

Key Points
NUP98-fusion proteins directly regulate leukemia-associated gene expression programs in AML.
CDK6 expression is under direct transcriptional control of NUP98-fusions, and NUP98-fusion AML is particularly sensitive to CDK6 inhibition.

Abstract
Fusion proteins involving Nucleoporin 98 (NUP98) are recurrently found in acute myeloid leukemia (AML) and are associated with poor prognosis. Lack of mechanistic insight into NUP98-fusion–dependent oncogenic transformation has so far precluded the development of rational targeted therapies. We reasoned that different NUP98-fusion proteins deregulate a common set of transcriptional targets that might be exploitable for therapy. To decipher transcriptional programs controlled by diverse NUP98-fusion proteins, we developed mouse models for regulatable expression of NUP98/NSD1, NUP98/JARID1A, and NUP98/DDX10. By integrating chromatin occupancy profiles of NUP98-fusion proteins with transcriptome profiling upon acute fusion protein inactivation in vivo, we defined the core set of direct transcriptional targets of NUP98-fusion proteins. Among those, CDK6 was highly expressed in murine and human AML samples. Loss of CDK6 severely attenuated NUP98-fusion–driven leukemogenesis, and NUP98-fusion AML was sensitive to pharmacologic CDK6 inhibition in vitro and in vivo. These findings identify CDK6 as a conserved, critical direct target of NUP98-fusion proteins, proposing CDK4/CDK6 inhibitors as a new rational treatment option for AML patients with NUP98-fusions.
Introduction
Recurrent translocations involving chromosome 11 lead to fusion of the Nucleoporin 98 (NUP98) gene to more than 25 different recipient loci in leukemia.1 Although the overall frequency of NUP98-fusions is low, they are significantly overrepresented in pediatric acute myeloid leukemia (AML), where the expression of these fusions defines a clinically and molecularly homogenous group of patients that have a particularly bad prognosis.2-4 Lack of a detailed molecular understanding of the mechanism of action of NUP98-fusion proteins has hampered the development of tailored approaches to efficiently target this leukemia subgroup.
NUP98/NSD1 and NUP98/JARID1A are the most frequent NUP98-fusion proteins in pediatric AML,3,4 representing hybrid proteins joining the NUP98–N-terminus with the catalytic domain of the lysine methyltransferase NSD1 (KMT3B) or the lysine demethylase JARID1A (KDM5A), respectively.5,6 In addition, NUP98-fusions with the RNA helicase DDX10, the DNA topoisomerases TOP1, or the adapter protein PSIP1 were identified.7 NUP98 can also be fused to members of the Homeodomain transcription factor family, such as HOXA9, HOXD13, or PMX2.7 Patients with NUP98-fusions often carry additional mutations in NRAS, KRAS, KIT, MYC, or FLT3 genes, indicating a potential functional cooperation with NUP98-fusions.1,8 In mouse models, NUP98-fusions recapitulate principal aspects of the human disease, including increased self-renewal of hematopoietic progenitor cells, inhibition of myeloid differentiation, and high expression of HOXA genes.9-13
The endogenous NUP98 protein is an integral part of the nuclear pore complex, mediating nucleocytoplasmic transport of macromolecules across the nuclear membrane.14 In addition, NUP98 is involved in transcriptional regulation through interactions between the NUP98–N-terminus with the histone-modifying enzymes CBP/p300 and HDAC1.15,16 In hematopoietic cells, NUP98 regulates H3K4me3 levels by recruiting the SET1A histone methyltransferase complex to promoters of highly expressed genes.17 Unlike endogenous NUP98, NUP98-fusion proteins do not localize to the nuclear pore, but are distributed across the nucleus.18,19
Given the involvement of endogenous NUP98 in transcriptional control and the structural and functional diversity of C-terminal fusion partners among NUP98-fusion proteins, it was hypothesized that chromatin targeting of NUP98-fusion proteins depends on the NUP98–N-terminus, whereas functional properties encoded in or recruited by the C-terminal fusion partners might mediate specific gene regulatory functions that drive leukemogenesis.17 For instance, both NUP98/NSD1 and NUP98/JARID1A fusions contain plant homeodomain (PHD) domains, which recognize methylated lysine residues in histone tails.9,10 Mutations of either the PHD domain or the catalytic methyltransferase domain of NUP98/NSD1 abolished its oncogenic potential.9 Similarly, the PHD domain of the NUP98/JARID1A fusion protein was critical for oncogenic transformation.10 This indicates that both chromatin targeting and aberrant histone modifications are required for leukemogenesis driven by NUP98/NSD1 and NUP98/JARID1A. Different molecular mechanisms were proposed for other NUP98-fusion proteins. For instance, mutations in the DDX10 helicase domain impaired the transforming potential of a NUP98/DDX10 fusion protein, but the molecular underpinnings of this effect remain unclear.20 Thus, given the structural heterogeneity and the absence of direct DNA-binding domains among non-homeodomain NUP98-fusion proteins, it is possible that they hijack different molecular mechanisms to induce leukemic transformation. However, as molecularly different NUP98-fusion proteins can induce leukemia in vivo, different molecular pathways likely converge on a conserved set of target genes that is critical for induction and maintenance of NUP98-fusion AML. Therefore, we reasoned that the identification of actionable gene products among overlapping target gene sets of different NUP98-fusion proteins might provide new insight into developing more efficient therapeutic strategies to combat AML driven by NUP98-fusions.
To identify critical gene targets that are shared by diverse NUP98-fusion proteins, we generated novel mouse models allowing for Doxycycline (Dox)-regulatable expression of NUP98/NSD1, NUP98/JARID1A, and NUP98/DDX10. The transcriptome of these model systems recapitulated specific characteristics of gene expression programs of human NUP98-fusion AML. Time-resolved profiling of the transcriptional response upon NUP98-fusion protein withdrawal in vivo combined with chromatin immunoprecipitation and DNA sequencing (ChIP-seq) analyses identified a surprisingly small, conserved core of genes whose expression is maintained by all NUP98-fusion proteins. Within this core of direct transcriptional targets, we identified the druggable kinase CDK6, which was highly expressed in murine and human NUP98-fusion AML. CDK6 expression was required for initiation and maintenance of NUP98-fusion AML. Consistently, murine and patient-derived NUP98-fusion AML cells were sensitive to the CDK4/6 inhibitor Palbociclib, which caused rapid induction of apoptosis and cell-cycle arrest in NUP98-fusion–expressing cells in vitro and in vivo. Our study defines the common core transcriptional targets of diverse NUP98-fusion proteins in AML and identifies CDK6 as an actionable critical target in this leukemia subtype.
Methods
GraphPad Prism 8.0 (San Diego, CA) was used for statistical analyses. If not stated differently, a 2-tailed Student t test was used for P-value determination. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Results
Different NUP98-fusion proteins drive common gene expression programs
To investigate shared transcriptional programs of distinct NUP98-fusion proteins in vivo, we focused on the most recurrent NUP98-fusion proteins NUP98/NSD1 and NUP98/JARID1A, as well as on the molecularly distinct NUP98/DDX10 fusion, which lacks an annotated DNA-binding or chromatin-interaction domain.20 We cotransduced murine hematopoietic stem and progenitor cells (HSPCs) with retroviral constructs driving tetracycline-dependent expression of green fluorescent protein (GFP) and NUP98-fusion proteins, as well as constitutive expression of the tetracycline transactivator protein, luciferase, and activated NrasG12D, as this genetic lesion is commonly found in NUP98-rearranged AML patients1,8 (Figure 1A). This strategy ensures that only coinfection of both constructs enables NUP98-fusion protein expression.
![Different NUP98-fusion proteins induce a phenotypically similar AML-like disease in mice. (A) Schematic outline of the experimental strategy for the generation of transplantation models for Dox-repressible expression of NUP98-fusion proteins. (B-D) Representative bone marrow histology (B), bone marrow cytospin, ×400 original magnification (C), and flow cytometric analysis (D) from a moribund mouse transplanted with NUP98/DDX10-transformed murine HSPCs. (E) Relative abundance of indicated cell populations in the bone marrow from moribund mice expressing indicated NUP98-fusion proteins (mean ± standard deviation [SD]; n ≥ 4). (F) Kaplan-Meier survival curves of mice transplanted with murine HSPCs expressing indicated NUP98-fusion proteins (n ≥ 4). (G) Principal component analysis of the distances between steady-state gene expression data of NUP98-fusion–protein-driven leukemia cells (n ≥ 3). (H) Venn diagram illustrating the overlap of overexpressed genes of NUP98-fusion–protein-driven leukemia cells vs cells expressing NUP98-5′ (>threefold upregulated, P < .01; n ≥ 3). (I) Gene Set Enrichment Analysis (GSEA) indicating conservation of the gene signature found in murine NUP98-fusion–protein-driven leukemia with AML patients harboring NUP98 rearrangements. FDR, false discovery rate; NES, normalized enrichment score.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/4/10.1182_blood.2019003267/3/m_bloodbld2019003267f1.png?Expires=1767700620&Signature=0NkfWPDyrfuh5bjlnSkoaQX3qV9u4-gQYkWPtcsZxbteAckNVLJXw4eBDGD-IbYG9qzw9SkYzj4UOJ8CupH-bAxA2gWb5NtBVzrEMJoxvhcrJXHRZeHX8IJGntvtRAKbOxD1uU~EeL3ThpyzIyQmLVI3HGM5cA-La6H1w~upo5WnnVErsI1Xn85zBObvrbIAE7xYeo859LRhmDefK8ffLoy7sh40IiU7vwXUxoZvg0CLAnTqUg95vK1b39ze4UiAg7UEm0Rkdd2s30wFhVesGH8s2E7xXqkZcuicq435ivS~RATeWOd-03Filq3AgKbOreNKI00SkqqeuZRxFfQxMw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Different NUP98-fusion proteins induce a phenotypically similar AML-like disease in mice. (A) Schematic outline of the experimental strategy for the generation of transplantation models for Dox-repressible expression of NUP98-fusion proteins. (B-D) Representative bone marrow histology (B), bone marrow cytospin, ×400 original magnification (C), and flow cytometric analysis (D) from a moribund mouse transplanted with NUP98/DDX10-transformed murine HSPCs. (E) Relative abundance of indicated cell populations in the bone marrow from moribund mice expressing indicated NUP98-fusion proteins (mean ± standard deviation [SD]; n ≥ 4). (F) Kaplan-Meier survival curves of mice transplanted with murine HSPCs expressing indicated NUP98-fusion proteins (n ≥ 4). (G) Principal component analysis of the distances between steady-state gene expression data of NUP98-fusion–protein-driven leukemia cells (n ≥ 3). (H) Venn diagram illustrating the overlap of overexpressed genes of NUP98-fusion–protein-driven leukemia cells vs cells expressing NUP98-5′ (>threefold upregulated, P < .01; n ≥ 3). (I) Gene Set Enrichment Analysis (GSEA) indicating conservation of the gene signature found in murine NUP98-fusion–protein-driven leukemia with AML patients harboring NUP98 rearrangements. FDR, false discovery rate; NES, normalized enrichment score.
Different NUP98-fusion proteins induce a phenotypically similar AML-like disease in mice. (A) Schematic outline of the experimental strategy for the generation of transplantation models for Dox-repressible expression of NUP98-fusion proteins. (B-D) Representative bone marrow histology (B), bone marrow cytospin, ×400 original magnification (C), and flow cytometric analysis (D) from a moribund mouse transplanted with NUP98/DDX10-transformed murine HSPCs. (E) Relative abundance of indicated cell populations in the bone marrow from moribund mice expressing indicated NUP98-fusion proteins (mean ± standard deviation [SD]; n ≥ 4). (F) Kaplan-Meier survival curves of mice transplanted with murine HSPCs expressing indicated NUP98-fusion proteins (n ≥ 4). (G) Principal component analysis of the distances between steady-state gene expression data of NUP98-fusion–protein-driven leukemia cells (n ≥ 3). (H) Venn diagram illustrating the overlap of overexpressed genes of NUP98-fusion–protein-driven leukemia cells vs cells expressing NUP98-5′ (>threefold upregulated, P < .01; n ≥ 3). (I) Gene Set Enrichment Analysis (GSEA) indicating conservation of the gene signature found in murine NUP98-fusion–protein-driven leukemia with AML patients harboring NUP98 rearrangements. FDR, false discovery rate; NES, normalized enrichment score.
Upon transplantation, recipient mice developed an aggressive AML-like disease that was characterized by leukocytosis, anemia, thrombocytopenia, splenomegaly, and infiltration of leukemic blasts into bone marrow, spleen, and liver21 (Figure 1B-C; supplemental Figure 1A-C, available on the Blood Web site). Leukemia cells in bone marrow and spleen expressed high levels of the mature myeloid surface markers Mac-1 and Gr-1, whereas only a minor fraction of blasts was positive for the progenitor marker c-Kit and the lymphoid marker B220 (Figure 1D-E; supplemental Figure 1D-F). Expression of NUP98/DDX10, NUP98/NSD1, and NUP98/JARID1A led to rapid development of a lethal, phenotypically highly similar disease in vivo (median survival 34 to 84 days). In contrast, expression of mutated NrasG12D together with the N-terminal NUP98-fusion moiety lacking a fusion partner (referred to as NUP98-5′) did not lead to any disease development within 300 days (Figure 1F).
To study the transcriptomes associated with these 3 NUP98-fusion–protein-driven leukemia models, we performed RNA-sequencing on ex vivo–isolated leukemia cells. Global gene expression patterns of leukemia driven by NUP98/DDX10, NUP98/NSD1, and NUP98/JARID1A were closely related and differed significantly from NUP98-5′–expressing bone marrow cells (Figure 1G). Integration of these data with transcriptomes of purified murine HSPC populations22 revealed that global gene expression patterns of NUP98-fusion–driven leukemia cells resembled common myeloid progenitors and granulocyte-monocyte progenitors (supplemental Figure 2A). Expression of 244 genes was significantly increased (>threefold, P < .01) in all 3 NUP98-fusion–protein-driven leukemia models compared with NUP98-5′–expressing cells (Figure 1H; supplemental Table 1). This core set of genes upregulated in NUP98-fusion leukemia was enriched for regulators of normal and aberrant self-renewal, including members of the Hoxa gene family as well as the transcription factor Meis123 (supplemental Figure 2B). Importantly, the gene expression programs in murine NUP98-fusion–protein-driven AML models recapitulated specific transcriptional signatures observed in AML patients with NUP98-rearrangements, highlighting the clinical relevance of our leukemia models (Figure 1I; supplemental Figure 2C-D).
These data show that expression of distinct NUP98-fusion proteins induce a phenotypically similar, aggressive AML-like disease in vivo that features characteristic transcriptional patterns of human AML with NUP98-fusion proteins.
Maintenance of NUP98-fusion leukemia is dependent on sustained oncogene expression
Next, we aimed to investigate whether expression of NUP98-fusion proteins is required for disease maintenance. Murine leukemia cells driven by NUP98/NSD1, NUP98/JARID1A, or NUP98/DDX10 induced a fatal disease with very similar characteristics in secondary recipients (supplemental Figure 3A-D). We used these tetracycline-responsive models to induce transcriptional shutdown of NUP98-fusion expression in established leukemias by Dox administration in vivo (Figure 2A). Bioluminescence imaging confirmed that the disease rapidly progressed in untreated secondary recipient mice (Figure 2B). In contrast, Dox-induced transcriptional shutdown of NUP98-fusion expression led to disease regression, translating into a significant survival benefit (Figure 2B-C; supplemental Figure 4A-B). The rapid, time-dependent decrease of GFP expression upon Dox treatment indicates that NUP98-fusion protein expression is downregulated with similar kinetics in leukemia cells, as GFP is translationally coupled to NUP98-fusions via an internal ribosome entry site (IRES) (Figure 2D; supplemental Figure 4C). Loss of NUP98-fusion protein expression induced higher levels of the myeloid surface markers Mac-1 and Gr-1, whereas levels of the progenitor cell marker c-Kit were decreasing within 5 days (Figure 2E). In line with this, Dox-treated leukemia cells showed strong morphological signs of terminal myeloid differentiation (Figure 2F). Likewise, in vitro cultured leukemia cells derived from the bone marrow of our in vivo models remained fully dependent on the expression of NUP98-fusion proteins, as Dox-induced downregulation of fusion protein expression resulted in growth inhibition and induction of apoptosis (supplemental Figure 4D-G).
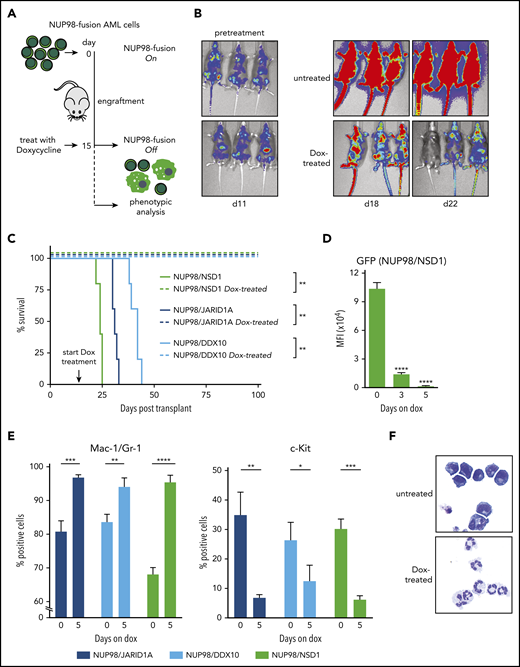
NUP98-fusion–protein-driven AML is highly dependent on sustained oncoprotein expression in vivo. (A) Schematic illustration of the experimental workflow to induce downregulation of oncogene expression in vivo. GFP/CD45.2-positive leukemia cells (1 × 106) were transplanted into sublethally irradiated secondary recipients. Animals were treated with Dox (4 mg/mL) after an engraftment period of 15 days. (B) Representative bioluminescence imaging of untreated vs Dox-treated mice transplanted with NUP98/NSD1-expressing leukemia cells. (C) Kaplan-Meier survival curves with statistical analyses using Log-rank tests of mice transplanted with leukemia cells expressing indicated NUP98-fusion proteins receiving either no treatment or Dox (4 mg/mL) (n ≥ 4). (D-E) Flow cytometric analyses of bone marrow–derived leukemia at indicated time points after Dox treatment of mice (mean ± SD; n = 3). (D) Quantification of mean fluorescence intensity (MFI) of GFP. (E) Quantification of the myeloid differentiation markers Mac-1/Gr-1 and the progenitor cell marker c-Kit. (F) Representative cytospin images of untreated vs Dox-treated (8 days) NUP98/JARID1A-expressing cells in vitro, ×400 original magnification. *P < .05, **P < .01, ***P < .001, ****P < .0001.
NUP98-fusion–protein-driven AML is highly dependent on sustained oncoprotein expression in vivo. (A) Schematic illustration of the experimental workflow to induce downregulation of oncogene expression in vivo. GFP/CD45.2-positive leukemia cells (1 × 106) were transplanted into sublethally irradiated secondary recipients. Animals were treated with Dox (4 mg/mL) after an engraftment period of 15 days. (B) Representative bioluminescence imaging of untreated vs Dox-treated mice transplanted with NUP98/NSD1-expressing leukemia cells. (C) Kaplan-Meier survival curves with statistical analyses using Log-rank tests of mice transplanted with leukemia cells expressing indicated NUP98-fusion proteins receiving either no treatment or Dox (4 mg/mL) (n ≥ 4). (D-E) Flow cytometric analyses of bone marrow–derived leukemia at indicated time points after Dox treatment of mice (mean ± SD; n = 3). (D) Quantification of mean fluorescence intensity (MFI) of GFP. (E) Quantification of the myeloid differentiation markers Mac-1/Gr-1 and the progenitor cell marker c-Kit. (F) Representative cytospin images of untreated vs Dox-treated (8 days) NUP98/JARID1A-expressing cells in vitro, ×400 original magnification. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Taken together, these data demonstrate that sustained expression of the driver oncogene is required for propagation of NUP98-fusion AML models in vivo and in vitro, and that loss of NUP98-fusion expression results in rapid terminal differentiation of leukemia cells and disease regression.
NUP98-fusion proteins regulate overlapping sets of transcriptional targets
To investigate global effects on gene expression upon transcriptional inactivation of NUP98-fusion expression, we isolated leukemia cells from secondary recipients 3 and 5 days after Dox administration and performed RNA-seq analysis (Figure 3A). Time-resolved analysis of conserved transcriptional kinetics after oncogene withdrawal in all 3 models revealed 4 consistent patterns of gene regulation: 2819 genes displayed early and pronounced transcriptional changes 3 days upon NUP98-fusion shutdown, following a pattern of steady down- or upregulation after oncogene withdrawal (Figure 3B left panels). In contrast, changes in the expression of 2847 genes were only observed after 5 days upon Dox-mediated loss of the driver oncogene (Figure 3B right panels). Although the first category likely contains the majority of direct gene targets of NUP98-fusion proteins, the latter category might reflect changes in gene expression that are further downstream of oncogene inactivation. Loss of NUP98-fusion proteins affected the differentiation trajectories of NUP98-fusion leukemias in the context of normal hematopoietic lineages. Oncogene shutdown in NUP98-fusion–transformed leukemia cells resulted in global transcriptional reprogramming toward mature monocytes/macrophages or granulocytes (supplemental Figure 5A). In line with this, we found gradual upregulation of gene sets characteristic of differentiated immune cells after downregulation of NUP98-fusion proteins, including Tnfa/NF-kB signaling, TLR activation, and interleukin signaling (Figure 3C). These data confirm that leukemia cells lose characteristics of immature hematopoietic progenitors after withdrawal of NUP98-fusion protein expression and initiate differentiation along monocytic and granulocytic lineages.
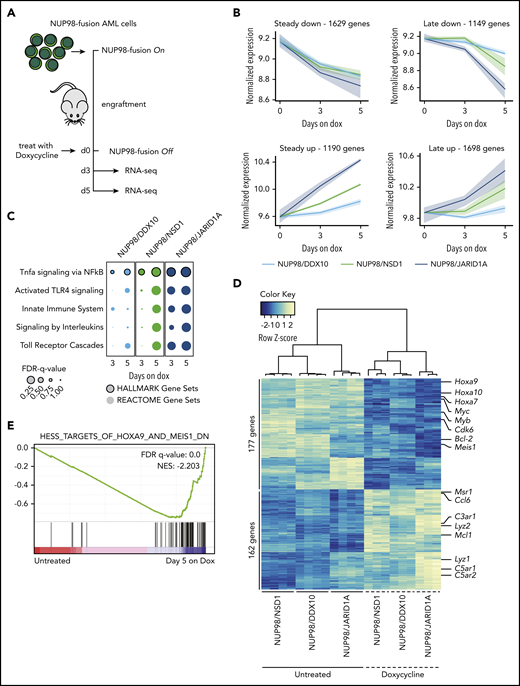
Different NUP98-fusion proteins regulate a common core of transcriptional targets. (A) Schematic illustration of the experimental workflow to investigate NUP98-fusion protein-dependent transcriptional programs. NUP98-fusion–protein-driven leukemia cells were transplanted into secondary recipient mice. Mice were treated with Dox for 3 or 5 days after an initial engraftment phase of 15 days; leukemia cells were sorted from the bone marrow based on GFP/CD45.2 expression, and gene expression was analyzed by RNA-seq (n ≥ 3). (B) Representation of dynamics of global gene expression changes after 3 and 5 days of Dox-induced NUP98-fusion–protein-repression. Each line represents the harmonized mean of the median expression of all genes within the cluster of distinct fusion-protein–driven cancer cells. Maximum/minimum medians are indicated by the colored area. (C) GSEA indicating induction of myeloid differentiation upon fusion protein withdrawal. (D) Heat map of 339 commonly up- and downregulated genes in all 3 NUP98-fusion–protein-driven models after 5 days of Dox-mediated fusion-protein repression (≥1.5-fold change, P < .01). (E) GSEA illustrating the enrichment of HOXA9/MEIS1-target genes in cells expressing NUP98/NSD1.
Different NUP98-fusion proteins regulate a common core of transcriptional targets. (A) Schematic illustration of the experimental workflow to investigate NUP98-fusion protein-dependent transcriptional programs. NUP98-fusion–protein-driven leukemia cells were transplanted into secondary recipient mice. Mice were treated with Dox for 3 or 5 days after an initial engraftment phase of 15 days; leukemia cells were sorted from the bone marrow based on GFP/CD45.2 expression, and gene expression was analyzed by RNA-seq (n ≥ 3). (B) Representation of dynamics of global gene expression changes after 3 and 5 days of Dox-induced NUP98-fusion–protein-repression. Each line represents the harmonized mean of the median expression of all genes within the cluster of distinct fusion-protein–driven cancer cells. Maximum/minimum medians are indicated by the colored area. (C) GSEA indicating induction of myeloid differentiation upon fusion protein withdrawal. (D) Heat map of 339 commonly up- and downregulated genes in all 3 NUP98-fusion–protein-driven models after 5 days of Dox-mediated fusion-protein repression (≥1.5-fold change, P < .01). (E) GSEA illustrating the enrichment of HOXA9/MEIS1-target genes in cells expressing NUP98/NSD1.
The extent of global gene expression changes 5 days upon Dox-mediated oncogene shutdown was significantly different (1110 genes for NUP98/DDX10, 2014 genes for NUP98/NSD1, 3304 genes for NUP98/JARID1A), and only a small set of genes was commonly regulated across different NUP98-fusion models. Among 2340 genes, 162 were jointly upregulated upon transcriptional shutdown of the 3 NUP98-fusions. Likewise, 177 of 2238 genes were downregulated after oncogene withdrawal in all 3 models. Thus, the expression of a core set of 339 genes is commonly regulated by NUP98-fusion proteins (Figure 3D; supplemental Figure 5B-C; supplemental Table 2). Among the 177 genes whose expression was maintained by all NUP98-fusion proteins, we found several known regulators of progenitor self-renewal, including Hoxa9, Meis1, Myb, and Bcl-223,24 (Figure 3D). Consistently, targets of HOXA9, MEIS1, and E2F transcription factors were downregulated upon NUP98-fusion protein withdrawal (Figure 3E; supplemental Figure 5D).
Altogether, time-resolved mapping of transcriptional responses upon oncogene withdrawal showed that different NUP98-fusion proteins control distinct and complex transcriptional programs that converge on a small set of common gene targets.
Cdk6 is a conserved direct transcriptional target of NUP98-fusion proteins
We next investigated whether NUP98-fusion proteins directly participate in the regulation of these genes. Coexpression of an HA-tagged NUP98/JARID1A variant and NrasG12D in mouse HSPCs induced an aggressive AML-like disease in vivo (supplemental Figure 6A-B). Leukemic blasts displayed high levels of Mac-1/Gr-1, together with low levels of c-Kit and B220 (supplemental Figure 6C-D). As these results show that the N-terminal HA-tag does not interfere with the oncogenic potential of NUP98/JARID1A, we analyzed global chromatin association of the NUP98/JARID1A-fusion protein by ChIP-seq using HA antibodies. ChIP-quantitative polymerase chain reaction (ChIP-qPCR) confirmed NUP98/JARID1A binding to the Hoxa7 promoter region10 (supplemental Figure 6E). Analysis of ChIP-seq data showed that 78% of NUP98/JARID1A-bound sites are located within promoters (2554 regions), whereas only 14% (439 regions) were found in intronic regions. Four percent (128 regions) localized to distal intergenic regions (Figure 4A; supplemental Table 3). Within promoter regions, NUP98/JARID1A binding was enriched around annotated transcriptional start sites (TSS) (Figure 4B-C). We detected NUP98/JARID1A binding in the promoters of genes that are important regulators of NUP98-fusion AML, such as the Hoxa cluster and Meis1 (Figure 4D).

NUP98-fusion proteins regulate the expression and bind the promoter of Cdk6. (A) Pie chart of HA-NUP98/JARID1A chromatin binding events (%) in the indicated chromatin contexts. (B) Signal distribution of HA-NUP98/JARID1A normalized to the input signal, between the TSS and transcription end sites (TES) ± 3 kb up- and downstream. (C) Heat map showing HA-NUP98/JARID1A chromatin binding within ± 3 kb of annotated TSS. All annotated murine genes are plotted on the y-axis. (D) Representative ChIP-seq tracks showing the binding of HA-NUP98/JARID1A within the Hoxa cluster (top) and the promoter region of Meis1 (bottom). (E) Venn diagram showing the overlap of genes with NUP98/JARID1A-occupied promoter regions, all downregulated genes upon repression of NUP98-fusion proteins, and all genes that are highly overexpressed in NUP98-fusion–protein-driven mouse models. (F) Western blot analysis of CDK6 levels in bone marrow of mice transplanted with leukemia cells expressing different NUP98-fusion proteins, compared with bone marrow of wild-type (WT) mice. (G) Expression kinetics of Cdk6 in ex vivo–derived leukemia cells from the bone marrow upon Dox-mediated repression of the indicated NUP98-fusion proteins (mean ± SD; n = 3). (H) Western blot analysis of CDK6 levels of in vitro cultured NUP98/JARID1A-driven leukemia cells at indicated time points during Dox-mediated fusion protein repression (0.5 µg/mL). (I) Representative ChIP-seq tracks showing the binding of indicated NUP98-fusion proteins within the promoter region of Cdk6. *P < .05, **P < .01, ***P < .001, ****P < .0001.
NUP98-fusion proteins regulate the expression and bind the promoter of Cdk6. (A) Pie chart of HA-NUP98/JARID1A chromatin binding events (%) in the indicated chromatin contexts. (B) Signal distribution of HA-NUP98/JARID1A normalized to the input signal, between the TSS and transcription end sites (TES) ± 3 kb up- and downstream. (C) Heat map showing HA-NUP98/JARID1A chromatin binding within ± 3 kb of annotated TSS. All annotated murine genes are plotted on the y-axis. (D) Representative ChIP-seq tracks showing the binding of HA-NUP98/JARID1A within the Hoxa cluster (top) and the promoter region of Meis1 (bottom). (E) Venn diagram showing the overlap of genes with NUP98/JARID1A-occupied promoter regions, all downregulated genes upon repression of NUP98-fusion proteins, and all genes that are highly overexpressed in NUP98-fusion–protein-driven mouse models. (F) Western blot analysis of CDK6 levels in bone marrow of mice transplanted with leukemia cells expressing different NUP98-fusion proteins, compared with bone marrow of wild-type (WT) mice. (G) Expression kinetics of Cdk6 in ex vivo–derived leukemia cells from the bone marrow upon Dox-mediated repression of the indicated NUP98-fusion proteins (mean ± SD; n = 3). (H) Western blot analysis of CDK6 levels of in vitro cultured NUP98/JARID1A-driven leukemia cells at indicated time points during Dox-mediated fusion protein repression (0.5 µg/mL). (I) Representative ChIP-seq tracks showing the binding of indicated NUP98-fusion proteins within the promoter region of Cdk6. *P < .05, **P < .01, ***P < .001, ****P < .0001.
As promoter binding by regulatory proteins often controls the expression of the corresponding gene, we reasoned that any direct transcriptional target gene of NUP98-fusion proteins would need to fulfill the following criteria: (i) it is overexpressed in NUP98-fusion AML compared with normal HSPCs; (ii) its expression is downregulated upon shutdown of fusion protein expression; and (iii) it exhibits NUP98-fusion protein binding in its promoter. Using this stringent approach, only 12 candidate genes classified as common direct transcriptional targets of NUP98-fusion proteins, including several genes of the Hoxa cluster and Meis1 (Figure 4E). However, the cyclin-dependent kinase 6 (Cdk6) gene particularly stood out. CDK6 was shown to be a critical target in MLL-fusion–expressing AML25 and is highly overexpressed in different AML subtypes (supplemental Figure 6F). Cdk6 was among the highest overexpressed genes in all 3 murine NUP98-fusion–protein-driven AML models (Figure 4F; supplemental Figure 6G), and its expression was rapidly downregulated upon loss of the driver fusion protein (Figure 4G-H). Cdk6 promoter binding was not restricted to NUP98/JARID1A, as reanalysis of published ChIP-seq datasets revealed that NUP98/TOP1 and NUP98/HOXD13 fusion proteins were also associated with the same genomic region19 (Figure 4I).
These data show that orthogonal integration of RNA-seq and ChIP-seq datasets efficiently identifies direct transcriptional targets of NUP98-fusion proteins and highlight Cdk6 as a regulated, direct target gene of multiple NUP98-fusion proteins.
Loss of Cdk6 impairs NUP98-fusion–protein-driven leukemogenesis
To investigate whether Cdk6 expression is required for development and maintenance of NUP98-fusion driven AML, we reengineered our transplantation-based leukemia models to combine constitutive NUP98-fusion expression with the presence of the reverse tet-transactivator (rtTA3) to be able to modulate Cdk6 expression by inducible RNAi26 (supplemental Figure 7A). Mice transplanted with cells expressing these vectors developed an aggressive AML-like disease that showed similar features as our other NUP98-fusion–protein-driven in vivo models (supplemental Figure 7B-D). NUP98/JARID1A-driven leukemia cells were transduced with lentiviral vectors enabling Dox-inducible expression of Cdk6-targeting short hairpin RNAs (shRNAs) (Figure 5A; supplemental Figure 8A). shRNA-mediated downregulation of Cdk6 caused a severe proliferative disadvantage of NUP98/JARID1A-driven cells in competitive growth assays in vitro and induced a reduction of actively cycling leukemia cells (Figure 5B; supplemental Figure 8B). Cdk6 knockdown strongly impaired the colony formation capacity of NUP98/JARID1A-expressing AML cells in semisolid media and was associated with increased apoptosis and elevated levels of the myeloid maturation markers Mac-1 and Gr-1 and lower expression of the progenitor marker c-Kit (Figure 5C-D; supplemental Figure 8C).
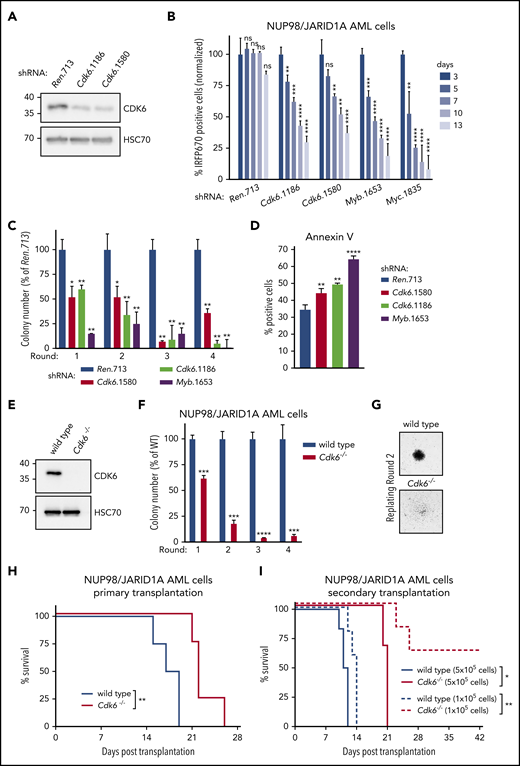
NUP98-fusion–protein-driven leukemia is dependent on CDK6 expression. (A) Western blot analysis of CDK6 levels in NUP98/JARID1A-expressing AML cells after 4 days of Dox-mediated Cdk6 shRNA induction. (B) Results of the competitive proliferation assay shown as the percentage of IRFP670-positive murine NUP98/JARID1A-driven leukemia cells expressing indicated shRNAs in the presence of Dox (0.5 µg/mL) over 13 days (mean ± SD; n = 3). The nontargeting shRNA (shRen.713) is used as neutral control, shRNAs targeting essential genes in hematopoietic cells Myb (shMyb.1653), or Myc (shMyc.1835) as positive controls. (C) Colony formation assay of G418-selected murine NUP98/JARID1A-driven leukemia cells expressing indicated shRNAs in the presence of Dox (0.5 µg/mL) over 4 rounds of replating. Colony numbers were normalized to cells expressing shRen.713 (mean ± SD; n = 3). (D) Quantification of AnnexinV-positive cells of murine NUP98/JARID1A-driven leukemia cells expressing indicated shRNAs after 2 rounds of replating (mean ± SD; n = 3). (E) Western blot analysis of CDK6 levels in wild-type vs Cdk6−/− fetal liver cells. (F) Colony formation assay of bone marrow–derived leukemia cells of mice transplanted with NUP98/NSD1-transformed Cdk6−/− or wild-type fetal liver cells. Colony numbers were normalized to wild-type NUP98/NSD1 leukemia cells (mean ± SD; n = 3). (G) Representative image of colonies from wild-type or Cdk6−/− NUP98/NSD1 leukemia cells, ×40 original magnification. (H) Kaplan-Meier survival curves with statistical analysis using the log-rank test of mice transplanted with wild-type vs Cdk6−/− fetal liver cells transformed with NUP98/NSD1 (n = 4). (I) Kaplan-Meier survival curves with statistical analyses using log-rank tests of secondary transplantations of bone marrow–derived wild-type or Cdk6−/− leukemia cells at indicated concentrations into sublethally irradiated (4.5 Gy) recipient mice (5 × 105 transplanted cells: n ≥ 3, 1 × 105 transplanted cells: n = 5). *P < .05, **P < .01, ***P < .001, ****P < .0001.
NUP98-fusion–protein-driven leukemia is dependent on CDK6 expression. (A) Western blot analysis of CDK6 levels in NUP98/JARID1A-expressing AML cells after 4 days of Dox-mediated Cdk6 shRNA induction. (B) Results of the competitive proliferation assay shown as the percentage of IRFP670-positive murine NUP98/JARID1A-driven leukemia cells expressing indicated shRNAs in the presence of Dox (0.5 µg/mL) over 13 days (mean ± SD; n = 3). The nontargeting shRNA (shRen.713) is used as neutral control, shRNAs targeting essential genes in hematopoietic cells Myb (shMyb.1653), or Myc (shMyc.1835) as positive controls. (C) Colony formation assay of G418-selected murine NUP98/JARID1A-driven leukemia cells expressing indicated shRNAs in the presence of Dox (0.5 µg/mL) over 4 rounds of replating. Colony numbers were normalized to cells expressing shRen.713 (mean ± SD; n = 3). (D) Quantification of AnnexinV-positive cells of murine NUP98/JARID1A-driven leukemia cells expressing indicated shRNAs after 2 rounds of replating (mean ± SD; n = 3). (E) Western blot analysis of CDK6 levels in wild-type vs Cdk6−/− fetal liver cells. (F) Colony formation assay of bone marrow–derived leukemia cells of mice transplanted with NUP98/NSD1-transformed Cdk6−/− or wild-type fetal liver cells. Colony numbers were normalized to wild-type NUP98/NSD1 leukemia cells (mean ± SD; n = 3). (G) Representative image of colonies from wild-type or Cdk6−/− NUP98/NSD1 leukemia cells, ×40 original magnification. (H) Kaplan-Meier survival curves with statistical analysis using the log-rank test of mice transplanted with wild-type vs Cdk6−/− fetal liver cells transformed with NUP98/NSD1 (n = 4). (I) Kaplan-Meier survival curves with statistical analyses using log-rank tests of secondary transplantations of bone marrow–derived wild-type or Cdk6−/− leukemia cells at indicated concentrations into sublethally irradiated (4.5 Gy) recipient mice (5 × 105 transplanted cells: n ≥ 3, 1 × 105 transplanted cells: n = 5). *P < .05, **P < .01, ***P < .001, ****P < .0001.
To investigate the role of CDK6 in leukemia initiation, we cotransduced wild-type or Cdk6−/− HSPCs with retroviral constructs for the expression of NUP98/NSD1 and NrasG12D (Figure 5E; supplemental Figure 8D). Cdk6 deficiency strongly abolished the serial replating capacity of NUP98/JARID1A-expressing AML cells and caused loss of blastlike colony morphology (Figure 5F-G). In vivo, disease latency was significantly increased in primary recipients of NUP98/NSD1-transformed Cdk6−/− cells as compared with mice receiving wild-type AML cells (Figure 5H). The reduced leukemogenic potential of NUP98/NSD1-transformed Cdk6−/− cells was even more pronounced in secondary transplantations despite similar levels of engraftment between NUP98/NSD1-transformed wild-type and Cdk6−/− cells (Figure 5I; supplemental Figure 8E-H).
Taken together, these data demonstrate that CDK6 is required for both the establishment and the maintenance of NUP98-fusion–protein-driven AML.
Pharmacologic CDK4/CDK6-inhibition efficiently targets NUP98-fusion AML
As NUP98-fusion AML is dependent on CDK6 expression, we reasoned that inhibition of CDK6 kinase activity might represent an attractive targeting strategy in this AML subtype. The small-molecule inhibitor Palbociclib inhibits CDK4/CDK6 and is approved for breast cancer therapy. We found that Palbociclib exerted significant dose- and time-dependent antiproliferative effects on murine AML cells driven by NUP98/JARID1A and NUP98/NSD1 (Figure 6A; supplemental Figure 9A). CDK4/CDK6 inhibition strongly impaired the replating capacity of NUP98/JARID1A-driven AML cells (Figure 6B-C). Palbociclib led to a reduction in actively proliferating cells (supplemental Figure 9B) and induced myeloid differentiation of NUP98/JARID1A-driven AML blasts, as measured by increased levels of Mac-1/Gr-1, lower levels of c-Kit, and loss of progenitor morphology (Figure 6D-E; supplemental Figure 9C). Finally, Palbociclib treatment caused a dose- and time-dependent increase in apoptosis (Figure 6F).

