

Key Points
E2A-PBX1, through direct interactions, serves as a coactivator for RUNX1 in the development of pre-B ALL.
The E2A-PBX1 transcriptome includes RUNX1, indicating an E2A-PBX1 role in pre-B ALL through enhancement of the RUNX1 autoregulatory loop.

Abstract
E2A, a basic helix-loop-helix transcription factor, plays a crucial role in determining tissue-specific cell fate, including differentiation of B-cell lineages. In 5% of childhood acute lymphoblastic leukemia (ALL), the t(1,19) chromosomal translocation specifically targets the E2A gene and produces an oncogenic E2A-PBX1 fusion protein. Although previous studies have shown the oncogenic functions of E2A-PBX1 in cell and animal models, the E2A-PBX1–enforced cistrome, the E2A-PBX1 interactome, and related mechanisms underlying leukemogenesis remain unclear. Here, by unbiased genomic profiling approaches, we identify the direct target sites of E2A-PBX1 in t(1,19)–positive pre-B ALL cells and show that, compared with normal E2A, E2A-PBX1 preferentially binds to a subset of gene loci cobound by RUNX1 and gene-activating machineries (p300, MED1, and H3K27 acetylation). Using biochemical analyses, we further document a direct interaction of E2A-PBX1, through a region spanning the PBX1 homeodomain, with RUNX1. Our results also show that E2A-PBX1 binding to gene enhancers is dependent on the RUNX1 interaction but not the DNA-binding activity harbored within the PBX1 homeodomain of E2A-PBX1. Transcriptome analyses and cell transformation assays further establish a significant RUNX1 requirement for E2A-PBX1–mediated target gene activation and leukemogenesis. Notably, the RUNX1 locus itself is also directly activated by E2A-PBX1, indicating a multilayered interplay between E2A-PBX1 and RUNX1. Collectively, our study provides the first unbiased profiling of the E2A-PBX1 cistrome in pre-B ALL cells and reveals a previously unappreciated pathway in which E2A-PBX1 acts in concert with RUNX1 to enforce transcriptome alterations for the development of pre-B ALL.
Introduction
The E2A proteins E47 and E12 belong to the E protein family of transcription factors (TFs), comprising E2A, E2-2, and HEB, and have previously been shown to regulate B- and T-lymphocyte development and differentiation through their effects on tissue-specific gene expression programs.1-4 In B-cell development, E2A proteins directly activate a subset of B lineage–specific genes,5 indicating their roles in potentiating development-related transcription networks. In support, E2A-null mice exhibit a severe blockage at the earliest stage of committed B-cell development.6,7 The transcription functions of E2A are mediated by 3 conserved N-terminal activation domains (ADs), designated AD1, AD2, and AD3, and a C-terminal basic helix-loop-helix DNA-binding domain (DBD).3,8 Although the ADs mediate E2A interactions with transcriptional coactivators p300 (through both AD1 and AD2)9 and TAF4 (through AD3),8 the DBD mediates formation of E2A homodimers or heterodimers with other basic helix-loop-helix factors that bind to cognate E-box–containing enhancers to activate associated target genes.10,11
In pre–B-cell or pro–B-cell acute lymphoblastic leukemias (ALLs), the chromosomal translocations t(1;19)(q23;p13) and t(17;19) (q22;p13) target the E2A locus and generate fusions between the composite activation domains of E2A and the C-terminal DBDs, respectively, of pre–B-cell leukemia transcription factor 1 (PBX1)12,13 and hepatic leukemia factor (HLF).14,15 The resultant chimeric proteins, E2A-PBX1 and E2A-HLF, strongly activate gene reporters carrying the PBX1- or HLF-binding sites.16-18 Therefore, apart from reducing normal E2A activity by disrupting one E2A allele, the chimeric proteins are believed to modulate specific oncogenic gene programs to mediate cellular transformation and leukemogenesis in ALL. Consistent with this notion, the intact E2A ADs were shown to be critical for cellular transformation by E2A chimeric proteins.18,19 In addition, a previous in vitro study reported that E2A-PBX1 dimerization with other homeodomain-containing TFs, particularly HOXB7, through a HOX cooperativity motif (HCM) within the PBX1 portion mediated both DNA binding and cell transformation20 ; this action implies that E2A-PBX1 may induce leukemogenesis through activities of an E2A-PBX1/HOX complex. However, the E2A-PBX1–enforced cistrome and its interactome have not been thoroughly examined and, as a result, the molecular mechanisms underlying E2A-PBX1–mediated ALL development remain largely elusive.
Here, we used the human t(1;19)(q23;p13)–positive pre–B-cell line to determine genome-wide binding sites of E2A-PBX1 and found, surprisingly, that they are most enriched with the DNA-binding motif of RUNX1 but not with that of PBX1. We further show that the physical interaction of E2A-PBX1 and RUNX1 leads to the recruitment of E2A-PBX1 onto the tested direct targets of RUNX1, which include the RUNX1 locus, and that this pathway is dependent on the RUNX1 interaction but not on the DNA-binding activity of E2A-PBX1. As a result, E2A-PBX1 is crucially involved in activation of RUNX1-related gene programs, both by direct activation of RUNX1 targets and by reinforcing the RUNX1 transcriptional autoregulatory loop, thereby contributing to ALL leukemogenicity. Collectively, our findings unveil a previously unappreciated synergy between E2A-PBX1 and RUNX1 and provide critical insights into molecular mechanisms underlying E2A-PBX1–induced leukemogenesis, which will shed light on future therapeutic targets.
Materials and methods
Antibodies
A human E2A fragment (aa 26-125) was used to generate the E2AN1 antibody in rabbits (Strategic Diagnostics). Commercial antibodies are described in the supplemental Data (available on the Blood Web site). All animals used in this study were approved by Animal Studies Core, the UNC Lineberger Comprehensive Cancer Center.
Cell culture
Human pre-B leukemia lines, 697 (DSMZ ACC42) and RCH-ACV (DSMZ AC-548), were grown in RPMI-1640 with 10% fetal bovine serum and antibiotic cocktail. Lines with doxycycline-inducible stable expression of E2A-HA-FLAG, designated 697(E2A-hf), or E2A-PBX1-HA-FLAG, designated 697(E2A-PBX1-hf), were established as previously described.21
Recombinant proteins, pull-down, and coimmunoprecipitation assays
Full-length complementary DNAs for E2A-PBX1, RUNX1, and EBF1 were cloned in-frame to an N-terminal FLAG tag into pFastBac (Thermo Scientific) and expressed in Sf9 cells. Expressed proteins were immunopurified and subjected to pull-down assay as previously described.8 Standard expressed protein coimmunoprecipitation (Co-IP) assays in HEK293T cells are detailed in the supplemental Data.
Short hairpin RNA knockdown
Short hairpin RNAs (shRNAs) (supplemental Data) were from the National RNAi Core Facility (Academia Sinica). Lentivirus-transduced cells were transduced, selected, and subjected to proliferation and colony-forming unit (CFU) assays or used for RNA extraction and cell lysate preparation as described previously.21,22
RT-qPCR and ChIP-qPCR assays
Real-time quantitative polymerase chain reaction (RT-qPCR) and chromatin immunoprecipitation (ChIP)-qPCR assays were performed as detailed elsewhere.21 They are described further in the supplemental Data.
ChIP-sequencing, RNA-sequencing, and data analysis
ChIP-sequencing (ChIP-seq) and RNA-sequencing (RNA-seq) analyses were performed as previously described.21 Illumina high-throughput sequencing was performed by the Sequencing Core Facility of National Yang-Ming University Genome Center and the Epigenomics Core Facility at Weill Cornell Medical College. A detailed data analysis protocol and statistics of experiments are given in the supplemental Data.
Bone marrow transformation and CFU assay
E2A-PBX1 (wild-type [WT] or mutant) was cloned into murine stem cell virus–based vectors. The hematopoietic stem and progenitor cell (HSPC) stimulation, retroviral infection, and assays for outgrowth and immortalization were as previously described.22-24 CFU assays for 697 cells were conducted in a methylcellulose-based medium as outlined elsewhere.22,24
Results
E2A-PBX1 and E2A exhibit common but largely distinctive binding patterns in t(1;19)-positive pre-B ALL cells
Because the genomic binding site of a TF is generally determined by its DBD, it has been believed that E2A-PBX1 can be targeted to sites carrying the PBX1 DNA-binding motif, a notion supported by previous studies using electrophoretic mobility shift assay,25 reporter assay,18 and ChIP-on-ChIP assay.26 However, an unbiased genome-wide profiling, such as ChIP-seq, for determining E2A-PBX1 binding in ALL cells has been lacking. In addition, although antibodies specific for E2A-PBX1 (ie, the fusion junction region) (Figure 1A) are available, we found them unsuitable for ChIP-seq assays (data not shown) (supplemental Figure 1) despite their efficient immunoprecipitation of endogenous E2A-PBX1 from leukemic lines. Furthermore, whereas ChIP-seq with antibodies against either the N-terminus or the C-terminus of E2A-PBX1 and E2A is possible, it would still be difficult to differentiate between E2A-PBX1 and endogenous E2A or PBX2/3/4 (supplemental Figure 2) occupancies.
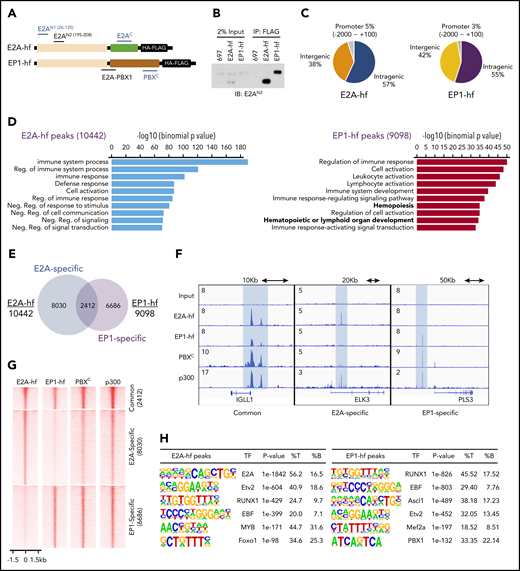
RUNX1 and EBF1 motifs are enriched at E2A-PBX1–bound genomic sites. (A) Schematic of C-terminal HA-FLAG–tagged E2A (E2A-hf) and E2A-PBX1 (EP1-hf) and the epitopes of antibodies used in ChIP-seq (blue) and immunoblotting (black) assays. (B) Immunoblots of E2A and E2A-PBX1 from stable doxycycline-induced 697 lines expressing either E2A-hf or EP1-hf. (C) Genomic distribution of E2A-hf (left) and E2A-PBX1 (EP1-hf, right) ChIP-seq peaks relative to gene loci. (D) Gene Ontology biological processes of E2A-hf– and E2A-PBX1-hf–associated ChIP-seq peaks using GREAT analysis.27 (E) Venn diagram showing the overlap between E2A-hf and EP1-hf ChIP-seq peaks. (F) ChIP-seq profiles of E2A-hf and EP1-hf in stable 697 lines and the C-terminal region of PBX1 (PBXC) and p300 antibodies in 697 lines at identified common or specific targets. (G) Heatmaps showing the ChIP-seq signals for E2A-hf and EP1-hf in stable 697 cell lines and for endogenous PBXC and p300 in the parental 697 line at identified common or specific sites in panel E. (H) Motif analyses of E2A-hf– and EP1-hf–occupied regions. %B, percentage of background sequences with motif of total background; %T, percentage of target sequences with motif of total targets.
RUNX1 and EBF1 motifs are enriched at E2A-PBX1–bound genomic sites. (A) Schematic of C-terminal HA-FLAG–tagged E2A (E2A-hf) and E2A-PBX1 (EP1-hf) and the epitopes of antibodies used in ChIP-seq (blue) and immunoblotting (black) assays. (B) Immunoblots of E2A and E2A-PBX1 from stable doxycycline-induced 697 lines expressing either E2A-hf or EP1-hf. (C) Genomic distribution of E2A-hf (left) and E2A-PBX1 (EP1-hf, right) ChIP-seq peaks relative to gene loci. (D) Gene Ontology biological processes of E2A-hf– and E2A-PBX1-hf–associated ChIP-seq peaks using GREAT analysis.27 (E) Venn diagram showing the overlap between E2A-hf and EP1-hf ChIP-seq peaks. (F) ChIP-seq profiles of E2A-hf and EP1-hf in stable 697 lines and the C-terminal region of PBX1 (PBXC) and p300 antibodies in 697 lines at identified common or specific targets. (G) Heatmaps showing the ChIP-seq signals for E2A-hf and EP1-hf in stable 697 cell lines and for endogenous PBXC and p300 in the parental 697 line at identified common or specific sites in panel E. (H) Motif analyses of E2A-hf– and EP1-hf–occupied regions. %B, percentage of background sequences with motif of total background; %T, percentage of target sequences with motif of total targets.
To overcome these issues, we constructed vectors encoding E2A and E2A-PBX1 proteins with C-terminal dual HA-Flag tags (E2A-hf and EP1-hf) (Figure 1A) and stably expressed them in 697 pre-B ALL cells in an inducible manner. Clonal lines were used in which E2A-PBX1 and E2A are expressed at close to endogenous levels (Figure 1B) and readily detected by anti-Flag immunoprecipitation. ChIP-seq with anti-HA antibody in these lines identified 10 442 and 9098 peaks for E2A-hf and EP1-hf (supplemental Dataset_1), respectively. Annotation of the E2A-hf– and EP1-hf–binding sites revealed typical patterns for TFs, with a vast majority of the binding sites found at intergenic and intragenic regions (Figure 1C). Genomic Regions Enrichment of Annotations Tool (GREAT)27 analyses indicated that E2A-PBX1–bound and E2A-bound regions are involved in hematopoietic processes or immune system functions (Figure 1D). Comparison of E2A-PBX1 and E2A target sites revealed that the 2 factors share a subset of cobound sites (at 20%-25% of sites) (Figure 1E). Similar to our previous ChIP-seq study with antibodies common to E2A-PBX1 and E2A,28 we identified sites cobound by E2A-PBX1 and E2A on genes, such as IGLL1, involved in pre–B-cell receptor signaling (Figure 1F).
Importantly, our current ChIP-seq was able to differentiate binding specific to E2A-PBX1 vs E2A and showed a largely distinctive binding pattern for the 2 factors (Figure 1G), as exemplified by ELK3 and PLS3 (Figure 1F). ChIP-seq with an antibody directed against the C-terminal region of PBX1 (Figure 1A; supplemental Figure 2) displays strong signals at the EP1-hf–specific peaks, indicating the potential binding of endogenous E2A-PBX1. Cooccupancy of p300, a well-known transcriptional coactivator that interacts with AD1 and AD2 within the E2A N-terminus, was also observed. Collectively, our analyses, for the first time, have delineated specific binding of E2A-PBX1 in pre-B ALL cells.
E2A-PBX1 directly binds to genomic sites enriched for RUNX1- and EBF1-, but not PBX1-, binding motifs
To gain further insights into the binding patterns of E2A-PBX1, we analyzed the identified binding sites for specific motifs. E2A-binding sites were enriched not only for the defined E2A motif (P = 1e-1842) (Figure 1H), as expected, but also for Etv2, RUNX1, and EBF1 motifs, in agreement with ChIP-seq analyses of E2A in murine precursor B cells.5 Surprisingly, the E2A-PBX1–binding sites were most enriched for RUNX1 and EBF1 motifs, whereas the PBX1 motif was also identified at a lower percentage. These results raised the possibility that E2A-PBX1 may either directly recognize these 2 consensus sequences or associate with these elements through direct interactions with bound RUNX1 and/or EBF1. Because it seemed unlikely that the DBD of PBX1 could recognize these 2 unrelated motifs/sequences, we further examined the possibility that E2A-PBX1 could indirectly bind to the RUNX1 and/or EBF1 sites via protein–protein interactions.
E2A-PBX1 is recruited to gene loci by RUNX1
To determine whether E2A-PBX1 interacts with RUNX1 or EBF1, we tested for Co-IP of ectopically expressed E2A-PBX1 and RUNX1 or EBF1 in HEK293T cells. We found that RUNX1, but not EBF1, coimmunoprecipitates with E2A-PBX1 (Figure 2A). Furthermore, pull-down assays with purified recombinant proteins confirmed strong direct interactions of RUNX1 with both E2A-PBX1 and PBX1, as well as a weaker interaction between E2A and RUNX1 (Figure 2B), indicating the possibility of multiple contacts between E2A-PBX1 and RUNX1. Previously, the HCM domain that flanks the PBX1 homeodomain was shown to be essential both for E2A-PBX1 interactions with HOX proteins and for gene activation using a PBX1 site-driven reporter.20 To further test whether the HCM domain contributes to the E2A-PBX1 interaction with RUNX1, a Y305E mutant known to disrupt HCM functions was used.20 Notably, Co-IP assays revealed that WT and Y305E mutant forms of E2A-PBX1 exhibit comparable binding to RUNX1 (Figure 2C), suggesting that the RUNX1 binding by E2A-PBX1 does not require its HCM function. To further examine E2A-PBX1 binding at RUNX1 targets, E2A-PBX1-hf WT and Y305E mutant proteins were inducibly expressed in stable 697 lines (Figure 2D) at comparable levels and subjected to ChIP-qPCR assays. Interestingly, comparable DNA binding of WT and Y305E mutant E2A-PBX1 was observed at a subset of examined sites that are directly bound by E2A-PBX1 and contain the consensus RUNX1 motif (Figure 2E), but no PBX1 motif; this finding indicates that the DNA-binding function of E2A-PBX1 is not required for genomic targeting. To further test the requirement of RUNX1 for genomic targeting of E2A-PBX1, shRNAs were used to deplete RUNX1 (Figure 2F), and significantly decreased binding of E2A-PBX1 to the selected cotargets of E2A-PBX1 and RUNX1 was detected (Figure 2G). These data support a role for RUNX1 in mediating the genomic binding of E2A-PBX1. Based on the aforementioned results, we conclude that E2A-PBX1 could be indirectly recruited to these examined gene targets by RUNX1.
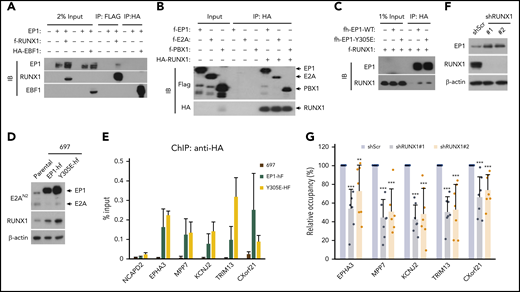
RUNX1 interacts directly with and mediates the DNA binding of E2A-PBX1. (A) Co-IP assays showing an interaction between E2A-PBX1 and RUNX1, but not EBF1, in transfected HEK293T cells. Immunoprecipitations were conducted with anti-FLAG or anti-HA beads, and bound proteins were visualized by immunoblotting with indicated antibodies. (B) Direct RUNX1 interactions with E2A-PBX1, E2A, and PBX1. Immunoprecipitation with purified proteins and anti-HA antibody followed by immunoblotting with indicated antibodies. (C) Co-IP assay showing RUNX1 binding to E2A-PBX1 WT and to a DNA-binding defective Y305E mutant. (D) Immunoblot showing the expression of exogenous EP1-hf and RUNX1 in stable 697 cells expressing E2A-PBX1 WT or Y305E mutant. (E) ChIP-qPCR of E2A-PBX1 WT or Y305E mutant at enhancers of indicated genes in stable 697 lines. (F) Immunoblot of endogenous E2A-PBX1 and RUNX1 in shScr- or shRUNX1-treated 697 lines. (G) ChIP-qPCR of E2A-PBX1 (HA) at enhancers of indicated genes in stable 697 line treated with shScr or shRUNX1. ChIP signals (y-axis) from 3 independent experiments, with 2 or 3 replicates, were compared with control (Scramble) and are presented as mean ± standard deviation (SD).
RUNX1 interacts directly with and mediates the DNA binding of E2A-PBX1. (A) Co-IP assays showing an interaction between E2A-PBX1 and RUNX1, but not EBF1, in transfected HEK293T cells. Immunoprecipitations were conducted with anti-FLAG or anti-HA beads, and bound proteins were visualized by immunoblotting with indicated antibodies. (B) Direct RUNX1 interactions with E2A-PBX1, E2A, and PBX1. Immunoprecipitation with purified proteins and anti-HA antibody followed by immunoblotting with indicated antibodies. (C) Co-IP assay showing RUNX1 binding to E2A-PBX1 WT and to a DNA-binding defective Y305E mutant. (D) Immunoblot showing the expression of exogenous EP1-hf and RUNX1 in stable 697 cells expressing E2A-PBX1 WT or Y305E mutant. (E) ChIP-qPCR of E2A-PBX1 WT or Y305E mutant at enhancers of indicated genes in stable 697 lines. (F) Immunoblot of endogenous E2A-PBX1 and RUNX1 in shScr- or shRUNX1-treated 697 lines. (G) ChIP-qPCR of E2A-PBX1 (HA) at enhancers of indicated genes in stable 697 line treated with shScr or shRUNX1. ChIP signals (y-axis) from 3 independent experiments, with 2 or 3 replicates, were compared with control (Scramble) and are presented as mean ± standard deviation (SD).
E2A-PBX1 potentiates the binding of coactivators (p300 and Mediator) and H3K27 acetylation at genomic sites cotargeted by RUNX1
To examine whether E2A-PBX1–occupied regions are indeed cooccupied by RUNX1 in E2A-PBX1+ pre-B ALL, we performed RUNX1 ChIP-seq profiling and identified 31 232 RUNX1 peaks (supplemental Dataset_1). Notably, a majority (56%) of E2A-PBX1 binding sites overlapped RUNX1 sites, which is significantly higher than the overlap with E2A sites (Figure 3A). GREAT analyses of the genomic regions cobound by E2A-PBX1 and RUNX1 revealed gene pathways involved in immune development (supplemental Figure 3), consistent with the reported role for RUNX1 in regulating the survival and development of precursor B cells.29 E2A-PBX1 retains the three N-terminal ADs that interact with coactivators, thereby potentiating gene activation. To further investigate whether the binding of E2A-PBX1 facilitates recruitment of coactivators, additional ChIP-seq profiling was conducted. We found that, relative to those binding sites unique to RUNX1, regions cobound by E2A-PBX1 and RUNX1 had significantly enhanced occupancies of p300, the cognate histone modification H3K27ac, and the MED1 subunit of the Mediator coactivator complex (Figure 3B). This is exemplified at the cooccupied MAP3K1 and WNT16 genes (Figure 3C) but not at the STK4 gene that is bound by RUNX1 only. Collectively, these analyses support the view that E2A-PBX1 is cotargeted to a subset of direct RUNX1 target genes, thereby potentiating local gene-active chromatin states.

ChIP-seq reveals elevated active marks and factors at E2A-PBX1–bound RUNX1 sites. (A) Venn diagram showing the overlap among E2A-PBX1, E2A, and RUNX1 ChIP-seq peaks. In the triple overlap region, there were 2337 peaks from E2A-hf and 2338 peaks from EP1-hf. (B) Metagene plots showing the averaged ChIP-seq signals for EP1-hf, p300, MED1, and H3K27Ac at identified RUNX1 only or at RUNX1 and E2A-PBX1 cobound sites. (C) ChIP-seq profiles of E2A and E2A-PBX1 in their stable 697 expression lines and those of RUNX1, p300, MED1, and H3K27ac in the parental 697 line at the indicated targets. Blue- and gray-colored boxes show overlap of EP1-hf and RUNX1 peaks.
ChIP-seq reveals elevated active marks and factors at E2A-PBX1–bound RUNX1 sites. (A) Venn diagram showing the overlap among E2A-PBX1, E2A, and RUNX1 ChIP-seq peaks. In the triple overlap region, there were 2337 peaks from E2A-hf and 2338 peaks from EP1-hf. (B) Metagene plots showing the averaged ChIP-seq signals for EP1-hf, p300, MED1, and H3K27Ac at identified RUNX1 only or at RUNX1 and E2A-PBX1 cobound sites. (C) ChIP-seq profiles of E2A and E2A-PBX1 in their stable 697 expression lines and those of RUNX1, p300, MED1, and H3K27ac in the parental 697 line at the indicated targets. Blue- and gray-colored boxes show overlap of EP1-hf and RUNX1 peaks.
Transcriptome analysis shows coregulatory roles for E2A-PBX1 and RUNX1 in a subset of RUNX1 target genes, thereby contributing to leukemic cell proliferation
To investigate cooperation between E2A-PBX1 and RUNX1 in gene expression, we performed RNA-seq assays in the E2A-PBX1–depleted and RUNX1-depleted 697 lines. Because the generation of shRNAs that specifically target the E2A-PBX1 fusion and not E2A or PBX1 is almost impossible, shRNAs targeting either the N-terminal (shE2A-N) or the C-terminal (shPBX1-C) portion of E2A-PBX1 were used. Two independent shRNAs for E2A-N and two shRNAs for PBX1-C were identified that efficiently deplete E2A-PBX1 (Figure 4A). Although E2A proteins were also efficiently depleted by shE2A-N treatment, no changes were detected in expression of tested E2A targets such as IGLL1, PAX5, VPREB3, or EBF1 (supplemental Figure 4), suggesting that other E proteins (HEB or E2-2) may compensate for the loss of E2A. Given that E2A-PBX1 and RUNX1 are positive regulators of gene activation, we focused on genes downregulated upon shRNA-mediated knockdown. RNA-seq analyses identified 46 transcripts that were significantly downregulated as a result of E2A-PBX1 or RUNX1 depletion (Figure 4B; supplemental Dataset_2). These genes were also generally downregulated using another set of shRNAs (shE2A-N#2, shPBX1-C#2, and shRUNX1#2) (Figure 4C). Furthermore, we found that 31 of the 46 downregulated genes are bound by both E2A-PBX1 and RUNX1, whereas 13 genes showed only RUNX1 binding. RT-qPCR assays confirmed that both E2A-PBX1 and RUNX1 are required for expression of tested cobound targets in E2A-PBX1+ leukemic lines (Figure 4D). Together, our findings indicate a striking association of E2A-PBX1 and RUNX1 in activating gene expression.
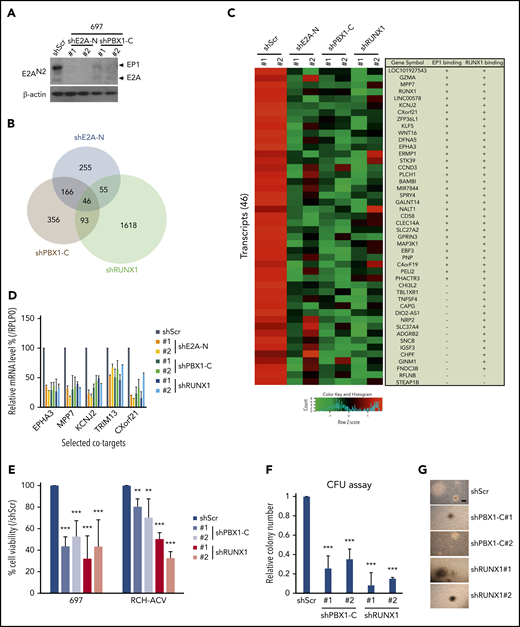
RUNX1 and E2A-PBX1 coregulate a subset of genes and are both required for cell growth in E2A-PBX1–positive leukemic lines. (A) Immunoblot of E2A/E2A-PBX1 in the 697 line treated with shScr, shE2A-N, or shPBX1-C. Actin was used as the loading control. (B) Venn diagram showing the overlap of downregulated genes, relative to Scramble, in the 697 line treated with shE2A-N (#1), shPBX1-C (#1), or shRUNX1 (#1). (C) Heatmap of RNA-seq results for 2 experiments with 2 differentially designed shRNAs (#1 and #2) revealing a subset of genes that are downregulated in either the E2A-PBX1 (by shE2A-N or shPBX1-C) or RUNX1-depleted 697 line. The gene symbols and binding by E2A-PBX1 and RUNX1 are denoted on the right. (D) RT-qPCR assays showing the relative mRNA levels of indicated genes in the 697 cell line treated with Scramble, shE2A-N, shPBX1-C, or shRUNX1. Depletion of E2A-PBX1 and RUNX1 reduces cell growth (E) and colony formation (F). Cell growth and colony numbers were determined after the shRNA-mediated knockdown. Data are presented as relative to Scramble-treated cells. Statistics by Student t test. **P < .01; ***P < .001. (G) Images of representative colonies from indicated treatments in panel F. Scale bar, 0.5 mm.
RUNX1 and E2A-PBX1 coregulate a subset of genes and are both required for cell growth in E2A-PBX1–positive leukemic lines. (A) Immunoblot of E2A/E2A-PBX1 in the 697 line treated with shScr, shE2A-N, or shPBX1-C. Actin was used as the loading control. (B) Venn diagram showing the overlap of downregulated genes, relative to Scramble, in the 697 line treated with shE2A-N (#1), shPBX1-C (#1), or shRUNX1 (#1). (C) Heatmap of RNA-seq results for 2 experiments with 2 differentially designed shRNAs (#1 and #2) revealing a subset of genes that are downregulated in either the E2A-PBX1 (by shE2A-N or shPBX1-C) or RUNX1-depleted 697 line. The gene symbols and binding by E2A-PBX1 and RUNX1 are denoted on the right. (D) RT-qPCR assays showing the relative mRNA levels of indicated genes in the 697 cell line treated with Scramble, shE2A-N, shPBX1-C, or shRUNX1. Depletion of E2A-PBX1 and RUNX1 reduces cell growth (E) and colony formation (F). Cell growth and colony numbers were determined after the shRNA-mediated knockdown. Data are presented as relative to Scramble-treated cells. Statistics by Student t test. **P < .01; ***P < .001. (G) Images of representative colonies from indicated treatments in panel F. Scale bar, 0.5 mm.
The functional roles of RUNX1 in E2A-PBX1 leukemias were further studied by using cell viability and colony-forming assays. Depletion of E2A-PBX1 or RUNX1 (supplemental Figure 5) in two E2A-PBX1+ leukemic lines correlated with significantly reduced cell viability (Figure 4E) compared with shScramble (shScr) treatment. In addition, the E2A-PBX1 and RUNX1 knockdowns resulted in similar reductions of colony numbers, but not sizes, in clonogenic assays (Figure 4F-G). Collectively, these results suggest that E2A-PBX1 and RUNX1 jointly contribute to the growth of E2A-PBX1+ leukemic cells.
The RUNX1 gene is a direct target of E2A-PBX1
Genetic aberrations of RUNX1 are frequent in several types of leukemias, including acute myeloid leukemia (AML), B-ALL, T-cell ALL, and myelodysplastic syndrome.30 However, studies also found that normal RUNX1 may contribute to AML induced by mixed lineage leukemia (MLL) fusion.31,32 Thus, considering the leukemogenic potential of RUNX1 in these leukemias, we further investigated the regulation of RUNX1 in E2A-PBX1+ leukemias. Notably, as in MLL-rearranged leukemias, we found that RUNX1 is significantly overexpressed in E2A-PBX1+ leukemia compared with other ALL subtypes (Figure 5A). Interestingly, our ChIP-seq analyses also revealed that E2A-PBX1, as well as RUNX1 itself, is specifically associated with upstream and intragenic enhancers of the RUNX1 locus (Figure 5B). Consistent with activation-related interactions, these two cobound regions are highly enriched for p300 and H3K27ac signals. By contrast, there are no, or lower, p300 and H3K27Ac signals at the 2 downstream regions occupied only by RUNX1. The indication that E2A-PBX1 plays a role in activating RUNX1 was established by 2 additional analyses. First, shRNA-mediated depletion of E2A-PBX1 resulted in loss of both RUNX1 mRNA (Figure 5B and 5C) and protein (Figure 5D) levels. Second, ectopic expression of E2A-PBX1 significantly upregulated both RUNX1 mRNA (Figure 5E) and protein (Figure 5F) levels, whereas ectopic E2A exhibited less of an effect.
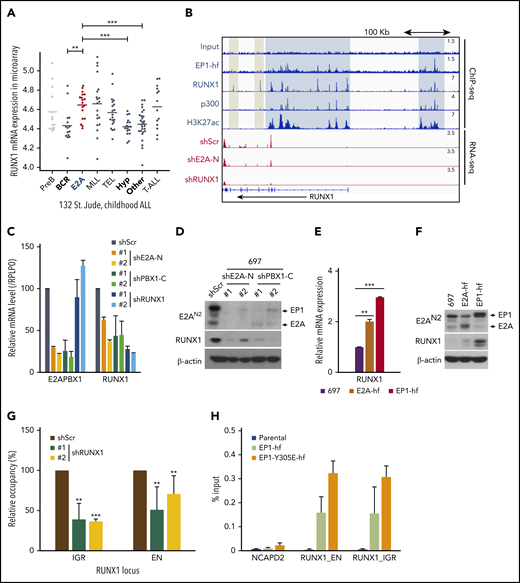
E2A-PBX1 directly binds to and activates the RUNX1 gene. (A) RUNX1 is significantly upregulated in E2A-PBX1+ and MLL-rearranged B-ALL. The expression levels of RUNX1 in different subtypes of B-ALL patients are shown. Data are from St. Jude Children's Research Hospital, n = 132 B-ALL patients.59 Statistics by two-sided Wilcoxon test. **P < .01; ***P < .001. (B) RUNX1 is a direct target of E2A-PBX1. ChIP-seq profiles (upper) of indicated factors at the RUNX1 locus. Blue bars denote the colocalization of E2A-PBX1 (EP1-hf), RUNX1 and p300, and associated H3K27 acetylation; gray bars denote the lack of active marks in the RUNX1-binding regions. RNA-seq profiles (bottom) reveal the reduction of RUNX1 expression in the 697 cell line treated with shE2A-N and shRUNX1. (C) RT-qPCR assays showing the mRNA levels of E2A-PBX1 and RUNX1 in the 697 cell line treated with shScr, shE2A-N, shPBX1-C, or shRUNX1. (D) Immunoblot of E2A/E2A-PBX1 (with E2AN2 antibody) and RUNX1 in the 697 cell line treated with shScr, shE2A-N, or shPBX1-C. Actin was used as the loading control. (E-F) RUNX1 is upregulated by ectopic E2A-PBX1. RT-qPCR (E) and immunoblot (F) showing the expression of RUNX1 in the stable 697 cell line inducibly expressing E2A or E2A-PBX1. Total RNA or cell lysates were collected 24 hours’ postinduction. (G) ChIP-qPCR of E2A-PBX1 (HA) at upstream (EN) and intragenic (IGR) enhancers of the RUNX locus in the stable 697 cell line treated with shScr or shRUNX1. ChIP signals (y-axis) from 3 independent experiments were compared with control (Scramble) and are presented as mean ± SD. Statistics by Student t test. **P < .01; ***P < .001. (H) ChIP-qPCR of WT and Y305E E2A-PBX1 (HA) at enhancers of the RUNX1 locus in stable 697 cell lines. The NCAPD2 locus was used as a negative control for E2A-PBX1 binding. ChIP signals (y-axis) from 3 independent experiments are presented as mean ± SD.
E2A-PBX1 directly binds to and activates the RUNX1 gene. (A) RUNX1 is significantly upregulated in E2A-PBX1+ and MLL-rearranged B-ALL. The expression levels of RUNX1 in different subtypes of B-ALL patients are shown. Data are from St. Jude Children's Research Hospital, n = 132 B-ALL patients.59 Statistics by two-sided Wilcoxon test. **P < .01; ***P < .001. (B) RUNX1 is a direct target of E2A-PBX1. ChIP-seq profiles (upper) of indicated factors at the RUNX1 locus. Blue bars denote the colocalization of E2A-PBX1 (EP1-hf), RUNX1 and p300, and associated H3K27 acetylation; gray bars denote the lack of active marks in the RUNX1-binding regions. RNA-seq profiles (bottom) reveal the reduction of RUNX1 expression in the 697 cell line treated with shE2A-N and shRUNX1. (C) RT-qPCR assays showing the mRNA levels of E2A-PBX1 and RUNX1 in the 697 cell line treated with shScr, shE2A-N, shPBX1-C, or shRUNX1. (D) Immunoblot of E2A/E2A-PBX1 (with E2AN2 antibody) and RUNX1 in the 697 cell line treated with shScr, shE2A-N, or shPBX1-C. Actin was used as the loading control. (E-F) RUNX1 is upregulated by ectopic E2A-PBX1. RT-qPCR (E) and immunoblot (F) showing the expression of RUNX1 in the stable 697 cell line inducibly expressing E2A or E2A-PBX1. Total RNA or cell lysates were collected 24 hours’ postinduction. (G) ChIP-qPCR of E2A-PBX1 (HA) at upstream (EN) and intragenic (IGR) enhancers of the RUNX locus in the stable 697 cell line treated with shScr or shRUNX1. ChIP signals (y-axis) from 3 independent experiments were compared with control (Scramble) and are presented as mean ± SD. Statistics by Student t test. **P < .01; ***P < .001. (H) ChIP-qPCR of WT and Y305E E2A-PBX1 (HA) at enhancers of the RUNX1 locus in stable 697 cell lines. The NCAPD2 locus was used as a negative control for E2A-PBX1 binding. ChIP signals (y-axis) from 3 independent experiments are presented as mean ± SD.
In agreement with our previous results, these ChIP-qPCR assays revealed that RUNX1 is required for E2A-PBX1 recruitment to RUNX1 enhancers (Figure 5G), whereas the DNA-binding function of E2A-PBX1 is dispensable (Figure 5H). Together, our results suggest that E2A-PBX1 directly targets and activates expression of RUNX1 in E2A-PBX1+ leukemia.
RUNX1 plays an essential role in E2A-PBX1–driven leukemogenesis
Our aforementioned results showed that chromatin targeting of E2A-PBX1 is mediated at least partially through RUNX1 in E2A-PBX1+ leukemic cells. To further examine if the DNA-binding function of E2A-PBX1 is crucial for leukemogenesis, we used a classic HSPC transformation assay to compare the transforming capabilities of the WT and DNA-binding defective mutant (Y305E) forms of E2A-PBX1. As expected, WT E2A-PBX1 efficiently transformed HSPCs. Notably, the Y305E mutant also exhibited efficient transformation, albeit at a lower level (Figure 6A). In addition, a CFU assay, a surrogate for scoring leukemia stem cells, showed that both WT and Y305E-mutant forms of E2A-PBX1 possess clonogenic abilities (Figure 6B), indicating that the DNA-binding function of E2A-PBX1 is also dispensable for cell transformation. In contrast, and most importantly, depletion of endogenous RUNX1 by shRNAs severely impaired both cell proliferation and the colony-forming abilities of E2A-PBX1–transduced HSPCs (Figure 6C-D), strongly supporting a role for RUNX1 in E2A-PBX1–mediated leukemogenesis.
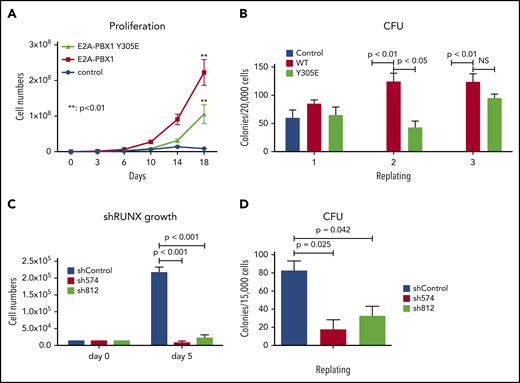
RUNX1 is essential for E2A-PBX1–mediated HSPC transformation in vitro. DNA-binding function of E2A-PBX1 is dispensable in HSPC transformation. Murine HPSCs transduced with control or indicated E2A-PBX1–expressing retroviruses were subjected to assays for (A) proliferation kinetics in a liquid culture and (B) in vitro serial replating to score CFUs in a semisolid culture. RUNX1 depletion significantly impairs the maintenance of E2A-PBX1-transformed HSPCs. (C) Cell proliferation and (D) CFU assays of E2A-PBX1–transformed HSPCs transduced with lentiviruses expressing Scramble (shControl) or RUNX1 (sh574 and sh812) shRNAs. Statistics by Student t test. **P < .01. NS, not significant.
RUNX1 is essential for E2A-PBX1–mediated HSPC transformation in vitro. DNA-binding function of E2A-PBX1 is dispensable in HSPC transformation. Murine HPSCs transduced with control or indicated E2A-PBX1–expressing retroviruses were subjected to assays for (A) proliferation kinetics in a liquid culture and (B) in vitro serial replating to score CFUs in a semisolid culture. RUNX1 depletion significantly impairs the maintenance of E2A-PBX1-transformed HSPCs. (C) Cell proliferation and (D) CFU assays of E2A-PBX1–transformed HSPCs transduced with lentiviruses expressing Scramble (shControl) or RUNX1 (sh574 and sh812) shRNAs. Statistics by Student t test. **P < .01. NS, not significant.
RUNX1 interaction, through the PBX1 homeodomain region, is essential for E2A-PBX1 recruitment and E2A-PBX1–mediated transcription
To confirm that the RUNX1 interaction is crucial for transcriptional activation by E2A-PBX1, we conducted domain mapping experiments with purified E2A-PBX1 fragments (Figure 7A). Biochemical analyses showed that a region spanning the PBX1 homeodomain (IH-HD) interacts strongly with RUNX1, whereas the N-terminal E2A ADs display no independent RUNX1 interaction (data not shown). Notably, deletion of the IH-HD region in E2A-PBX1 completely abolished the RUXN1 interaction in a cell-based Co-IP assay (Figure 7B). Furthermore, the IH-HD–deleted E2A-PBX1 exhibited diminished binding and transcription functions at tested cobound targets that included RUNX1 (Figure 7C-D). Interestingly, Calvo et al33 found that the IH-HD domain is crucial for both murine 3T3 and bone marrow cell transformation by E2A-PBX1. Together, these results strongly suggest that the RUNX1 interaction, through the IH-HD domain, is essential for E2A-PBX1 transcription and oncogenic functions.
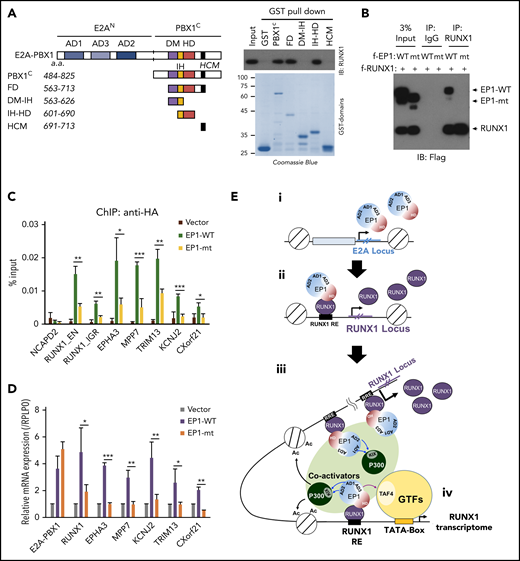
RUNX1 interaction is essential for E2A-PBX1–enhanced gene activation. (A) Schematic of PBX1 domains (left). GST pull-down assays (right) showing direct RUNX1 binding by the IH-HD domain of C-terminal region of PBX1 (PBX1C). (B) Co-IP assay showing RUNX1 binding to WT but not the IH-HD domain-deleted (mt) E2A-PBX1. (C) ChIP-qPCR with anti-HA at enhancers and (D) RT-qPCR of indicated genes in 697 cells transduced by lentivirus expressing WT or IH-HD–deleted (dIH-HD) E2A-PBX1. The NCAPD2 locus was used as a negative control for E2A-PBX1 binding. Data from 3 independent experiments are presented as mean ± SD. Statistics by Student t test. *P < .05; **P < .01; ***P < .001. (E) Model for the coactivator function of E2A-PBX1 in regulating the expression of RUNX1 and RUNX1 programs in pre–B-ALL. Based on previous and current results, the following working model for E2A-PBX1–mediated gene activation is proposed: (i) the E2A-PBX1 fusion protein is expressed under the control of the E2A promoter; (ii) E2A-PBX1, by interaction with RUNX1, is recruited to the RUNX1 locus and sustains high-level expression of RUNX1; (iii) RUNX1 mediates recruitment of E2A-PBX1, which in turn recruits p300 through E2A AD1/2-p300 KIX domain interactions and TFIID through E2A-AD3-TFIID TAF4 interactions; and (iv) these interactions, and potentially other cofactor interactions of E2A-PBX1 and/or RUNX1, enhance expression of a subset of RUNX1 targets that lead to leukemogenesis. DM, dimerization domain; GTFs, general transcription factors; HCM, Hox cooperative motif; HD, homeodomain; IH, inhibitory helix.
RUNX1 interaction is essential for E2A-PBX1–enhanced gene activation. (A) Schematic of PBX1 domains (left). GST pull-down assays (right) showing direct RUNX1 binding by the IH-HD domain of C-terminal region of PBX1 (PBX1C). (B) Co-IP assay showing RUNX1 binding to WT but not the IH-HD domain-deleted (mt) E2A-PBX1. (C) ChIP-qPCR with anti-HA at enhancers and (D) RT-qPCR of indicated genes in 697 cells transduced by lentivirus expressing WT or IH-HD–deleted (dIH-HD) E2A-PBX1. The NCAPD2 locus was used as a negative control for E2A-PBX1 binding. Data from 3 independent experiments are presented as mean ± SD. Statistics by Student t test. *P < .05; **P < .01; ***P < .001. (E) Model for the coactivator function of E2A-PBX1 in regulating the expression of RUNX1 and RUNX1 programs in pre–B-ALL. Based on previous and current results, the following working model for E2A-PBX1–mediated gene activation is proposed: (i) the E2A-PBX1 fusion protein is expressed under the control of the E2A promoter; (ii) E2A-PBX1, by interaction with RUNX1, is recruited to the RUNX1 locus and sustains high-level expression of RUNX1; (iii) RUNX1 mediates recruitment of E2A-PBX1, which in turn recruits p300 through E2A AD1/2-p300 KIX domain interactions and TFIID through E2A-AD3-TFIID TAF4 interactions; and (iv) these interactions, and potentially other cofactor interactions of E2A-PBX1 and/or RUNX1, enhance expression of a subset of RUNX1 targets that lead to leukemogenesis. DM, dimerization domain; GTFs, general transcription factors; HCM, Hox cooperative motif; HD, homeodomain; IH, inhibitory helix.
Discussion
E2A-PBX1 and E2A-HLF fusion proteins lead, respectively, to pre–B-cell and pro–B-cell ALLs. Although good to excellent outcomes have been achieved by more recent therapeutic protocols, there are still long-term sequelae and risk of central nervous system relapse in pediatric patients,34 who respond less effectively than adults. Mechanistic studies are required to characterize and target these oncogenic proteins for developing better treatment strategies. Toward this goal, we determined the genome-wide binding and transcription signatures of the oncogenic E2A-PBX1 in pre-B ALL–derived cell lines. We found that E2A-PBX1, through an interaction with RUNX1, preferentially binds to a subset of RUNX1-occupied enhancers. We also found that p300, H3K27ac, and MED1 enrichments are a feature of E2A-PBX1–targeted RUNX1 sites that could promote the expression of associated genes. Interestingly, E2A-PBX1 not only binds to RUNX1-controlled genes but also targets the positive feedback loop of the RUNX1 locus. Hence, the positive impact of E2A-PBX1 on activation of the RUNX1 locus and downstream RUNX1 targets may contribute to a sustained RUNX1 transcriptome in pre–B cells that leads to leukemogenesis. In support of this idea, we observed that RUNX1 is upregulated in E2A-PBX1+ patients and required for E2A-PBX1–mediated transformation of murine HSPCs. Based on previous and current results, we present a previously unappreciated model for the oncogenic function of E2A-PBX1 through its role as a transcriptional coactivator, in a feed-forward mechanism, for RUNX1 in pre-B ALL (Figure 7E).
DNA-binding preferences of E2A-PBX1 in vitro and in vivo and a transcriptional coactivator function
PBX1 belongs to the TALE (three amino acid loop extension) class of TFs and binds to DNA in association with other TALE proteins that include HOX, MEIS, and Prep. Thus, E2A-PBX1 was also believed to dimerize with other TALE proteins for DNA binding. Indeed, previous biochemical and cell-based reporter assays suggested that E2A-PBX1, together with HOX proteins, was likely to activate transcription of HOX/PBX1 target genes.20,25,33,35,36 However, our RNA-seq profiling revealed that only three HOX proteins, HOX A3, A5, and A6, are expressed in 697 pre-B ALL cells. Furthermore, modulation of these HOX proteins did not affect the chromatin association of E2A-PBX1 at identified enhancers (data not shown). Interestingly, our analyses further showed that the leukemic cells also express dominant levels of PBX2/3 (supplemental Figure 2) that may compete for the limited HOX proteins. Another unexpected but highly important finding, especially in view of an intact DBD within the PBX1 portion of E2A-PBX1, was that E2A-PBX1 preferentially binds to a subset of RUNX1 site–containing target genes in a RUNX1-dependent fashion. The mechanism underlying the selective E2A-PBX1 targeting of only a small fraction of RUNX1-bound regions is still unclear but must reflect as yet unknown context effects. Toward identification of an alternative mechanism for E2A-PBX1 recruitment to target genes, our biochemical analyses showed a direct interaction, through the IH-HD domain of PBX1, between E2A-PBX1 and RUNX1. Consistent with this indication of a transcriptional coactivator function, rather than a conventional direct DNA-binding TF function, the gene-specific recruitment, transcription functions, and transformation properties (murine HSPCs) of E2A-PBX1 are independent of the previously identified HOX-interacting function (lost in the active E2A-PBX1 Y305E mutant) but dependent on the RUNX1 and the RUNX1-interacting homeodomain region of E2A-PBX1 (as shown with the IH-HD–defective E2A-PBX1 mutant). Interestingly, investigations have also shown that several hematopoietic regulators, such as FLI1, RUNX1, and SPI1, are able to mediate the DNA binding of other oncogenic fusion proteins, including AML1-ETO,37,38 MLL-AF4/AF9,32,34 and PML/RARalpha.39 Together, these findings strongly suggest that leukemogenic fusion proteins such as E2A-PBX1 may be targeted to specific DNA sites in vivo through other hematopoietic TFs.
Given its direct interactions with the site-specific DNA-binding RUNX1, its selective association with a subset of RUNX1 sites relative to PBX1 sites in leukemic cells, and its activity in the absence of a functional DBD, E2A-PBX1 can be viewed mechanistically as a transcriptional coactivator rather than a conventional site-specific DNA binding TF (Figure 7E). In relation to its actual mechanism of action on target genes, resident E2A AD1 and AD2 have been shown to interact with p300,40-42 whereas AD3 has been shown to interact physically and functionally with the TAF4 subunit of transcription initiation factor TFIID to facilitate TFIID recruitment.8 In this regard, and for purposes of elucidating potential therapeutic targets, it will be important to assess the significance of these interactions for E2A-PBX1 function in the generation of t(1;19) leukemia.
RUNX1 and E2A-PBX1 coregulate a subset of targets in pre-B ALL
By searching for dysregulated genes that could help to explain the essential roles of both E2A-PBX1 and RUNX1 in cell proliferation, we identified 46 genes whose mRNA levels were significantly reduced upon knockdown of RUNX1 and E2A-PBX1 in pre-B ALL. Notably, 31 genes were also identified with prominent RUNX1 and E2A-PBX1 binding to their genomic loci. Intriguingly, our list includes the following: (1) potential tumor-promoting factors, such as KCNJ2,43 STK39,44 and SPRY4,45 reflecting the oncogenic function of E2A-PBX1; (2) genes, such as CCND3, that control the proliferation and expansion of B-lineage cells,46 MAP3K1,47 and WNT16, a previously described E2A-PBX1 target48,49 ; and (3) self-renewal factors, such as KLF550 and MPP7.51 Another important target gene is the ephrin receptor EPHA3, which is specifically expressed and considered as an antibody immunotherapeutic target in E2A-PBX1+ ALL.52 Overall, our study identified a subset of E2A-PBX1 and RUNX1 cotargets that could be further examined as potential therapeutic targets in E2A-PBX1+ ALL.
RUNX1 is also implicated in E2A-PBX1+ pre-B ALL
Our ChIP-seq profiling revealed that E2A-PBX1 targets not only a subset of RUNX1-occupied genes but also 2 enhancers in the RUNX1 locus. Gene expression analyses further confirmed that E2A-PBX1 directly controls RUNX1 activation in pre-B ALL. RUNX1 acts as a master regulator in the establishment of hematopoietic stem cells (HSCs).53-55 Although RUNX1 is expressed in HSCs, myeloid cells, and lymphoid cells,56 genetic inactivation in a mouse model has shown that Runx1 is absolutely required only during the initial generation of HSCs.57 Recent studies using a conditional mouse model have found that the RUNX1 program is necessary for governing the survival and development of progenitor B cells.29 In addition to playing an essential role in hematopoiesis, RUNX1 is a frequent target of chromosomal translocations in several types of acute myeloid and lymphoid leukemias,53-55 indicating either a tumor suppressor function for WT RUNX1 or an oncogenic function for derived chimeric proteins such as the RUNX1-ETO protein generated by the t(8;21) translocation.58 In contrast, studies in MLL revealed that MLL fusions (MLL-AF9 and MLL-AF4) activate RUNX1 expression and RUNX1-mediated gene programs in leukemogenesis.31,32 Taken together, these findings suggest that high RUNX1 levels and RUNX1 gene programs may suspend the differentiation of hematopoietic progenitor cells and lead to leukemogenesis. Our studies further extend the pathological function of RUNX1 to the development of pre-B ALL in the context of E2A-PBX1.
All sequence reads in this article have been deposited in the Gene Expression Omnibus database (accession number GSE138031).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
All reagents used in this study can be requested by e-mail to the corresponding author. The deposited high-throughput datasets will be available to public.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the Sequencing Core Facility of National Yang-Ming University Genome Center and the Epigenomics Core Facility at Weill Cornell Medical College for the use of the NGS services and facilities.
This work was supported by MOST (107-2320-B-010-024-MY3, W.-Y.C.) and Yen Tjing Ling Medical Foundation (CI-106-12, W.-Y.C.) grants and by National Institutes of Health, National Cancer Institute grants (CA178765 and CA163086, R.G.R.). W.-Y.C. is also supported by the Cancer Progression Research Center, National Yang-Ming University from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education in Taiwan. W.-C.P. was funded by the Biomedical Industry PhD Program from National Yang-Ming University and CHUNGHWA Chemical Synthesis & Biotech Co., Ltd. G.G.W. is an American Society of Hematology Scholar in basic research, an American Cancer Society Research Scholar, and a Leukemia & Lymphoma Society Scholar.
Authorship
Contribution: W.-C.P. and W.-Y.C. planned the project and designed and conducted the biochemical and cell-based experiments; J.W., R.L., and D.L. designed and performed the HSPC experiments; J.-W.L. performed the biochemical pull-down assays; M.S. and Y.-L.L. performed the RNA-seq analysis; H.G. performed the bioinformatics analysis; W.-C.P., G.G.W., W.-Y.C., and R.G.R. wrote the manuscript with input from all coauthors; and W.-Y.C. and R.G.R. conceived and supervised the overall project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wei-Yi Chen, National Yang-Ming University, 155, Sec 2, Linong St Taipei, Taiwan; e-mail: chenwy@ym.edu.tw; and Robert G. Roeder, The Rockefeller University, 1230 York Ave, New York, NY 10065; e-mail: roeder@rockefeller.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal