

TO THE EDITOR:
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell (HSC) diseases characterized by increased proliferation of erythroid, megakaryocytic, and/or myeloid lineages.1 The JAK2-V617F mutation can be found in >95% of polycythemia vera (PV) patients, and also in approximately one-half of patients with essential thrombocythemia (ET) or primary myelofibrosis (PMF).2-5 Somatic mutations in exon 12 of JAK2 are found in 3% to 5% of PV patients.6 Quantification of the JAK2-mutant allele burden, also called variant allele frequency (VAF), in DNA from peripheral blood granulocytes is used to monitor the size of the mutant clone. ET patients have lower JAK2 VAF than PMF or PV patients.7 Interestingly, some MPN patients display very low VAF, which calls into question why they develop MPNs if the clone is apparently unable to expand. We therefore studied MPN patients with JAK2 VAF ≤20%.
In our cohort of 205 patients with JAK2-V617F+ MPNs, we identified 56 patients with a VAF ≤20% in purified granulocyte DNA (supplemental Figure 1A, available on the Blood Web site). Survival of MPN patients with low JAK2-V617F VAF was not significantly altered compared with other MPN patients (supplemental Figure 1B-D). The contribution of the JAK2-mutant clone to peripheral blood lineages was previously shown to be highly variable between individual MPN patients,8,9 and low VAF correlated with lineage-restricted clonal distribution.9 We therefore determined the JAK2 VAF in platelets and reticulocytes, representing the lineages most relevant to ET and PV, respectively. The purification procedures are described in supplemental Figures 2 and 3. Because platelets and reticulocytes do not contain DNA, we established a quantitative polymerase chain reaction assay to measure JAK2-V617F in RNA (supplemental Figure 2C).
RNA from purified platelets was available from 44 of 56 patients (79%) with low JAK2-V617F VAF (supplemental Figure 4). One patient (P021) simultaneously also carried a JAK2 exon 12 mutation and we included 2 additional patients with a JAK2 exon 12 mutation in our study. Most patients with ET and PMF, as well as 15 of 17 patients with PV (88%), had higher JAK2 VAF in platelets compared with granulocytes (Figure 1A). We were able to obtain fresh blood and to purify reticulocytes by fluorescence-activated cell sorting (FACS) from 22 of 46 patients (48%) with available platelet RNA. All patients with PV and PMF and, surprisingly, 8 of 11 patients with ET (72%) had higher JAK2 VAF in reticulocytes compared with granulocytes (Figure 1B). This increase in allele burden compared with granulocytes was restricted to platelets and reticulocytes (Figure 1C). We can distinguish 4 patterns of lineage contribution of the JAK2-mutant clone (Figure 1D): a “platelet-biased” pattern, with increased mutant allele burden solely in platelets; a “red cell–biased” pattern, with a selective increase in reticulocytes; a “platelet and red cell–biased” pattern, with increase in both platelets and reticulocytes; and a “persistently low” pattern, with very low allele burden in granulocytes, platelets, and reticulocytes.
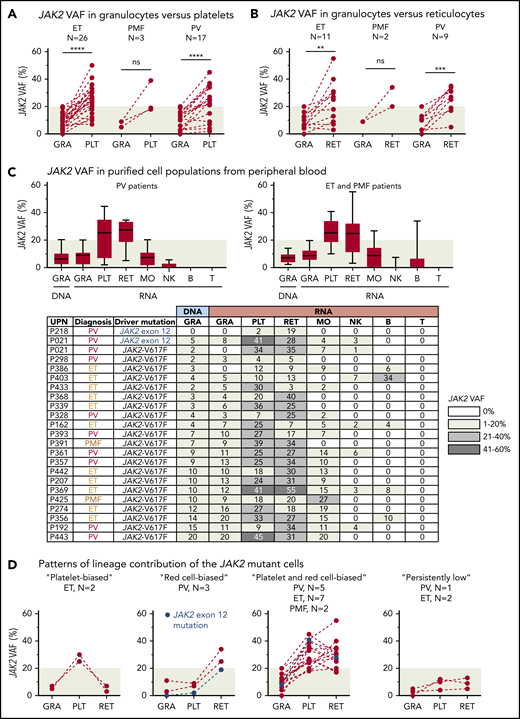
Analysis of JAK2 VAFs in peripheral blood. (A) Comparison of JAK2 VAF measured in RNA from granulocytes (GRA) vs platelets (PLT). Dashed lines connect data points from the same patient. (B) Comparison of JAK2 VAF measured in RNA from granulocytes vs reticulocytes (RET). (C) JAK2 VAF in purified cell populations from peripheral blood. Boxes represent 50% of the measured values; whiskers indicate the range; and horizontal lines indicate the median. The JAK2 VAF of 22 patients measured in DNA or RNA from different peripheral blood lineages is shown below. Numbers in the cells of the table indicate the percentages of JAK2 VAF; the shading of boxes corresponds to the ranges shown on the right. (D) Patterns of lineage contribution of the JAK2-mutant cells derived from the data presented in panels A and B. **P < .01; ***P < .001; ****P < .0001. B, B cell; MO, monocyte; NK, natural killer cell; ns, not significant; T, T cell; UPN, unique patient number.
Analysis of JAK2 VAFs in peripheral blood. (A) Comparison of JAK2 VAF measured in RNA from granulocytes (GRA) vs platelets (PLT). Dashed lines connect data points from the same patient. (B) Comparison of JAK2 VAF measured in RNA from granulocytes vs reticulocytes (RET). (C) JAK2 VAF in purified cell populations from peripheral blood. Boxes represent 50% of the measured values; whiskers indicate the range; and horizontal lines indicate the median. The JAK2 VAF of 22 patients measured in DNA or RNA from different peripheral blood lineages is shown below. Numbers in the cells of the table indicate the percentages of JAK2 VAF; the shading of boxes corresponds to the ranges shown on the right. (D) Patterns of lineage contribution of the JAK2-mutant cells derived from the data presented in panels A and B. **P < .01; ***P < .001; ****P < .0001. B, B cell; MO, monocyte; NK, natural killer cell; ns, not significant; T, T cell; UPN, unique patient number.
To define at which stages of the hematopoietic development the expansion of the JAK2-mutant clone occurs, we determined the JAK2 VAF by genotyping single myeloid and erythroid progenitors (Figure 2A; supplemental Figure 5). As expected, the derived VAF in granulocyte and granulocyte/monocyte colonies (colony-forming unit granulocyte/granulocyte-macrophage [CFU-G/GM]) was low and comparable with VAF in peripheral blood granulocytes. Burst-forming unit–erythroid (BFU-E) colonies also showed very low VAF, but, in most patients, VAF substantially increased in peripheral blood reticulocytes, suggesting that the expansion of the mutant clone occurs at late stages of erythroid development. The late erythroid expansion of the JAK2-mutant clone in PV patients could be favored by low serum erythropoietin levels and hypersensitivity of JAK2-V617F–expressing progenitors to erythropoietin.10
![Analysis of JAK2 VAFs in hematopoietic progenitors. (A) Analysis of single colonies grown in methylcellulose. Single erythroid colonies (burst-forming unit–erythroid [BFU-E]) and granulocyte or granulocyte/monocyte colonies (colony-forming unit granulocyte/granulocyte-macrophage [CFU-G/GM]) were picked, and the percentage of JAK2-mutant colonies was converted into VAF by taking into account the heterozygous and homozygous state of each colony. (B) JAK2 VAF in FACS-sorted progenitor cells and mature blood cells. Data from 17 MPN patients are shown. Data points connected by solid lines were obtained from FACS-sorted progenitor cells. Dashed lines connect the progenitors with their corresponding mature cells isolated from peripheral blood. (C) Model depicting the stages of hematopoiesis in which the clonal expansion occurred in MPN patients with low JAK2 VAF. (D) Schematic illustration of dynamics in a branched population structure. (E) Modeling of a common platelet and red cell–biased pattern using the branched compartmental model of hematopoiesis. The distribution of JAK2-mutant cells observed in MPN patients with low VAF can be reproduced by altering the division rate (b), self-renewal (a), and death (d) probabilities, and the differentiation bias (c) at specific stages of hematopoietic development. For the clonal expansion during terminal stages of megakaryopoiesis, Δa signifies the probability that megakaryocytes repeatedly undergo endomitosis resulting in higher ploidy, and the division rate Δb stands for the number of endomitoses per time unit, for example, per day. Hematopoietic cell compartments are indicated on the x-axis. **P < .01; ****P < .0001. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; EP, erythroid progenitor; HSPC, hematopoietic stem and progenitor cell; Meg, megakaryocyte; MEP, megakaryocyte-erythroid progenitor.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/22/10.1182_blood.2019002943/2/m_bloodbld2019002943f2.png?Expires=1769091893&Signature=o-eMC6VzOWyvP-JitIw2YiSket3DhoX4ClCouSXtWyZLebh82SIvVPlNdlztBfpbkaG7BqeTyniH77L5UdhR6vJjSwFgCarCI6HY85b~HJl9q43E81BRXPXxxELxmhdhqM-Lzn95UN7DRUqZmkhLRn-j155cgDKP-sUpS01SY7644MNsstQc6UX9sMIidbGqAfweaTintBqUULwP5u0uHa1UaFlWOcgqxQjUE3NkFl1bKw3hVA5gHszbQS~1y71AL4ZQ3TG4t9Uet-9hNAzZQ6FevfOvVBKDGyho3Et1i3BXGew6Ap5YRcl4vocRl8N6eTmPJUEas6TCCiG25Nh60A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Analysis of JAK2 VAFs in hematopoietic progenitors. (A) Analysis of single colonies grown in methylcellulose. Single erythroid colonies (burst-forming unit–erythroid [BFU-E]) and granulocyte or granulocyte/monocyte colonies (colony-forming unit granulocyte/granulocyte-macrophage [CFU-G/GM]) were picked, and the percentage of JAK2-mutant colonies was converted into VAF by taking into account the heterozygous and homozygous state of each colony. (B) JAK2 VAF in FACS-sorted progenitor cells and mature blood cells. Data from 17 MPN patients are shown. Data points connected by solid lines were obtained from FACS-sorted progenitor cells. Dashed lines connect the progenitors with their corresponding mature cells isolated from peripheral blood. (C) Model depicting the stages of hematopoiesis in which the clonal expansion occurred in MPN patients with low JAK2 VAF. (D) Schematic illustration of dynamics in a branched population structure. (E) Modeling of a common platelet and red cell–biased pattern using the branched compartmental model of hematopoiesis. The distribution of JAK2-mutant cells observed in MPN patients with low VAF can be reproduced by altering the division rate (b), self-renewal (a), and death (d) probabilities, and the differentiation bias (c) at specific stages of hematopoietic development. For the clonal expansion during terminal stages of megakaryopoiesis, Δa signifies the probability that megakaryocytes repeatedly undergo endomitosis resulting in higher ploidy, and the division rate Δb stands for the number of endomitoses per time unit, for example, per day. Hematopoietic cell compartments are indicated on the x-axis. **P < .01; ****P < .0001. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; EP, erythroid progenitor; HSPC, hematopoietic stem and progenitor cell; Meg, megakaryocyte; MEP, megakaryocyte-erythroid progenitor.
Analysis of JAK2 VAFs in hematopoietic progenitors. (A) Analysis of single colonies grown in methylcellulose. Single erythroid colonies (burst-forming unit–erythroid [BFU-E]) and granulocyte or granulocyte/monocyte colonies (colony-forming unit granulocyte/granulocyte-macrophage [CFU-G/GM]) were picked, and the percentage of JAK2-mutant colonies was converted into VAF by taking into account the heterozygous and homozygous state of each colony. (B) JAK2 VAF in FACS-sorted progenitor cells and mature blood cells. Data from 17 MPN patients are shown. Data points connected by solid lines were obtained from FACS-sorted progenitor cells. Dashed lines connect the progenitors with their corresponding mature cells isolated from peripheral blood. (C) Model depicting the stages of hematopoiesis in which the clonal expansion occurred in MPN patients with low JAK2 VAF. (D) Schematic illustration of dynamics in a branched population structure. (E) Modeling of a common platelet and red cell–biased pattern using the branched compartmental model of hematopoiesis. The distribution of JAK2-mutant cells observed in MPN patients with low VAF can be reproduced by altering the division rate (b), self-renewal (a), and death (d) probabilities, and the differentiation bias (c) at specific stages of hematopoietic development. For the clonal expansion during terminal stages of megakaryopoiesis, Δa signifies the probability that megakaryocytes repeatedly undergo endomitosis resulting in higher ploidy, and the division rate Δb stands for the number of endomitoses per time unit, for example, per day. Hematopoietic cell compartments are indicated on the x-axis. **P < .01; ****P < .0001. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; EP, erythroid progenitor; HSPC, hematopoietic stem and progenitor cell; Meg, megakaryocyte; MEP, megakaryocyte-erythroid progenitor.
To complement the data from single colonies, we isolated megakaryocyte progenitors (MkPs) and other progenitor cell populations from peripheral blood by FACS (supplemental Figure 6). Consistent with the data obtained in the analysis of single colonies, in the vast majority of patients, the JAK2-mutant VAF was low in MkPs and other sorted progenitor subsets (Figure 2B; supplemental Figure 7). In contrast, a large percentage of erythroid progenitors was positive for the JAK2 mutation in previous reports, when patients with higher JAK2 allele burden were analyzed.9,11,12 Thus, the expansion of the JAK2-mutant clone at terminal stages of erythroid and/or megakaryocytic development appears to be typical for the low allele burden subset of MPN patients (Figure 2C).
Lineage-biased HSCs and long-term progenitors have been described.13-16 We hypothesized that MPN patients with low VAF might acquire the JAK2 mutation in such progenitors and we expected that single platelet-biased or red cell–biased patterns would be the most frequent. However, our data do not favor such a model. The frequent occurrence of the platelet and red cell–biased pattern in MPN patients with low VAF fits with the proposed model of a “biological continuum” between the phenotypes of JAK2-V617F+ ET and PV.17 However, none of the PV or ET patients with low VAF displayed a dominant homozygous subclone (supplemental Figure 5), which is typically found in the majority of PV patients.11,12,18
To examine the role of additional somatic mutations, we analyzed 104 cancer-related genes using targeted next-generation sequencing.19 Additional somatic mutations were detected in 12 of the 46 patients with low VAF (26%) (supplemental Figure 8; supplemental Table 1). The presence of additional somatic mutations had no relevant impact on the mutant allele distributions (supplemental Figure 8D). We determined the clonal architecture in 11 of 12 patients with additional somatic gene mutations by genotyping single hematopoietic colonies (supplemental Figure 8E). A sequential pattern of acquisition of mutations was observed in 4 of 11 patients (36%). In all of these patients, the additional somatic mutation was acquired before JAK2-V617F. In the remaining 7 patients (64%), a biclonal pattern was observed, with the JAK2 mutation and the additional somatic gene mutation representing separate clones. Thus, interestingly, none of the patients with low VAF displayed JAK2 mutation as the first event, and the frequency of the biclonal pattern appears to be higher than the reported frequency of 30% in overall MPNs.19,20
To derive hypotheses that could explain the observed clonal structures, we generated an online app based on a compartmental mathematical model of hematopoiesis (https://ibz-shiny.ethz.ch/ashcroft/lowJAK2/app/; supplemental Methods). This model approximates the continuous process of hematopoietic development and assumes that the cells in each compartment are homogeneous and indistinguishable. All analysis is based on the steady state of the system.21-23 The dynamics of cells in each compartment can be described by 4 parameters: self-renewal probability (ai), division rate (bi), lineage bias (cij), and probability of death (di) (Figure 2D). The influence of independently varying the mutant’s parameters on the mutant allele burden is summarized in supplemental Figure 9. By modifying the parameters at different stages of hematopoietic development, we can qualitatively reproduce all observed patterns of clonal expansion in MPN patients with low VAF (Figure 2E; supplemental Figure 10). We therefore hypothesize that interindividual differences in these 4 parameters are sufficient to explain the different patterns of mutant allele burden observed in MPN patients. Indeed, differences in expression levels of JAK2 and MPL protein have been shown to determine thrombopoietin-induced megakaryocyte proliferation vs differentiation,24 and, on mouse models, resulted in switching between ET and PV phenotypes.25 Additional factors contributing to interindividual differences in humans could be the availability and activity of downstream signaling components and differences in genetic background.
For original data, please e-mail the corresponding author, Radek C. Skoda, at radek.skoda@unibas.ch.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Nils Hansen for helpful comments on the manuscript. The authors also thank Hui Hao-Shen, Gabriele Mild-Schneider, and Hélène Mereau for valuable technical support; Danny Labes and Emmanuel Traunecker for cell sorting; Christian Beisel, Genomics Facility Basel, for conducting next-generation sequencing; and Susanne Erpel, Nano Imaging Laboratory, Swiss Nanoscience Institute (SNI), University of Basel, for performing the scanning electron microscopy.
This work was supported by grants from the Swiss Cancer League (KLS-2950-02-2012 and KFS-3655-02-2015), SystemsX.ch (Medical Research and Development grant 2014/266), and the Swiss National Science Foundation (31003A-147016/1 and 31003A_166613) (R.C.S.) and from the Swiss National Science Foundation through a joint research project (SCOPES IZ73Z0 152420/1; V.C. and R.C.S.).
Authorship
Contribution: R.N. performed research, analyzed data, and wrote the manuscript; P.A. and S.B. analyzed data and performed mathematical modeling; J.Z. and S.R. performed research; T.N.R. designed research; B.D., S.C.M., J.R.P., D.L., and V.C. collected data; P.L. designed research and analyzed data; and R.C.S. designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Radek C. Skoda, Department of Biomedicine, University Hospital Basel and University of Basel, Hebelstrasse 20, Basel, 4031 Switzerland; e-mail: radek.skoda@unibas.ch.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal