic cell death is the common feature of chemorefractory cancers, the inhibition of LUBAC would represent a promising strategy for the treatment of multiple types of cancer.
Presented in abstract form at the 60th annual meeting of the American Society of Hematology, San Diego, CA, 3 December 2018.
For original data, please contact nishikor@kuhp.kyotou.ac.jp.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Y. Fuseya and H. Fujita for insightful discussions, Y. Sugahara for assistance with animal care, and I. Kuwahara for assistance with HTS for small-molecule inhibitors of LUBAC. They also thank A. Reddy and S. S. Dave (Duke University, Durham, NC) for providing DLBCL sequencing data. The results published here are in whole or part based upon data generated by the TCGA Research Network (https://www.cancer.gov/tcga). Preparation of paraffin-embedded sections was supported by the Anatomic Pathology Center of the Graduate School of Medicine of Kyoto University.
This work was supported in part by P-DIRECT, a Grant-in-Aid to K.I. from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; grants from AMED to S.O. (18cm0106501h0003) and A.T.-K. (18ck0106250h0002 and 17fk0108040h0002); and grants from KAKENHI from Japan Society for the Promotion of Science or MEXT to M.N. (15K09474), A.T.-K. (18H03992), M.Y. (18H05503), and K.I. (24112002, 25253019, 26670154, 17H06174, and 18H05499).
Authorship
Contribution: T.J., M.N., Y.S., K.I., and A.T.-K. conceived and designed the project; T.J. performed most of the experiments; S.M. provided essential experimental support; A.S. and H.K. supported generation of transgenic mice; Y.K., K.K., and S.O. performed whole-exome sequencing analyses of the lymphomas derived from transgenic mice; H.A. performed analyses of clinical RNA-seq data; N.K., T.O., K. Shin-ya, and M.Y. developed and performed the HTS for small-molecule inhibitors for LUBAC; T.N. and F.I. performed experiments on aureothricin and thiolutin; K. Sasaki advised on experimental design; and T.J., M.N., K.I., and A.T-K. wrote the manuscript with contributions from all other authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Momoko Nishikori, Department of Hematology and Oncology, Graduate School of Medicine, Kyoto University, 54 Shogoin Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan; e-mail: nishikor@kuhp.kyoto-u.ac.jp; and Kazuhiro Iwai, Department of Molecular and Cellular Physiology, Graduate School of Medicine, Kyoto University, Yoshida-konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan; e-mail: kiwai@mcp.med.kyoto-u.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal