

In this issue of Blood, describe their rationale for a combinatorial approach to fighting cytokine storm syndrome (CSS) with a one-two punch of glucocorticoids and the JAK/STAT inhibitor ruxolitinib (RUX) (see figure).1
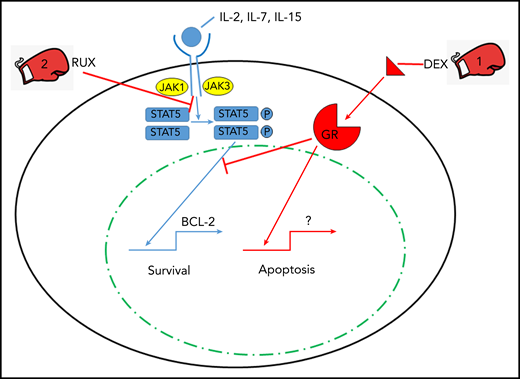
The one-two punch of dexamethasone (DEX) and RUX in treating CSS. Cytokines interleukin-2 (IL-2), IL-7, and IL-15 signal through lymphocyte cell surface cytokine receptors activating JAK proteins which leads to phosphorylation (P) of STAT5. Phosphorylated STAT5 homodimers enter the nucleus and transcriptionally activate prosurvival genes such as BCL-2. DEX triggers apoptosis via engagement of the glucocorticoid receptor (GR) and its nuclear signaling, and RUX inhibits JAK/STAT signaling to provide a one-two punch with DEX to overcome cytokine-induced survival signals that allow for lymphocyte cell death via apoptosis.
The one-two punch of dexamethasone (DEX) and RUX in treating CSS. Cytokines interleukin-2 (IL-2), IL-7, and IL-15 signal through lymphocyte cell surface cytokine receptors activating JAK proteins which leads to phosphorylation (P) of STAT5. Phosphorylated STAT5 homodimers enter the nucleus and transcriptionally activate prosurvival genes such as BCL-2. DEX triggers apoptosis via engagement of the glucocorticoid receptor (GR) and its nuclear signaling, and RUX inhibits JAK/STAT signaling to provide a one-two punch with DEX to overcome cytokine-induced survival signals that allow for lymphocyte cell death via apoptosis.
CSS is a frequently fatal hyperinflammatory state that arises during a number of underlying conditions such as autoimmune diseases and hematopoietic malignancies and can be triggered by viral infections.2 The pathophysiology of CSS is variable, but the most common mechanism relates to genetic defects in lymphocyte (natural killer and CD8 T cells) cytolysis via perforin3 and genes critical for delivering perforin to the immunologic synapse between the lytic lymphocyte and the infected antigen-presenting cell.4 The ineffective lysis of the innate immune cell by the cytolytic adaptive immune cell leads to a prolonged interaction of the cells that triggers an overly abundant array of proinflammatory cytokines5 and results in multi-organ failure.6,7 Treatment of CSS often includes glucocorticoids, including DEX, and etoposide, but patients can be refractory to this approach.
By using a murine perforin-deficient lymphocytic choriomeningitis virus–induced model of CSS, specifically hemophagocytic lymphohistiocytosis (HLH),8 Meyer and colleagues explored the reversal of cytokine-induced resistance of cytotoxic CD8 T cells to apoptosis mediated by DEX. The authors demonstrated ex vivo that CSS-associated IL-2, IL-4, IL-7, and IL-15 provide resistance to DEX-induced lymphocyte apoptosis through STAT5 triggering downstream of JAK activation via the cytokine receptors. Specifically, STAT5 phosphorylation correlates with increased transcripts for antiapoptotic proteins such as BCL-2. This is countered by the addition of RUX to IL-2 (representative STAT5 activator) and DEX-stimulated CD8 T cells, which results in decreased STAT5 signaling and subsequent reduction in lymphocyte resistance to DEX-induced apoptosis. One might argue with the choice of IL-2 as the primary STAT5 activator during HLH, but other dominant HLH/CSS cytokines, IL-6, and interferon-γ did not activate or phosphorylate STAT5 in the Meyer et al study. Moreover, IL-2 exposure did not result in increased phosphorylation of either STAT1 or STAT3, which provides some specificity to the experimental conditions. Thus, the combination of RUX and DEX was explored in the perforin-deficient HLH model of disease.
By using a predetermined suboptimal dose of DEX that induced only a modest reduction of HLH manifestations without controlling HLH disease (all mice died within 2 weeks), the addition of RUX resulted in 100% survival during the 18-day course of treatment. Indeed much of the benefit afforded these mice is attributable to RUX alone, with modest benefit in the presence of DEX. However, the combination of DEX and RUX decreased CD8 T-cell infiltration of the liver and resulted in the lowest levels of soluble CD25 (an HLH-04 and HScore diagnostic criterion).2 An added strength of this study was the exploration of human HLH plasma samples that demonstrated increased levels of cytokines (IL-2, IL-4, IL-7, IL-9, and IL-15) that signal through STAT5, although only levels of IL-7 and IL-15 reached statistical significance compared across the cohort to controls. Moreover, IL-2, IL-7, and IL-15 all conferred DEX resistance of human CD8 T cells to apoptosis in vitro in proportion to STAT5 phosphorylation. Human CD8 T cells exposed to HLH patient plasma, but not to control plasma, exhibited DEX resistance to apoptosis, thus providing a rationale for giving RUX combined with DEX for treating CSS.
A novel approach to treating CSS is timely because the planet is currently struggling with the disturbing scourge of the COVID-19 pandemic. The viral cause of COVID-19, SARS-CoV-2, has killed more than 350 000 individuals worldwide, many of whom suffered a CSS.9,10 The World Health Organization recommends against the use of glucocorticoids in treating COVID-19, but there are a variety of targeted approaches to inflammatory cytokines currently be explored in clinical trials. At present, it is unclear which is the best possible treatment of COVID-19 CSS, but there are several options available, including JAK/STAT inhibitors like RUX.9,10 In select COVID-19 populations (those with evidence of CSS), treatment with a combination of DEX and RUX during the hyperinflammatory stage of the illness may be an effective combination of therapies to knock out the deadly CSS.
Conflict-of-interest disclosure: R.Q.C. is co-principal investigator on an investigator-initiated clinical trial that is exploring anakinra treatment of CSS funded by Sobi. R.Q.C. has also received consulting fees from Sobi, Novartis, and Pfizer.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal