


Abstract
Recognition that germline mutations can predispose individuals to blood cancers, often presenting as secondary leukemias, has largely been driven in the last 20 years by studies of families with inherited mutations in the myeloid transcription factors (TFs) RUNX1, GATA2, and CEBPA. As a result, in 2016, classification of myeloid neoplasms with germline predisposition for each of these and other genes was added to the World Health Organization guidelines. The incidence of germline mutation carriers in the general population or in various clinically presenting patient groups remains poorly defined for reasons including that somatic mutations in these genes are common in blood cancers, and our ability to distinguish germline (inherited or de novo) and somatic mutations is often limited by the laboratory analyses. Knowledge of the regulation of these TFs and their mutant alleles, their interaction with other genes and proteins and the environment, and how these alter the clinical presentation of patients and their leukemias is also incomplete. Outstanding questions that remain for patients with these germline mutations or their treating clinicians include: What is the natural course of the disease? What other symptoms may I develop and when? Can you predict them? Can I prevent them? and What is the best treatment? The resolution of many of the remaining clinical and biological questions and effective evidence-based treatment of patients with these inherited mutations will depend on worldwide partnerships among patients, clinicians, diagnosticians, and researchers to aggregate sufficient longitudinal clinical and laboratory data and integrate these data with model systems.
Introduction
Familial predisposition to hematological malignancy (FHM) has been investigated phenotypically for >100 years, and genetically for decades, leading to the identification of germline RUNX1 mutations associated with familial platelet disorder with predisposition to myeloid malignancy (FPD-MM) 20 years ago.1 This discovery was followed by identification of additional myeloid malignancy predisposition germline mutations in CEBPA and GATA2.2,3 Germline contribution to hematological malignancy (HM) is incompletely defined, with multitumor cohorts suggesting ≥15% germline contribution4 and recent phenotype-driven studies from large cohorts identifying significant familial clustering across all HM subtypes.5 More than 14 genes are known to predispose to autosomal-dominant FHM.6 In the most recent World Health Organization classification of myeloid neoplasms and acute leukemia, familial HMs were recognized as an entity, “Myeloid neoplasms with germline predisposition,” with annotation of several predisposition syndromes, including those described in this review.7
RUNX1, CEBPA, and GATA2 predisposition disorders differ from many other cancer predisposition disorders in the limited syndromic features associated with heterozygous carriers of mutations to aid in their diagnosis (Table 1). Independent families with germline mutations in RUNX1 (∼130 families) and CEBPA (∼25 families) are rare, and frequent GATA2 (de novo) mutations affecting pediatric patients and phenotypic heterogeneity complicate their accurate aggregation.8-10 Increasingly, germline mutations in these transcription factors (TFs) are routinely identified in clinical genetic laboratories worldwide and classified in concordance with the American College of Medical Genetics and Genomics guidelines for reporting back to the referring clinician and patients.11 However, the generated genetic data remain in local institutions and are not routinely shared in publications or databases (eg, ClinVar). There is also the clinical challenge of recognizing FHM without taking an adequate family history, the occurrence of small families due to decreased reproductive fitness, and families with atypical clinical presentation of HM and solid tumors (“pan-cancer” families).12,13
Clinical characteristics secondary to germline mutations affecting the TFs RUNX1, GATA2, and CEBPA
| RUNX1 | GATA2 | CEBPA | |
|---|---|---|---|
| HM | MDS, AML, T-ALL, T-NHL, CLL, HCL | MDS, AML, aCML, CMML | AML |
| Cytopenia | Thrombocytopenia | Monocytopenia, dendritic cell, B and NK lymphoid deficiency, chronic neutropenia | No preleukemic cytopenias |
| Other presentations | Easy bruising, epistaxis, eczema, petechiae, psoriasis | Lymphedema; pulmonary alveolar proteinosis; recurrent bacterial, fungal, and viral infections associated with immunodeficiencies; deafness; urogenital tract anomalies; behavioral problems | No preleukemic phenotype |
| Germline mutation types | Whole and partial gene deletions, intragenic deletions, truncating, missense (mainly in RUNT domain), splicing mutations | Whole and partial gene deletions, intronic deletions, truncating, missense and indels (zinc finger 2), intronic enhancer | N-terminal frameshift, C-terminal missense |
| Technology considerations for germline mutation detection | Coding SNVs and small indels: WGS, WES, NGS panels, Sanger, AFLP (CEBPA), MLPA. Larger CNVs: WGS, MLPA, SNP microarray. Noncoding variants: WGS, custom NGS panels, RNA sequencing (splicing alterations and expression) | ||
| Mutation-specific phenotype correlations | Dominant-negative mutations: earlier onset and increased penetrance of HM? | De novo LoF mutations common in pediatric MDS | N-terminal mutations: 90% penetrance of AML |
| T354M: mostly early-onset MDS/AML | C-terminal mutations: 50% penetrance of AML | ||
| R396Q and R398W: mostly immune defects and MDS | |||
| Common second hits (Figure 1D-F) | Somatic RUNX1 [including UPD 21, +21(q)], PHF6, BCOR, NOTCH1, EZH2 | −7, +8, ASXL1, NRAS, WT1, STAG2, KRAS, SETBP1 | Somatic CEBPA, WT1, GATA2, KIT, TET2, EZH2 |
| Mutation spectrum | Overlap between germline and somatic mutations | Distinct mutational pattern for germline (truncating, ZF2 missense and indels, intronic enhancer) and somatic (mostly ZF1 missense and some ZF2 missense) mutations | Overlap between germline and somatic mutations (N- and C-terminal regions; however poor sequence coverage in past may have masked some mutations) |
| Mode of germline mutation | Inherited predominant, de novo infrequent (except large chromosome deletions) | Inherited and de novo mutations frequently reported | Wholly inherited, with no published reports of de novo mutation |
| NGS Gene coverage gnomAD | Complete (canonical transcript) | Complete | Poor (<20× for ∼70% of coding region) |
| Age of onset, median, y | 29 | 19 | 23 |
| Age of onset, range (y), % | |||
| <10 | 17.1 | 13.4 | 25.9 |
| 11-20 | 13.0 | 39.7 | 22.4 |
| 21-30 | 14.6 | 21.5 | 17.3 |
| 31-40 | 11.4 | 10.6 | 15.5 |
| 41-50 | 19.5 | 9.0 | 8.6 |
| 51-60 | 16.3 | 3.9 | 8.6 |
| >61 | 8.1 | 1.9 | 1.7 |
| Presymptomatic monitoring and treatment | Patient monitoring depends on local guidelines and expert opinion | ||
| Chemotherapy (GATA2, RUNX1, CEBPA), followed by HSCT (GATA2, RUNX1). Evidence-based conclusions on best treatments and monitoring options not available because of small cohort sizes and lack of data aggregation for rare disorders. | |||
| Prognosis | Not well defined for germline. Poor for sporadic/somatic RUNX1 mutated. | Poor for leukemic patients. HSCT for immune deficiency and leukemia prevents progression with favorable outcome. | Generally favorable long-term outcomes with chemotherapy alone |
| RUNX1 | GATA2 | CEBPA | |
|---|---|---|---|
| HM | MDS, AML, T-ALL, T-NHL, CLL, HCL | MDS, AML, aCML, CMML | AML |
| Cytopenia | Thrombocytopenia | Monocytopenia, dendritic cell, B and NK lymphoid deficiency, chronic neutropenia | No preleukemic cytopenias |
| Other presentations | Easy bruising, epistaxis, eczema, petechiae, psoriasis | Lymphedema; pulmonary alveolar proteinosis; recurrent bacterial, fungal, and viral infections associated with immunodeficiencies; deafness; urogenital tract anomalies; behavioral problems | No preleukemic phenotype |
| Germline mutation types | Whole and partial gene deletions, intragenic deletions, truncating, missense (mainly in RUNT domain), splicing mutations | Whole and partial gene deletions, intronic deletions, truncating, missense and indels (zinc finger 2), intronic enhancer | N-terminal frameshift, C-terminal missense |
| Technology considerations for germline mutation detection | Coding SNVs and small indels: WGS, WES, NGS panels, Sanger, AFLP (CEBPA), MLPA. Larger CNVs: WGS, MLPA, SNP microarray. Noncoding variants: WGS, custom NGS panels, RNA sequencing (splicing alterations and expression) | ||
| Mutation-specific phenotype correlations | Dominant-negative mutations: earlier onset and increased penetrance of HM? | De novo LoF mutations common in pediatric MDS | N-terminal mutations: 90% penetrance of AML |
| T354M: mostly early-onset MDS/AML | C-terminal mutations: 50% penetrance of AML | ||
| R396Q and R398W: mostly immune defects and MDS | |||
| Common second hits (Figure 1D-F) | Somatic RUNX1 [including UPD 21, +21(q)], PHF6, BCOR, NOTCH1, EZH2 | −7, +8, ASXL1, NRAS, WT1, STAG2, KRAS, SETBP1 | Somatic CEBPA, WT1, GATA2, KIT, TET2, EZH2 |
| Mutation spectrum | Overlap between germline and somatic mutations | Distinct mutational pattern for germline (truncating, ZF2 missense and indels, intronic enhancer) and somatic (mostly ZF1 missense and some ZF2 missense) mutations | Overlap between germline and somatic mutations (N- and C-terminal regions; however poor sequence coverage in past may have masked some mutations) |
| Mode of germline mutation | Inherited predominant, de novo infrequent (except large chromosome deletions) | Inherited and de novo mutations frequently reported | Wholly inherited, with no published reports of de novo mutation |
| NGS Gene coverage gnomAD | Complete (canonical transcript) | Complete | Poor (<20× for ∼70% of coding region) |
| Age of onset, median, y | 29 | 19 | 23 |
| Age of onset, range (y), % | |||
| <10 | 17.1 | 13.4 | 25.9 |
| 11-20 | 13.0 | 39.7 | 22.4 |
| 21-30 | 14.6 | 21.5 | 17.3 |
| 31-40 | 11.4 | 10.6 | 15.5 |
| 41-50 | 19.5 | 9.0 | 8.6 |
| 51-60 | 16.3 | 3.9 | 8.6 |
| >61 | 8.1 | 1.9 | 1.7 |
| Presymptomatic monitoring and treatment | Patient monitoring depends on local guidelines and expert opinion | ||
| Chemotherapy (GATA2, RUNX1, CEBPA), followed by HSCT (GATA2, RUNX1). Evidence-based conclusions on best treatments and monitoring options not available because of small cohort sizes and lack of data aggregation for rare disorders. | |||
| Prognosis | Not well defined for germline. Poor for sporadic/somatic RUNX1 mutated. | Poor for leukemic patients. HSCT for immune deficiency and leukemia prevents progression with favorable outcome. | Generally favorable long-term outcomes with chemotherapy alone |
aCML, atypical chronic myeloid leukemia; AFLP, amplified fragment length polymorphism; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; CNV, copy number variant; HCL, hairy cell leukemia; HSCT, hematopoietic stem cell transplantation; LoF, loss of function; MDS, myelodysplastic syndrome; MLPA, multiplex ligation-dependent probe amplification; NGS, next-generation sequencing; NK, natural killer; SNP, single nucleotide polymorphism; SNV, single nucleotide variant; T-ALL, T-cell acute lymphoblastic leukemia; T-NHL, T-cell non-Hodgkin lymphoma; WES, whole-exome sequencing; WGS, whole-genome sequencing; ZF, zinc finger.
From somatic testing in blood cancers, adequate distinction between germline vs somatic mutations is also not performed routinely, and the standard diagnostic analyte for blood cancers is blood or bone marrow, in which somatic mutations may appear indistinguishable from germline variants without the testing of other tissues. Other limiting factors are our incomplete knowledge of the genes involved in FHM, the often poor recognition of intronic or synonymous or missense variants that affect splicing, and the challenge of detecting copy number variants from gene panels or whole-exome sequencing.14 Further, causal intronic and promoter/enhancer variants and those affecting mRNA stability are not routinely assayed by the most commonly used technologies and prediction algorithms. Thus, a genetic test without a positive result should not be overinterpreted as negative, and a range of different technologies should be considered to detect all mutation types (Table 1). Equally, identification of a single variant in an appropriate gene does not automatically imply causality to that variant, regardless of clinical urgency, such as the selection of sibling donor transplants. Limited reports indicate that transplanting a predisposed or preleukemic marrow results in a poor clinical outcome, which may include poor engraftment or subsequent donor cell leukemia.15-20
Therefore, it is extremely difficult to estimate the total number of families and patients diagnosed with germline mutations, and the numbers presented here are likely to be a substantial underestimate of true FHM disease incidence, including that caused by RUNX1, CEBPA, and GATA2. Here, we discuss what is known about relevant biological, clinical, and genetic aspects of FHM associated with germline mutations in these 3 TFs.
RUNX1 mutations in FPD-MM
RUNX1 is arguably the founding member of the FHM gene collection,1 with germline mutations described in >200 families worldwide,21,22 leading to the autosomal dominantly inherited FPD-MM (Online Mendelian Inheritance in Man [OMIM] 601399).23,24 RUNX1 encodes a TF that is a master regulator of hematopoiesis, with knockout animal models demonstrating a failure of definitive hematopoiesis.25 RUNX1 mediates its transcriptional effects through heterodimerization with the core binding factor subunit β, which interacts with the DNA binding RUNT homology domain.26,27 Three major protein isoforms have been characterized as being expressed from the RUNX1 locus, controlled by 2 promoters. The P1 promoter controls expression of the longest isoform (RUNX1c; NM_001754.4), whereas the P2 promoter produces transcripts for isoforms RUNX1b (NM_001001890.3) and RUNX1a (NM_001122607.2), the latter of which lacks the transactivation domain and may act as a dominant-negative regulator.28,29 RUNX1c is the predominantly expressed isoform in adult hematopoiesis; it is a marker of definitive hematopoiesis and is highly expressed in hematopoietic stem cells (HSCs).28,30,31 Interestingly, several FPD-MM families have been described with deletion of P1 and/or the RUNX1c-specific exons 1-2, leaving RUNX1a,b intact, which suggests that dysregulation of RUNX1c may be the major mediator of the FPD-MM phenotype.32-35
Clinical presentation and penetrance of RUNX1-associated HM
Platelet counts in FPD-MM are most often mild to moderately low (70-145 × 109/L), but they can be lower or within the low-normal range in mutation carriers.36-38 Platelet functional studies frequently show an aspirin-like defect on platelet aggregometry and α/δ-granule deficits.37,39,40 Carriers can show evidence of premalignant bone marrow abnormalities, most commonly dysmegakaryopoiesis36,38,41,42 ; in some cases, they have aberrant expression of cell surface markers, such as CD123, which is a characterized marker of leukemic stem cells.41,43 Few nonhematological phenotypes have been described, with the exception of eczema/psoriasis, which is emerging as a possible recurrent feature.35,44-47
Despite having the highest median age of onset of the TFs discussed here (Table 1; 29 years), childhood-onset (<18 years) malignancy is observed in 50% of families. In our local cohort of RUNX1 families, we found a 43% cumulative incidence of HM by 50 years, increasing to 79% by age 70 years (age range at diagnosis, 3-65 years; A.L.B., C.N.H., H.S.S., unpublished data).
Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are the most frequent HMs that develop, but ∼25% of FPD-MM families also report lymphoid malignancies of varying subtypes, with T-cell acute lymphoblastic leukemia being most common (Table 1; see Brown et al21 ).1,15,21,35,36,48-54 Analysis of the intersection of different germline mutations with HM subtype has not revealed any association of particular mutations or mutation types with HM phenotype.21 This is likely due to the large intrafamily heterogeneity for age of onset and HM phenotype, which complicates the counseling of individuals within a family, even though they carry the same germline RUNX1 mutation.
Spectrum of RUNX1 germline mutations
Germline mutations in RUNX1 encompass partial and whole gene deletions and frameshift, stop-gain, and missense mutations (ClinGen recommends using RUNX1c for variant annotation),55 indicating that pathogenic disruption of RUNX1 activity may occur through different mutational mechanisms.21 Although frameshift and stop-gain mutations occur throughout the protein, missense mutations are primarily confined to the RUNT domain and frequently affect DNA binding residues.21,23 Complete deletion of the RUNX1 locus suggests haploinsufficiency as a mechanism of predisposition; however, not all mutations fit this pattern, because some may also have a dominant-negative capacity.37,56,57 For example, RUNT domain missense mutations, such as R201Q, can have a loss of DNA binding activity (ie, loss of function [LoF]) but still retain core binding factor subunit β binding, which may further subvert or impact the activity of RUNX1-associated functions in a dominant-negative manner, as demonstrated in in vitro transactivation assays.37,56 Whether different mutation types predispose more or less strongly to malignancy is still an open question.37,56,57 The frequent somatic mutation of the second RUNX1 allele in progression to HM supports the concept that a further reduction in activity below haploinsufficiency may be more innately leukemogenic.37,57
Germline RUNX1-associated somatic mutations
Aggregating tumor genomic information secondary to germline RUNX1 mutations has shown that somatic mutation of the second RUNX1 allele is frequently associated with malignancy (including duplication of the germline mutation through trisomy 21 or uniparental disomy). Other recurrent somatically mutated genes include PHF6, BCOR, WT1, and TET2 (Figure 1D). Compared with somatic RUNX1 comutation in sporadic myeloid disease, PHF6 and BCOR are also reported to be frequently mutated with RUNX158-61 ; conversely ASXL1 is frequently comutated with RUNX1 in sporadic HM, but it is not the dominant somatic comutation in FPD-MM (Figure 1D). Somatic mutation in RUNX1 has not been observed in carriers prior to the development of malignancy, whereas premalignant somatic mutations have been reported in TET2, DNMT3A, KRAS, and SRSF2.51,52,62-64 Mechanistically, acquisition of secondary pathogenic somatic mutations may be the result of increased mutagenic processes in germline RUNX1 carriers,63,65 characterized as early-onset clonal hematopoiesis, and potentially a result of dysregulated DNA repair pathways associated with RUNX1 mutation.66
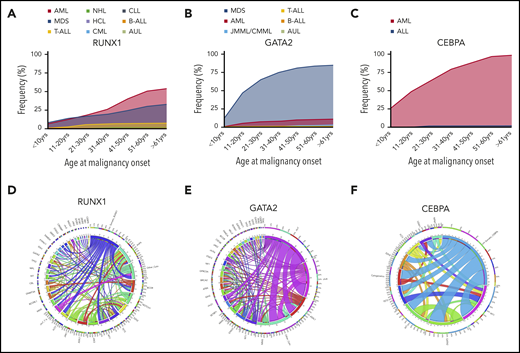
Incidence of different HMs and frequently co-occurring mutations associated with germline TF mutations. Incidence of HMs (first presentation) by age group associated with germline mutations in RUNX1 (n = 123) (A), GATA2 (n = 312) (B), and CEBPA (n = 58) (C). Circos plot showing the relative percentages of frequently co-occurring somatic alterations (mutations and cytogenetic abnormalities) in patients with germline mutations in RUNX1 (D), GATA2 (E), and CEBPA (F). The variables are arranged clockwise in descending order from the most frequent to the least frequent and are distinguished by different colors. The inner circle shows the absolute number of samples with mutations in each gene as indicated. The outer circle shows the percentage of cases with comutation of other genes (indicated by designated gene color from inner circle) for each gene. Co-occurring alterations are also shown as paths emerging from 1 to the other with widths proportional to the number of cases. ALL, acute lymphoblastic leukemia (including T- and B-cell subtypes); AUL, acute undifferentiated leukemia; B-ALL, B-cell acute lymphoblastic leukemia; chr5, del5q; chr7, monosomy 7, del 7q or der (1;7); chr8, trisomy 8; chr21, trisomy 21; Cyto, cytogenetic changes; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; HCL, hairy cell leukemia; JMML, juvenile monocytic leukemia; NHL, non-Hodgkin lymphoma; T-ALL, T-cell acute lymphoblastic leukemia. See supplemental References (available on the Blood Web site).
Incidence of different HMs and frequently co-occurring mutations associated with germline TF mutations. Incidence of HMs (first presentation) by age group associated with germline mutations in RUNX1 (n = 123) (A), GATA2 (n = 312) (B), and CEBPA (n = 58) (C). Circos plot showing the relative percentages of frequently co-occurring somatic alterations (mutations and cytogenetic abnormalities) in patients with germline mutations in RUNX1 (D), GATA2 (E), and CEBPA (F). The variables are arranged clockwise in descending order from the most frequent to the least frequent and are distinguished by different colors. The inner circle shows the absolute number of samples with mutations in each gene as indicated. The outer circle shows the percentage of cases with comutation of other genes (indicated by designated gene color from inner circle) for each gene. Co-occurring alterations are also shown as paths emerging from 1 to the other with widths proportional to the number of cases. ALL, acute lymphoblastic leukemia (including T- and B-cell subtypes); AUL, acute undifferentiated leukemia; B-ALL, B-cell acute lymphoblastic leukemia; chr5, del5q; chr7, monosomy 7, del 7q or der (1;7); chr8, trisomy 8; chr21, trisomy 21; Cyto, cytogenetic changes; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; HCL, hairy cell leukemia; JMML, juvenile monocytic leukemia; NHL, non-Hodgkin lymphoma; T-ALL, T-cell acute lymphoblastic leukemia. See supplemental References (available on the Blood Web site).
Reflecting the diverse germline phenotypic manifestations, RUNX1 is also somatically mutated across a spectrum of sporadic HM subtypes, including MDS/AML (10%),58,59 chronic myeloid leukemia blast crisis (40%),67 therapy-related myeloid neoplasm (16%),68,69 T-cell acute lymphoblastic leukemia (18%)70 and HM transformation in patients with severe congenital neutropenia (64%)71 or Fanconi anemia (20%).72 Interestingly, the spectrum of somatic mutation types mirrors that seen in germline predisposition, making deconvolution of germline vs somatic mutations in a patient, being screened at malignancy presentation, complex.
Germline GATA2 mutations in GATA2 deficiency syndrome
A range of phenotypes, resulting from heterozygous autosomal dominantly inherited and de novo germline GATA2 mutations, began to be described in 2011.3,73,74 Now called GATA2 deficiency syndrome (OMIM: 137295 covers the majority of symptoms), there are >100 families and 122 individual cases (7 pediatric cases confirmed de novo).8,9,75-86 GATA2 is critical for normal adult hematopoiesis by regulating maintenance and self-renewal of HSCs, as well as differentiation to blood progenitors and mature blood cells (myeloid cells, B and natural killer [NK] lymphocytes, megakaryocytes, and mast cells). It plays an important role in endothelial to hematopoietic transition and definitive hematopoiesis during development. GATA2 expression is high in endothelial cells, HSCs, and myeloerythroid progenitors.87 GATA2 is also crucial in the generation of the lymphatic system, especially in lymphatic valve development.74,88,89
The GATA2 gene has separate hematopoietic-specific and nonhematopoietic promoters that generate transcripts with different 5′UTRs, yet generate the same coding sequence. The protein contains N-terminal and C-terminal zinc finger (ZF1 and ZF2) domains, a nuclear localization signal, and poorly defined transactivation domains. Although both ZFs are important for DNA binding, ZF2 along with R396 and R398 residues directly contacts the consensus DNA sequence (A/T)GATA(A/G) within regulatory regions of target genes.90,91 GATA2 is a chromatin decondensing “pioneer” TF providing access to other TFs. It associates with TFs, such as RUNX1, SCL/TAL1, PU.1, MYB, and androgen receptor.92-95 Notably, GATA2 interacts with androgen receptor to activate GATA2-dependent androgen-responsive genes without directly binding DNA. Different GATA2 mutations may impact interactions with ≥1 of these binding partners, contributing to differences in predisposition or penetrance.
Clinical presentation and penetrance of GATA2-associated HM
Germline pathogenic variants in GATA2 predispose to a range of malignant and nonmalignant phenotypes and have been described as a protean disorder of hematopoiesis, lymphatics, and immunity.10 Phenotypes may incorporate hematological, neoplastic, infectious, pulmonary, vascular, lymphatic, auditory, and dermatological features. These can be partially explained by associated immunological phenotypes (OMIM 614172) encompassing viral, bacterial, and fungal infections associated with reduced B and NK lymphocytes, NK cells, and dendritic cells (monocytopenia and mycobacterial infection [MonoMAC] and dendritic cell, monocyte, B and NK lymphocyte [DCML] deficiency).73,96,97 The broad range of phenotypes and penetrance implicate the involvement of stressors (eg, environmental) that increase the likelihood of developing particular phenotypes.10 Obvious (human papillomavirus) infection(-associated) phenotypes, such as warts and genital warts, are well known, whereas other disease associations are not as evident. The combination of HSC exhaustion and hematopoietic stress from repeated infections may drive clonal evolution, leading to the development of malignancies in patients. Although patients may present with single or multiple cytopenias (eg, B cells, NK cells, monocytes, dendritic cells), a subset of patients presents with MDS/AML without obvious prior cytopenia.3,8
A majority (75%) of individuals develop myeloid neoplasms (MDS, AML, chronic myelomonocytic leukemia), although there is likely to be ascertainment bias.9,86 MDS is the most common first malignancy seen in GATA2 carriers, unlike CEBPA carriers, in whom AML is predominant (Figure 1B-C). Unlike RUNX1 and CEBPA, there is a noticeable peak in the onset of myeloid malignancy in GATA2 carriers in the second decade of life (Figure 2).
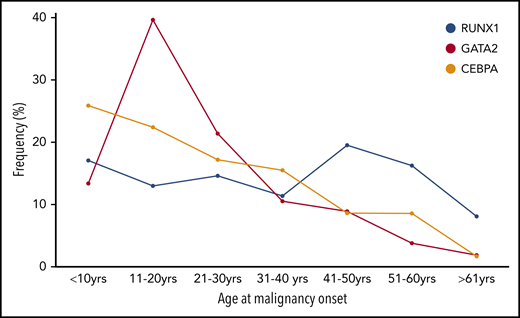
Age at onset of first HM in individuals with germline TF mutations. The frequency (%) of the total number of individuals who developed HM (first diagnosis) is plotted for each 10-year age range with germline RUNX1, GATA2), and CEBPA.
Age at onset of first HM in individuals with germline TF mutations. The frequency (%) of the total number of individuals who developed HM (first diagnosis) is plotted for each 10-year age range with germline RUNX1, GATA2), and CEBPA.
Spectrum of GATA2 germline mutations
Since the first literature on germline GATA2 mutations as a cause of hematological disease, truncating mutations and GATA2 gene deletions have indicated an LoF (haploinsufficiency) disease mechanism.3,8,73,88 Truncating mutations are distributed throughout the gene, in contrast to missense mutations, which are clustered in ZF2 and the C-terminal region (particularly R396 and R398). No germline missense mutations have been described to date within the N terminus up to and including the ZF1 region. Noncoding mutations affecting the enhancer element in intron 4 are observed in ∼10% of patients with GATA2 deficiency.86 Further, a recurrent synonymous p.T117 = (c.351C>G) variant activates a cryptic splice donor site, resulting in a premature termination (p.V118Qfs*55).98
Comparing disease phenotypes associated with missense mutations or truncating mutations indicates that the missense mutations are more likely causing AML, whereas there is no difference for MDS.9 Several lines of evidence suggest that haploinsufficiency predisposes to all GATA2 deficiency phenotypes, whereas some variants, such as T354M, with partial LoF for DNA binding and transactivation, leads to HM and immunodeficiency but not lymphedema (>50 carriers reported).3,90 Lymphedema appears to require haploinsufficiency, which includes GATA2 large deletions, truncating mutations, and missense mutations (such as R396Q), leading to complete LoF and intronic enhancer element mutations.90
Although numbers are small, analysis of recurrent germline GATA2 missense mutations (T354M, R396Q, and R398W) affecting ZF2 and the C-terminal domain revealed that these variants may predispose differently to HM or immunodeficiency.90 T354M was primarily seen in MDS/AML or AML, whereas R396Q and R398W were more common in immunodeficiency with MDS or chronic myelomonocytic leukemia.
T354M displays LoF of DNA binding and transactivation while binding more efficiently to the master hematopoietic differentiation regulator PU.1 (SPI1).90 It is not clear whether T354M impacts downstream targets of GATA2 and PU.1 in driving leukemia development. Interestingly, a subsequent study noted downregulation of a PU.1 target gene signature in individuals carrying T354M and displaying monoallelic expression.99
Allelic imbalance was first reported for GATA2 in normal karyotype AML and was linked to increased DNA methylation of the lower expressed allele.100 Recently, in a family carrying a germline T354M variant, allele-specific expression was linked to increased GATA2 promoter DNA methylation and deposition of histone H3 lysine 4 tri-methylation (H3K4me3) (closed chromatin).99 Intriguingly, expression of both alleles was seen in 2 unaffected carriers, whereas re-expression of the wild-type allele was linked with a spontaneous improvement in monocytopenia and neutropenia in another individual. Further, a study looking at allelic imbalance in 499 AML samples across genes recurrently mutated in AML identified GATA2 as having the greatest allelic imbalance, although most samples did not carry GATA2 mutations.101 It is possible that factors driving allelic imbalance of GATA2 play unrecognized roles in predisposing to or protecting against the detrimental leukemogenic effects of mutated alleles.
Germline GATA2-associated somatic mutations
Germline CEBPA mutations and AML predisposition
Germline mutations in CCAAT enhancer binding protein α (CEBPα) predisposing to AML (OMIM: 601626) were first described in 2004,2 yet patients and families identified with germline CEBPA mutations are probably very rare (0.65% of AML patients),104 and only 68 patients from 26 families have been described in the literature.2,104-119
The single-exon gene CEBPA encodes CEBPα, which is the founder of the 6-CEBP family of TFs.120 All CEBP TFs contain a basic leucine zipper (bZIP) domain at the C terminus and form a subgroup within the leucine zipper family of TFs.121 The CEBPα zipper domain is required for dimerization, and the adjacent basic region is responsible for DNA binding, thereby promoting transcription of target genes.122-124 The N terminus is unique to CEBPα, containing 2 transactivation domains that regulate transcription control and protein interaction.123
CEBPα generates 2 isoforms from alternative initiation codons: the long isoform (p42) is 358 aa, and the short isoform (p30) is 239 aa and lacks a transactivation domain.125-128 The p30 isoform maintains dimerization and DNA binding capacities and, hence, is able to inhibit p42 activity.126 Both isoforms are coexpressed in a range of tissues, with p42 generally being more abundant.126,128
Myeloid differentiation is driven by p42 inducing the expression of target genes (eg, SPI1, CSF3R, IL6R, GFI1B, KLF1, and KLF5).120,121,129 CEBPα-induced gene expression drives the cell fate of hematopoietic stem and progenitor cells towards the myeloid lineage.120 Expression of CEBPα decreases as cells mature toward becoming granulocytes and neutrophils.
Clinical presentation and penetrance of CEBPA-associated HM
Unlike the recognizable preleukemic clinical manifestations associated with RUNX1 and GATA2,130 mutations in CEBPA predispose to de novo AML without a prior phenotype. Almost irrespective of DNA sequencing technique, the high guanine-cytosine (GC)-rich regions in the middle of the CEBPA coding exon have biased germline and somatic mutation analyses. Combined with the reduced penetrance observed in the limited families described as having C-terminal missense mutations, this complicates clinical recognition of familial CEBPA AML.113 Early-onset AML would be the main indication for a possible germline-causing disease, because 81% of patients were diagnosed with AML before the age of 40 years (Figure 1C; Table 1).
Spectrum of CEBPA germline mutations
Germline and somatic mutations in CEBPA are clustered at the N terminus or within the C-terminal bZIP domain. Commonly, the germline (often protein-truncating) mutation affects the N terminus, whereas the acquired mutation arises in the C-terminal bZIP region (predominantly missense or in-frame indels).121 Although numbers are limited, families with germline N-terminal mutations display a higher degree of penetrance (90%)112 compared with the families with germline C-terminal mutations (50%).113
The CEBPA mutations that predispose to AML are generally considered to have a dominant-negative effect. The N-terminal truncating mutations destroy p42 but enable expression of p30 (16-21% of transactivation activity of p42). The C-terminal mutations abolish DNA binding (basic region) or dimerization (bZIP domain).121,122
There are minimal data describing mutations outside the N terminus or C terminus of CEBPA, which may result from the high (75%) GC content and a trinucleotide repeat in the coding region, leading to poor median coverage for polymerase chain reaction–based enrichment approaches and sequencing.131 Coverage statistics from exomes in gnomAD show that there is insufficient median coverage (>20 fold) to reliably call and interpret variants for ∼70% of the CEBPA coding region.132
Germline CEBPA-associated somatic mutations
Identifying common trends for AML patients with CEBPA mutations remains challenging because of small cohort sizes.121 Nonetheless, the majority of germline CEBPA-mutated AML patients have 2 (biallelic) mutations: 1 N-terminal mutation and 1 bZIP mutation. Similarly to RUNX1, but in contrast to GATA2, biallelic CEBPA mutations in myeloid malignancies can occur somatically (ie, without a germline predisposing CEBPA mutation). In addition to biallelic CEBPA AML, somatic mutations in combination with germline CEBPA mutations are recurrently reported in GATA2, WT1, KIT, and TET2 (Figure 1F).112 Comparison of CEBPA double-mutant and single-mutant AML patients revealed distinct RNA expression profiles and favorable disease outcome for those with double-mutant disease.133
Functional models for TF-associated predisposition and progression to HM
Although mouse genetic models have demonstrated the importance of Runx1 and Gata2 as master regulators of hematopoiesis, they have been less amenable to the study of leukemia development or treatment, because heterozygous RUNX1 and GATA2 LoF mutation mouse models do not develop a bleeding disorder or leukemia, and biallelic mutations (LoF and missense) are embryonically lethal when present in germline configuration.56,94,134,135 In contrast, homozygous Cebpa LoF mice survive until birth and have a lack of neutrophils as a result of defective granulocyte colony-stimulating factor signaling, but they die shortly thereafter from a nonhematological metabolism defect.136 In adult hematopoiesis, stem cell stress through competitive reconstitution has shown that Gata2-heterozygous mice have significantly fewer stem cells and a larger proportion of quiescent cells and perform poorly in competitive reconstitution assays.137 In short-term assays, homozygous conditional Runx1 deficiency has little effect on HSC reconstitution ability; however, over time, mice display age-related stem cell exhaustion, greater susceptibility to leukemia induced by secondary mutations, and, in some cases, spontaneous development of myeloproliferative/myelodysplastic and lymphoma phenotypes.135,138-140 In contrast to Gata2 and Runx1 deficiency models, Cebpa-deficient HSCs show increased competitive repopulation activity.141 Interestingly, these different HSC reconstitution abilities/stress adaptation responses correlate with the level of antecedent cytopenias (or lack thereof for CEBPA) in the human germline disorders, suggesting that a different HSC response to stress may be an important determinant of disease course.
In vitro models, in particular induced pluripotent stem cells (iPSC), are potentially more amenable to rapid genetic manipulation. GATA2−/− human iPSC have almost completely abolished hematopoietic differentiation with a significant reduction in hematopoietic stem and progenitor cells (HSPCs), whereas GATA2−/+ lines have a smaller reduction in HSPCs.142 For RUNX1, iPSC generated from patients with different mutations confirmed the haploinsufficient baseline megakaryocyte defects that manifest as frequent thrombocytopenia in humans carriers, and cells with duplication of the RUNX1 mutation (trisomy 21) or “dominant- negative” missense mutations were associated with increased HSCs and granulocyte-monocyte progenitors, respectively, suggesting an additional functional step toward leukemia.57,143 This supports that the degree of reduction in gene activity is an important contributor to the predisposition and progression to leukemia, a concept that is clearly illustrated by the frequency of second somatic mutations in the leukemic progression of germline RUNX1- and CEBPA-mutated disorders. However, the accumulated somatic profiling of these FHMs indicates that mutation of multiple genes and pathways can contribute to the development of malignancy; future in vivo and in vitro models need to be more sophisticated and incorporate mutations in multiple genes to properly model disease progression. In addition, as outlined as part of ongoing risk assessment and monitoring (Table 2), longitudinal studies of carriers to monitor the appearance of additional mutations in real time will be crucial for our full understanding of the natural history of progression to malignancy and the development of new models and therapies.
Challenges and suggested improvements for identification, monitoring, and treatment of familial HMs
| Detection and classification | Population- and cohort-specific frequency of FHM gene defects |
| Comprehensive personal and family history for all patients at diagnosis, with urgent attention if SCT is needed | |
| Comprehensive and integrative genomics screening: capturing indels and noncoding variants, as well as frank coding variants | |
| Appropriate germline reference material for interpretation of tumor molecular screening results (hair bulbs, MSC, fibroblasts) | |
| Continued development and correct application of expert panel gene-specific ACMG guidelines (eg, ClinGen RUNX1 rules) | |
| Appropriate expert review (MDT) of variants before clinical notification: focus on variants of uncertain significance or variants with existing classification discordance | |
| Risk assessment and monitoring | Gene-specific longitudinal cohort studies, including comprehensive phenotype screening and monitoring protocols |
| Comprehensive phenotyping: age of onset in family, including evidence of anticipation, history of infections (HSC stress), partial penetrance, asymptomatic carriers | |
| Cohort aggregation of genomics data (eg, RUNX1db) for analysis of germline mutation–specific associations, acquired mutations in blood/marrow, germline modifiers, epigenetics and gene expression (eg, allelic imbalance) | |
| High-depth molecular monitoring of blood/marrow from asymptomatic carriers for progression mutations | |
| New and effective treatments | Mutation-specific in vivo and in vitro systems for disease modeling, including progression, drug screening, and preclinical studies |
| Premalignant interventions (eg, ameliorate HSC stress, target early somatic drivers) | |
| International clinical trial consortia for rapid testing of new therapeutic options for rare FHM disorders |
| Detection and classification | Population- and cohort-specific frequency of FHM gene defects |
| Comprehensive personal and family history for all patients at diagnosis, with urgent attention if SCT is needed | |
| Comprehensive and integrative genomics screening: capturing indels and noncoding variants, as well as frank coding variants | |
| Appropriate germline reference material for interpretation of tumor molecular screening results (hair bulbs, MSC, fibroblasts) | |
| Continued development and correct application of expert panel gene-specific ACMG guidelines (eg, ClinGen RUNX1 rules) | |
| Appropriate expert review (MDT) of variants before clinical notification: focus on variants of uncertain significance or variants with existing classification discordance | |
| Risk assessment and monitoring | Gene-specific longitudinal cohort studies, including comprehensive phenotype screening and monitoring protocols |
| Comprehensive phenotyping: age of onset in family, including evidence of anticipation, history of infections (HSC stress), partial penetrance, asymptomatic carriers | |
| Cohort aggregation of genomics data (eg, RUNX1db) for analysis of germline mutation–specific associations, acquired mutations in blood/marrow, germline modifiers, epigenetics and gene expression (eg, allelic imbalance) | |
| High-depth molecular monitoring of blood/marrow from asymptomatic carriers for progression mutations | |
| New and effective treatments | Mutation-specific in vivo and in vitro systems for disease modeling, including progression, drug screening, and preclinical studies |
| Premalignant interventions (eg, ameliorate HSC stress, target early somatic drivers) | |
| International clinical trial consortia for rapid testing of new therapeutic options for rare FHM disorders |
ACMG, American College of Medical Genetics and Genomics; MDT, multidisciplinary team; MSC, mesenchymal stem cell.
Current treatment approaches and novel therapeutic opportunities
Caveats on using family members as donors aside, for all forms of FHM, hematopoietic stem cell transplantation (HSCT) is the only curative therapy, with controversial discussions around when and if it would be appropriate to use it preemptively.144 In sporadic myeloid malignancies, somatic mutation of RUNX1 is associated with high-risk disease,7 with the acquisition of a second mutation conferring an increasingly poor prognosis.58 Although a systematic study of outcome of malignancy in germline RUNX1 carriers has not been done, it is anecdotally consistent, in our experience, that similarly high-risk HM occurs as a result of FPD-MM, ∼30% of which are biallelic RUNX1 mutations (Figure 1D). For GATA2 mutation, HSCT is used for treatment of immunodeficiency/bone marrow failure and HM, with a T-cell–depleted reduced-intensity regimen recently outlined to treat infectious and respiratory complications of GATA2 mutation.145 Biallelic CEBPA malignancies, when sporadic, fall into a favorable risk category; consistent with this, FHM with CEBPA mutations can have durable remission responses to chemotherapy, with HSCT reserved for complex cases.146,147 Moving beyond the standard options of chemotherapy and stem cell transplantation, new approaches have recently implicated new compounds for the treatment of FHM. For example, recent preclinical studies have identified addition of RUNX1-mutated cells to residual RUNX1 activity, which could be exploited with the use of BET inhibitors for a synthetic lethal result.148 Similarly, a number of in vitro drug screens in RUNX1-mutated AML have identified glucocorticoids, phosphatidylinositol 3-kinase inhibitors, and JAK inhibitors as selectively effective.61,149 In colorectal cancer and lung cancer cells, downregulation of GATA2 expression was shown to be synthetic lethal for oncogenic RAS mutations.150,151 RAS mutations are not uncommon in germline GATA2 individuals with myeloid malignancy (Figure 1E), opening up possibilities for new approaches to treatment.
Conclusions and future directions
Although the advent of next-generation sequencing has advanced the field greatly in terms of identifying individuals with germline variants conferring a risk for HM, the relative rarity of individual predisposition disorders means that accumulation of significant bodies of data in the literature takes time and global effort. As a result, optimal conditions for the identification and management of individuals with predisposition to HM are still primarily provided by expert opinion, rather than official guidelines.6,23,147,152,153
Here, we have compared and contrasted the information that is available on malignancy development secondary to germline mutations in RUNX1, GATA2 and CEBPA, including technical considerations for successful detection of germline variants, through to the availability of information on malignant and nonmalignant phenotypes, penetrance, and preleukemic states, as well as the tools and models for furthering our understanding of these disorders. In Table 2, we have summarized the key challenges and opportunities inherent in each of these aspects to encourage new ideas for research and collaboration to advance the field.
To overcome the difficulty of collecting and aggregating a sufficient weight of clinical information upon which to base cogent recommendations, international collaborative efforts to collate genetic and clinical information into global FHM cohorts will be key for the next phase of these disorders. Important for facilitating this are the increasing numbers of clinical centers specializing in FHM and tools available for online data sharing, curation, and analysis. Collectively, these initiatives will provide a detailed understanding of molecular progression to malignancy, as well as the refinement of risk assessment and monitoring, and provide a platform to design and test therapeutic interventions to ideally prevent malignancy.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank patients and family members worldwide for willingness to participate in research. They also thank past and present laboratory members and colleagues; in particular, Peer Arts, Parvathy Venugopal, Claire Homan, Nur Hezrin Shahrin, Julia Dobbins, and Jesse Cheah helped to collate information, generate text, and design tables and diagrams.
This work was supported by the Leukaemia Foundation of Australia, the Cancer Council SA’s Beat Cancer Project on behalf of its donors, and the State Government of South Australia through the Department of Health (project grant APP1125849 and Principal Research Fellowship to H.S.S.). The authors are supported by grants from the National Health and Medical Research Council of Australia (project grants APP1145278 and APP1164601 and Principal Research Fellowship APP1023059 [H.S.S.]) and the Royal Adelaide Hospital Research Foundation.
Authorship
Contribution: All authors performed background research, analyzed and interpreted data, and wrote, critically reviewed, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hamish S. Scott, Department of Genetics and Molecular Pathology, Centre for Cancer Biology, SA Pathology, PO Box 14, Rundle Mall, Adelaide, SA 5000, Australia; e-mail: hamish.scott@sa.gov.au.