NUP98-fusion–protein-driven leukemia is highly sensitive to pharmacological CDK4/CDK6 inhibition. (A) Proliferation curves of murine NUP98/JARID1A-expressing leukemia cells in the presence of indicated concentrations of Palbociclib (mean ± SD; n = 3). (B) Colony formation assay of murine NUP98/JARID1A-expressing leukemia cells in the presence of dimethyl sulfoxide (DMSO) or Palbociclib. Colony numbers were normalized to DMSO (mean ± SD; n = 3). (C) Representative image of colonies in the presence of DMSO or Palbociclib after 3 rounds of replating, ×40 original magnification. (D) Flow cytometric analyses of Mac-1/Gr-1 surface marker expression in NUP98/JARID1A-expressing AML cells treated with indicated concentrations of Palbociclib (mean ± SD; n = 3). (E) Representative Cytospin images illustrating the morphology of NUP98/JARID1A-expressing AML cells 7 days after DMSO or Palbociclib treatment, ×400 original magnification. (F) Flow cytometric analyses of AnnexinV expression in NUP98/JARID1A-expressing leukemia cells treated with indicated concentrations of Palbociclib (mean ± SD; n = 3). (G-H) Dose-response curves of Palbociclib-mediated growth inhibition of murine leukemia cells (G) and primary patient cells (H) driven by different fusion proteins (mean ± standard error of the mean; n = 3). (I) Kaplan-Meier survival curves with statistical analysis using the log-rank test of C57BL/6 Ly5.1 recipients transplanted with murine NUP98/NSD1-driven leukemia cells after vehicle or Palbociclib treatment (50 mg/kg; n = 4). (J) Kaplan-Meier survival curves with statistical analysis using the log-rank test of NSG mice transplanted with NUP98/NSD1-PDX AML cells after vehicle or Palbociclib treatment (50 mg/kg; n = 4). *P < .05, **P < .01, ***P < .001, ****P < .0001.
NUP98-fusion–protein-driven leukemia is highly sensitive to pharmacological CDK4/CDK6 inhibition. (A) Proliferation curves of murine NUP98/JARID1A-expressing leukemia cells in the presence of indicated concentrations of Palbociclib (mean ± SD; n = 3). (B) Colony formation assay of murine NUP98/JARID1A-expressing leukemia cells in the presence of dimethyl sulfoxide (DMSO) or Palbociclib. Colony numbers were normalized to DMSO (mean ± SD; n = 3). (C) Representative image of colonies in the presence of DMSO or Palbociclib after 3 rounds of replating, ×40 original magnification. (D) Flow cytometric analyses of Mac-1/Gr-1 surface marker expression in NUP98/JARID1A-expressing AML cells treated with indicated concentrations of Palbociclib (mean ± SD; n = 3). (E) Representative Cytospin images illustrating the morphology of NUP98/JARID1A-expressing AML cells 7 days after DMSO or Palbociclib treatment, ×400 original magnification. (F) Flow cytometric analyses of AnnexinV expression in NUP98/JARID1A-expressing leukemia cells treated with indicated concentrations of Palbociclib (mean ± SD; n = 3). (G-H) Dose-response curves of Palbociclib-mediated growth inhibition of murine leukemia cells (G) and primary patient cells (H) driven by different fusion proteins (mean ± standard error of the mean; n = 3). (I) Kaplan-Meier survival curves with statistical analysis using the log-rank test of C57BL/6 Ly5.1 recipients transplanted with murine NUP98/NSD1-driven leukemia cells after vehicle or Palbociclib treatment (50 mg/kg; n = 4). (J) Kaplan-Meier survival curves with statistical analysis using the log-rank test of NSG mice transplanted with NUP98/NSD1-PDX AML cells after vehicle or Palbociclib treatment (50 mg/kg; n = 4). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Next, we compared the Palbociclib sensitivity of NUP98/JARID1A-, NUP98/NSD1-, and NUP98/DDX10-driven AML samples with leukemia cells driven by the fusion proteins MLL/AF9 and AML1/ETO9a, which were both shown to be sensitive to CDK4/CDK6 inhibition.24,27 All 3 murine NUP98-rearranged cell lines were very sensitive to Palbociclib treatment (50% effective concentration [EC50] values ranging from 53 to 147 nM). MLL/AF9-driven cells showed comparable inhibitor sensitivity (EC50: 144 nM), whereas AML1/ETO9a-expressing AML tolerated higher concentrations (EC50: 1271 nM) (Figure 6G). To validate these findings in human AML, we examined 6 distinct primary, patient-derived leukemia samples expressing different NUP98-fusion proteins (supplemental Table 4) for their Palbociclib sensitivity. Also in NUP98/NSD1-expressing primary human AML cells, CDK4/CDK6 inhibition induced cell-cycle arrest and apoptosis (supplemental Figure 9D-E). Four of 6 NUP98-rearranged patient samples were highly sensitive to CDK4/CDK6 inhibition (EC50: 18 to 508 nM), whereas 2 samples tolerated slightly higher doses (EC50: 2300 to 2400 nM). However, all 6 NUP98-fusion AML samples were more sensitive than a primary AML sample expressing AML1/ETO9a (EC50: 4820 nM) and a BCR/ABL1-expressing CML sample (EC50: 17501 nM) (Figure 6H; supplemental Figure 9F). To investigate whether Palbociclib displays antileukemic activity in vivo that can be harnessed to efficiently combat NUP98-fusion–driven AML, we transplanted NUP98/NSD1-driven leukemia cells into recipient mice and initiated Palbociclib treatment as soon as leukemia cells were detectable by bioluminescence imaging (supplemental Figure 9G). CDK4/CDK6 inhibition delayed leukemia progression, caused increased myeloid differentiation of leukemic blasts, and led to a significant survival benefit in vivo (Figure 6I; supplemental Figure 9G-I).
Finally, we employed a patient-derived xenograft model of NUP98/NSD1-rearranged AML (supplemental Figure 9J-K). Palbociclib monotherapy was initiated 80 days after transplantation. Strikingly, Palbociclib treatment caused a highly significant prolongation of survival compared with the vehicle-treated control cohort (median survival 152 vs 103 days) (Figure 6J).
In summary, these data show that NUP98-rearranged AML is sensitive toward CDK4/CDK6 inhibition in vivo and in vitro. Therefore, we propose Palbociclib may be a new rational treatment option for patients suffering from this disease.
Discussion
Targeting critical effectors of oncogenes represents an attractive strategy in cancer therapy, particularly when the opportunity to inhibit the driver oncoprotein itself is limited. NUP98-fusion–protein-driven AML is a devastating disease with very poor prognosis. No convincing targeting approach has been developed for this leukemia subtype. We established novel mouse models for controllable expression of 3 distinct NUP98-fusion proteins and used RNA-seq and ChIP-seq to identify conserved transcriptional targets. These studies revealed CDK6 as a highly expressed, directly regulated target of NUP98-fusion proteins and demonstrate that genetic and pharmacologic interference with CDK6 results in cell-cycle arrest, myeloid differentiation, and apoptosis in vitro and in vivo. We thus propose CDK4/6 inhibition as a rational strategy to target AML with NUP98-fusions.
The cyclin-dependent kinase CDK6 is highly expressed in AML patient samples and represents a promising target in MLL-fusion expressing- and FLT3-ITD-positive leukemia.27,28 In addition, a recent report identified deregulation of cell-cycle control via aberrant regulation of cyclin D2/CDK6 as a critical feature of RUNX1/ETO-driven AML.27 The canonical role of the serine/threonine kinase CDK6 and its homolog CDK4 is to regulate cell-cycle progression through association with D-type cyclins.29 As CDK6 but not CDK4 is amplified in human hematopoietic malignancies, it was hypothesized that CDK6 exerts distinct functions in addition to cell-cycle control. In fact, CDK6 acts as a transcriptional regulator in cancer cells. Although the expression of genes important for proliferation, survival, and cytokine production depends on its kinase activity, CDK6-dependent regulation of angiogenesis and stem cell functions are kinase independent.28,30-33 As NUP98-fusion–protein-driven leukemia was sensitive to treatment with the CDK4/CDK6 inhibitor Palbociclib, it is likely that targeting the kinase-dependent role of CDK6 is of particular importance within the context of NUP98-fusion AML. Loss of RB1 expression or the acquisition of TP53 mutations causes resistance to CDK4/6 inhibition.33,34 Thus, future efforts should focus on the development of specific CDK6 inhibitors and on the identification of targets that synergize with CDK6 inhibition to maximize therapy response.
In our study, structurally distinct NUP98-fusion proteins induce oncogenic transformation in mouse models. Although we cannot exclude that different NUP98-fusion proteins employ unrelated molecular pathways to induce leukemia, we show that all NUP98-fusions share common sets of target genes that are critical for their oncogenicity. As several partner genes among NUP98-fusion proteins lack an annotated DNA-binding- or chromatin-interaction domain, it is likely that chromatin targeting by the aminoterminus of NUP98, which is shared between all distinct fusion partners, is essential for leukemogenesis. Fusion proteins exert their oncogenic activity in the context of large protein complexes.35-38 NUP98-fusion proteins interact with the MLL1/NSL complexes, and MLL1 function is critical for NUP98-fusion–dependent leukemogenesis.19,39 It is thus appealing to speculate that the NUP98-moiety is required for chromatin targeting, whereas the C-terminal fusion partner exerts critical functions in gene control during leukemogenesis. This concept may also explain how fusions without annotated DNA-binding capacity, such as NUP98/DDX10, still result in aberrant expression of defined genes.
The 2 most common NUP98-fusion proteins, NUP98/NSD1 and NUP98/JARID1A, but also the molecularly distinct NUP98/DDX10-fusion protein share several highly overexpressed target genes in addition to Cdk6. A common core signature of 244 genes whose expression is high in our NUP98-fusion AML mouse models was strongly enriched in patients harboring NUP98-rearrangements. This signature contained many genes with critical roles in normal and malignant hematopoiesis. For instance, genes of the Hoxa cluster and Meis1 are critical factors in murine and human cells transformed by NUP98-fusion proteins and AML patients harboring NUP98 gene rearrangements.2,9,11 Among the 12 shared direct targets of NUP98-fusion–potein-driven AML, there are several interesting candidates with potential relevance for leukemia biology. For instance, the C-type lectin CD93 is highly expressed in leukemia-initiating subpopulations of MLL-rearranged AML cells and was proposed to regulate self-renewal by downregulating the tumor suppressor gene CDKN2B.40 Moreover, CD93 expression has been shown to discriminate leukemia cells in the monitoring of minimal residual disease.41 Screening for CD93 could thus be useful in the detection of minimal residual disease among AML patients with NUP98-fusion proteins. Furthermore, the RING domain E3 ubiquitin ligase RNF220 was shown to stabilize b-catenin, resulting in increased Wnt signaling,42 an essential pathway in AML cells.43
NUP98-fusions also regulate a number of genes whose expression is not particularly high in our murine AML-like model systems but whose functions are still pivotal in leukemia biology, such as the transcription factors Myc and Myb.44 Interestingly, CDK6 was shown to be a target gene of MYB, indicating that CDK6 expression could be regulated at multiple levels downstream of NUP98-fusion proteins.45 Furthermore, the promoter of the antiapoptotic Bcl-2 gene is bound by NUP98/JARID1A in the steady state, and Bcl-2 expression was rapidly downregulated upon oncoprotein shut down. BCL2 inhibition by the small molecule Venetoclax has recently been approved for AML therapy in a combinatorial treatment regimen.46 Combined inhibition of CDK6 and BCL2 might thus be an attractive approach in the therapy of AML patients with NUP98-rearrangements, as both genes are direct targets of NUP98-fusion proteins.
In summary, our findings show that combining advanced modeling of NUP98-fusion–dependent leukemogenesis in mice with integrated transcriptomic analyses provides valuable insight into shared fusion-protein–driven transcriptional circuitries. The identification of CDK6 as a direct target in NUP98-fusion–protein-driven AML has potential clinical implications, as CDK6 inhibition represents a rational therapeutic intervention strategy for this subgroup of leukemia patients with particularly poor prognosis.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE134784).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Please contact the corresponding author for additional information and original data.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all members of the F.G. and J.Z. laboratories for stimulating discussions, in particular Mareike Roth. Julian Jude is acknowledged for providing help with molecular cloning, Michaela Fellner for excellent technical assistance, and Matthias Muhar for providing murine AML1/ETO9a-driven cells. The authors thank Gerald Schmauss and Marietta Weninger for expert FACS sorting, Manuela Telsnig for animal care, and the Molecular Biology Services at the Vienna Biocenter (VBC). Irina Sadovnik, Rene Gaupmann, Gabriele Stefanzl, Barbara Peter, Daniela Berger, and Karin Nebral are acknowledged for excellent technical assistance. R.I. and A. Puissant are indebted to Jihène Benlagha and Jean-Michel Cayuela from the Saint-Louis Hospital Cell Bank. Next-generation sequencing was performed at the VBCF NGS Unit (www.viennabiocenter.org/facilities).
This project has received funding from the European Research Council under the European Union's Horizon 2020 research and innovation programme grant agreements 636855 (F.G.) and 336860 (J.Z.). This work was supported by Austrian Science Fund the SFB grants F4710 (J.Z.), F4704-B20 (P.V.), F4707 (R.M. and H.T.T.P.), and SFB-F06105 (R.M. and H.T.T.P.). J.S. and L.S. are recipients of a DOC Fellowship of the Austrian Academy of Sciences at the University of Veterinary Medicine and the Ludwig Boltzmann Institute for Cancer Research, respectively. Research at the Research Institute of Molecular Pathology is generously supported by Boehringer Ingelheim.
Authorship
Contribution: J.S., I.A.M.B., J.Z., and F.G. conceptualized the work; J.S., I.A.M.B., T.E., J.Z., and F.G. provided the methodology; J.S., I.A.M.B., T.E., T.B., L.S., B.M., S.T., H.T.T.P., M.S., J.E., G.M., E.A., S.T.-Z., C.V.d.V., G.H., N.D., A. Petit, H.L., A. Puissant, R.I., R. Moriggl, M.H., R. Meisel, P.V., V.S., J.Z., and F.G. performed the investigating; and J.S. and F.G. wrote the paper.
Conflict-of-interest disclosure: P.V. has received research grants and/or honoraria from Pfizer, Celgene, and Novartis. The remaining authors declare no competing financial interests.
Correspondence: Florian Grebien, Institute for Medical Biochemistry, University of Veterinary Medicine Vienna, Veterinaerplatz 1, Vienna A-1210, Austria; e-mail: florian.grebien@vetmeduni.ac.at.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal