

In this issue of Blood, 1 provide compelling evidence in a mouse model that enhanced activity of the linear ubiquitin chain assembly complex (LUBAC) can drive B-cell lymphomagenesis and may offer a new therapeutic target for the activated B-cell–like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL).
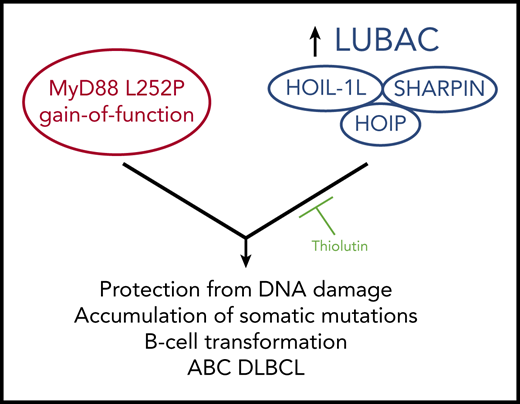
Jo et al demonstrate that an increase in LUBAC activity via elevated levels of HOIP cooperates with the MyD88 L252P activating mutation in mice to induce B lymphomas that resemble ABC DLBCL. The lymphomas are thought to arise in part from a HOIP-mediated cellular protection from DNA damage events, leading to the accumulation of somatic mutations that likely play a role in B-cell transformation. The authors identify Thiolutin as an inhibitor of HOIP E3 ligase activity in vitro that can inhibit lymphoma growth in vivo.
Jo et al demonstrate that an increase in LUBAC activity via elevated levels of HOIP cooperates with the MyD88 L252P activating mutation in mice to induce B lymphomas that resemble ABC DLBCL. The lymphomas are thought to arise in part from a HOIP-mediated cellular protection from DNA damage events, leading to the accumulation of somatic mutations that likely play a role in B-cell transformation. The authors identify Thiolutin as an inhibitor of HOIP E3 ligase activity in vitro that can inhibit lymphoma growth in vivo.
In recent years, the DLBCL field has seen substantial progress in mapping the network of signaling pathways, components, and intermediates involved in the chronic, oncogenic signaling observed in this lymphoma. The translation of these discoveries into better patient care will require answering how many of the signaling pathway components really drive B-cell transformation, and of these, how many can be targeted in novel therapeutic approaches. Genomic studies have made it abundantly clear that key signaling components are frequently mutated in human DLBCL biopsies with alterations that induce gain or loss of function, overexpression, or deletion.2 However, the high number and variety of mutations present in each biopsy make it difficult to pinpoint which genetic lesions are driving the disease. Mouse models are extremely valuable in this regard, offering the opportunity, first, to examine direct effects of single-gene lesions with appropriate syngeneic controls; second, to directly address synergistic functional interactions between 2 or more defined lesions; and third, to offer a preclinical platform to test new drugs.
Jo et al build upon previous work3-6 that highlighted the role of the LUBAC in normal and dysregulated antigen receptor signaling to NF-κB, a transcription factor of which inappropriate sustained activation is a signature feature of ABC DLBCL.7 LUBAC is an enzymatic complex, composed classically of HOIP, HOIL-1L, and Sharpin, which functions as an E3 ubiquitin ligase to conjugate substrates with linear ubiquitin chains at requisite steps in several pathways that signal to NF-κB, including the B-cell receptor pathway that is chronically activated by a variety of mechanisms in ABC DLBCL.8 Jo et al generated mice that overexpress the HOIP catalytic subunit of LUBAC specifically in the B-cell lineage. Although the increase in HOIP was insufficient to induce lymphoma by itself, HOIP elevation shortened lifespan and exacerbated the DLBCL-like disease elicited by B-cell–specific expression of MyD88 L252P, a gain-of-function mutation in a signaling protein that is frequently observed in human DLBCL and is known to constitutively signal to NF-κB.
In the presence of MyD88 L252P, HOIP elevation in B-cell lymphomas increased the observed number of genome-wide somatic mutations, many of which resembled known or predicted targets of aberrant somatic hypermutation induced by activation-induced cytidine deaminase, a critical player in normal antibody class-switch recombination and affinity maturation. The authors explain this observation by demonstrating that increased HOIP and LUBAC activity provide enhanced cellular protection from DNA damage, which likely allows genome-wide mutations to accumulate to a larger extent. Impressively, the authors go on to identify small molecules that can inhibit LUBAC activity in vitro and then demonstrate an inhibitory effect of one of them on tumor growth in vivo following transplantation into a recipient mouse. Thus, increased signaling from HOIP and MyD88 can cooperate to induce B-cell lymphoma, and agents that block HOIP E3 ligase activity can reduce the growth of tumors induced by that cooperation (see figure).
Several interesting possibilities are raised by these results. First, it will be fruitful to better understand how MyD88 L252P and HOIP elevation cooperate in lymphomagenesis. Both proteins signal to NF-κB. Does the outcome of their cooperation result merely from an overall enhanced level of signaling in B cells or does each factor provide a unique functional specificity? HOIP elevation appears to be specifically required to signal protection from genotoxic stress, but how precisely does the E3 ligase activity promote this program and what are its relevant substrates in this process? Clearly, HOIP elevation alone is insufficient to induce lymphoma. This likely reflects the fact that HOIP and LUBAC even when elevated in quantity are subject to regulation that must determine when the complex is enzymatically active and accessible to substrates, including the regulation imposed by CARD11 and its signaling cofactors.9 HOIP elevation likely poises cells to respond to signals that induce LUBAC action with amplified output. It will be beneficial to better understand how LUBAC activity is normally regulated in lymphocytes and how the oncogenic environment may alter this regulation.
Second, the promising preclinical effects of HOIP inhibitors are intriguing and suggest that further work should optimize the compounds described. Compounds that target LUBAC assembly may also be beneficial, as may those that target the action of key LUBAC substrates in signaling to NF-κB, including polyubiquitinylated Bcl10, polyubiquitinylated IKKγ, free linear polybuiquitin chains, and likely others.6,8,10 Although Jo et al show that LUBAC targeting is beneficial for lymphomas that elevate HOIP and harbor the MyD88 L252P mutation, other lymphomas that do not express MyD88 mutations or high HOIP levels should also be examined for susceptibility to LUBAC inhibitors. ABC DLBCLs that proliferate in response to chronic BCR signaling via a variety of molecular mechanisms will likely also depend on LUBAC to induce the quantity and quality of dysregulated NF-κB activity that drives lymphoma proliferation and survival.
Finally, the time course of the appearance of lymphomas in mice with elevated HOIP and MyD88 L252P suggests that other genetic lesions must be cooperating with these two to transform B cells. Jo et al identify a collection of genes mutated in the lymphomas they observe, some of which are frequently mutated in human DLBCL. Their study in the context of ABC DLBCL offers exciting prospects for future understanding and therapy for this disease.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal