


Abstract
Acquired aplastic anemia (AA) and paroxysmal nocturnal hemoglobinuria (PNH) are pathogenically related nonmalignant bone marrow failure disorders linked to T-cell–mediated autoimmunity; they are associated with an increased risk of secondary myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Approximately 15% to 20% of AA patients and 2% to 6% of PNH patients go on to develop secondary MDS/AML by 10 years of follow-up. Factors determining an individual patient’s risk of malignant transformation remain poorly defined. Recent studies identified nearly ubiquitous clonal hematopoiesis (CH) in AA patients. Similarly, CH with additional, non-PIGA, somatic alterations occurs in the majority of patients with PNH. Factors associated with progression to secondary MDS/AML include longer duration of disease, increased telomere attrition, presence of adverse prognostic mutations, and multiple mutations, particularly when occurring early in the disease course and at a high allelic burden. Here, we will review the prevalence and characteristics of somatic alterations in AA and PNH and will explore their prognostic significance and mechanisms of clonal selection. We will then discuss the available data on post-AA and post-PNH progression to secondary MDS/AML and provide practical guidance for approaching patients with PNH and AA who have CH.
Introduction
Aplastic anemia (AA) is a nonneoplastic autoimmune bone marrow failure (BMF) syndrome caused by T-cell–mediated destruction of hematopoietic stem and progenitor cells (HSPCs) that is closely associated with clonal hematopoietic diseases (Figure 1).1,2 Paroxysmal nocturnal hemoglobinuria (PNH) is a closely related BMF disorder caused by somatic mutations in the PIGA gene leading to clonal expansion of hematopoietic stem cells deficient in glycosylphosphatidylinositol (GPI)-anchored proteins (GPI-APs).3 The lack of complement-regulatory GPI-APs CD55 and CD59 leads to intravascular hemolysis and frequent thrombotic complications, which are characteristic clinical features of PNH4 (Figure 2). The greatest risk factor for PNH is immune-mediated AA, and the close association between the 2 disorders has led to the hypothesis that PNH cells arise as a means of immune escape in AA.5-8
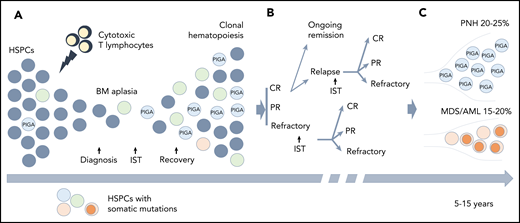
Clonal hematopoiesis (CH) in AA. (A) A schematic diagram depicting the relationship between the autoimmune pathogenesis of AA and the emergence of CH. The autoimmune attack (lightning symbol) on HSPCs (gray circles) by cytotoxic T lymphocytes leads to bone marrow (BM) aplasia. After the patients achieve hematopoietic recovery following immunosuppressive therapy (IST), they may develop the clonal expansion of cells bearing somatic mutations (circles of different colors) due to a relative growth or survival advantage conferred by somatic mutations. (B) A schematic diagram illustrating various treatment outcomes that may affect clonal evolution, including achievement of complete remission (CR), partial remission (PR), or having refractory disease following IST. Approximately one-third of the patients will experience a relapse. Some of the patients with relapsed and refractory disease may be salvaged by a second round of IST. (C) With long-term follow-up, 15% to 20% of patients will acquire additional genetic alterations (dark orange circles) and will progress to secondary myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML), whereas 20% to 25% of patients will develop hemolytic paroxysmal nocturnal hemoglobinuria (PNH) over the course of their disease (caused by PIGA mutation).
Clonal hematopoiesis (CH) in AA. (A) A schematic diagram depicting the relationship between the autoimmune pathogenesis of AA and the emergence of CH. The autoimmune attack (lightning symbol) on HSPCs (gray circles) by cytotoxic T lymphocytes leads to bone marrow (BM) aplasia. After the patients achieve hematopoietic recovery following immunosuppressive therapy (IST), they may develop the clonal expansion of cells bearing somatic mutations (circles of different colors) due to a relative growth or survival advantage conferred by somatic mutations. (B) A schematic diagram illustrating various treatment outcomes that may affect clonal evolution, including achievement of complete remission (CR), partial remission (PR), or having refractory disease following IST. Approximately one-third of the patients will experience a relapse. Some of the patients with relapsed and refractory disease may be salvaged by a second round of IST. (C) With long-term follow-up, 15% to 20% of patients will acquire additional genetic alterations (dark orange circles) and will progress to secondary myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML), whereas 20% to 25% of patients will develop hemolytic paroxysmal nocturnal hemoglobinuria (PNH) over the course of their disease (caused by PIGA mutation).
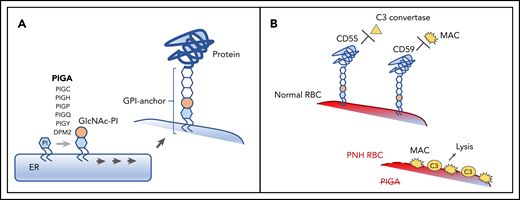
Pathogenesis of PNH. (A) A schematic diagram depicting the normal biosynthesis of glycophosphatidylinositol (GPI)-anchored proteins. The first enzymatic step in GPI-anchor biosynthesis, the addition of N-acetylglucosamine (GIcNAc, tan circle) to phosphatidylinositol (PI, blue hexagon), is catalyzed on the cytoplasmic side of the endoplasmic reticulum (ER) by a multisubunit complex that requires PIGA. Subsequent enzymatic steps (denoted by multiple arrows) add sugar moieties (white hexagons) to form the GPI anchor, which is then added to the precursor proteins on the luminal side of the ER. (B) The clinical symptoms of PNH are characterized by complement-mediated intravascular hemolysis, which occurs due to the deficiency of 2 GPI-anchored complement-regulatory proteins, CD55 and CD59. Under normal conditions, CD55 and CD59 on the surface of the red blood cell (RBC) inhibit C3 convertase and the membrane attack complex (MAC), respectively, protecting RBCs from complement-mediated lysis. In PNH, the defect in the PIGA gene leads to the absence of all GPI-anchored proteins, including CD55 and CD59, making the PNH RBC susceptible to complement activation and lysis.
Pathogenesis of PNH. (A) A schematic diagram depicting the normal biosynthesis of glycophosphatidylinositol (GPI)-anchored proteins. The first enzymatic step in GPI-anchor biosynthesis, the addition of N-acetylglucosamine (GIcNAc, tan circle) to phosphatidylinositol (PI, blue hexagon), is catalyzed on the cytoplasmic side of the endoplasmic reticulum (ER) by a multisubunit complex that requires PIGA. Subsequent enzymatic steps (denoted by multiple arrows) add sugar moieties (white hexagons) to form the GPI anchor, which is then added to the precursor proteins on the luminal side of the ER. (B) The clinical symptoms of PNH are characterized by complement-mediated intravascular hemolysis, which occurs due to the deficiency of 2 GPI-anchored complement-regulatory proteins, CD55 and CD59. Under normal conditions, CD55 and CD59 on the surface of the red blood cell (RBC) inhibit C3 convertase and the membrane attack complex (MAC), respectively, protecting RBCs from complement-mediated lysis. In PNH, the defect in the PIGA gene leads to the absence of all GPI-anchored proteins, including CD55 and CD59, making the PNH RBC susceptible to complement activation and lysis.
It has long been recognized that patients with AA and PNH have an increased risk of developing myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).9-11 Incidence of secondary MDS/AML at 10 years can reach 15% to 20% in patients with AA,12-18 and 2% to 6% in patients with PNH19,20 (Tables 1 and 2). Here, we will review current data on clonal evolution in AA and PNH, describe risk factors for post-AA and PNH secondary MDS/AML, and provide practical guidance for approaching AA and PNH patients with CH.
Evolution to secondary MDS/AML in AA
| Study/Year | Patients, n | Median age (range), y | Location, study period, design | Treatment regimen | Median follow-up (range), y | No. of patients transformed to MDS/AML and, if reported, the median time to transformation | Projected incidence of transformation to MDS/AML |
|---|---|---|---|---|---|---|---|
| de Planque et al12 /1989 | 209* | 23 (1-73) | Europe, 1975-1985, retrospective | ATG, CsA, steroids | 4.1 (2.0-10.9) | Total (MDS or AML): n = 12; | MDS or AL: 15% at 7 y |
| MDS: n = 11, 4.6 (2.5-7.5) y; | |||||||
| AL: n = 5 (4 with prior MDS), 5.0 (2.8-7.6) y | |||||||
| Socié et al10 /1993† | 860 | 29‡ (0.3-83) | Europe, 1971-1991, prospective | ATG + MP ± androgens | 3.3‡ (0.1-14) | Total (MDS or AML): n = 34; | MDS: 9.6% (95% CI, 5.5% to 16.5%) at 10 y; |
| MDS: n = 28, 4.3 (0.2-10.2) y; | AL: 6.6% (95% CI, 3.6% to 11.8%) at 10 y | ||||||
| AL: n = 15 (9 with prior MDS), 3.9 (0.6-9.6) y | |||||||
| Socie et al95 /2007 | 840 | 26.8 (IQR, 15.4-46.6) | Europe, 1990-2002, retrospective | ATG + CsA, ± G-CSF | 4.7 (NR) | MDS: n = 24, 2.5 (IQR, 2-5.6) y; | MDS: 4.3% at 10 y |
| AML: n = 26, 2.2 (IQR, 0.9-5.4) y | AML: 4.6% at 10 y | ||||||
| Tichelli et al13 /1988 | 129 | 22 (1-73) | Germany, 1976-1992, prospective | ALG, norethandrolone | NR | Total (MDS or AML): n = 13; | MDS: 26% (±8%) at 15 y |
| Tichelli et al14 /1994 | MDS: n = 13, 5 (1.8-13) y; | ||||||
| AL: n = 8 (all with prior MDS) | |||||||
| Tichelli et al15 /2011 | 192 | 46 (2-81) | Germany, 2002-2008, prospective | ATG/CsA ± G-CSF | 11.7 (10.9-12.5) | Total (MDS or AML): n = 9 | 8.5% (±3%) for G-CSF group; |
| Tichelli et al139 /2019 | 8.2% (±3%) for non–G-CSF group at 15 y | ||||||
| Führer et al97 /1998 | 86 | 9 (0.9-15) | Germany/Austria, 1993-1997, prospective | ALG, CsA, ± G-CSF | NR | MDS: n = 1; | NR |
| AL: n = 4; | |||||||
| NR (0.3-1.6) | |||||||
| Huang et al98 /2015 | 100§ | 31 (5-76) | China, 2012-2014, prospective | CsA ± ATG | NR | Total (MDS or AML): n = 9; | NR |
| MDS: n = 9, 3 (0.6-7.2) y; | |||||||
| AL: n = 2 (both with prior MDS) | |||||||
| Li et al99 /2011 | 802 | 23 (3-74) | China, 1991-2007, prospective | ATG, CsA, G-CSF, androgens | 6.8 (1-28.4) | Total (MDS or AML): n = 19, 2.8 (1-20) y; | 2.5% (95% CI, 1.5% to 4.1%) at 10 y |
| MDS: n = 14; | |||||||
| AML: n = 6 (1 with prior MDS) | |||||||
| Kojima et al100 /2002 | 113 | 9 (1-18) | Japan, 1992-1997, prospective | ATG, CsA, danazol ± G-CSF | 5.3 (3.75-8.9) | Total (MDS or AML): n = 12, 4.6 (0.8-6.8) y | 13.7% (±3.9%) at 8 y |
| Ohara et al101 /1997 | |||||||
| Frickhofen et al102 /1991 | 84 | 32 (2-80) | Germany, 1986-1989, prospective | ATG, MP ± CsA | 11.3 (9.4-13.4) | Total (MDS or AML): n = 4, | 8% at 11.3 y |
| Frickhofen et al17 /2003 | NR (6.6-9.5 y) | ||||||
| Rosenfeld et al16 /2003 | 122 | 35 (NR) | USA, 1989-1998, prospective | ATG, CsA, steroids | 7.2 (NR) | MDS: n = 10||; | NR |
| AML: n = 3 | |||||||
| Paquette et al103 /1995 | 77* | 42 (16-82) | USA, 1977-1988, prospective | ATG ± androgens, steroids, cyclosporine | ∼5¶ (NR) | Total (MDS or AML): n = 5, 4.1 (2.5-10.5) y | 13% (±7%) at 10.5 y |
| Locasciulli et al104 /2001 | 87 (IST + G-CSF) | 19 (1-72) | Italy, 1978-1991, retrospective | IST + G-CSF | 3.8 (0.5-10.8) | Total (MDS or AML): n = 5, 2.1 (0.5-4.4) y; | 9% at 5 y |
| MDS: n = 3; | |||||||
| AL: n = 2 | |||||||
| Locasciulli et al104 /2001 | 57 (IST alone) | 26 (7-70) | Italy, 1978-1991, retrospective | IST (ATG, steroids, androgens, cyclosporine) | 8.6 (0.4-19) | Total (MDS or AML): n = 7, 1.0 (0.3-9.1) y; | 7% at 5 y |
| MDS: n = 5; | |||||||
| AL: n = 2 | |||||||
| Townsley et al105 /2017 | 92 | 32 (3-82) | USA, 2012-2015, prospective | ATG + CsA + eltrombopag | 1.9 (0.2-3.9) | MDS: n = 3; | N/A |
| AML: n = 1 | |||||||
| Rogers et al77 /2019 | 314 | 9.8 (1-20.3) | North America, 2002-2014, retrospective | ATG + CsA | 5 (NR) | Total (MDS or AML): n = 6 | NR |
| Doney et al106 /1997 | 227 | 25 (1-74) | USA, 1978-1986, retrospective | ATG, oxymetholone, G-CSF | NR (at least 5 y) | Total (MDS or AML): n = 20 | NR |
| Bacigalupo et al107 /2000 | 100 | 16 (1-72) | Italy, NR, prospective | 4-drug: ALG, CsA, MP, G-CSF | 3.9 (0.2-7.9) | MDS: n = 4; | NR |
| AL: n = 2 |
| Study/Year | Patients, n | Median age (range), y | Location, study period, design | Treatment regimen | Median follow-up (range), y | No. of patients transformed to MDS/AML and, if reported, the median time to transformation | Projected incidence of transformation to MDS/AML |
|---|---|---|---|---|---|---|---|
| de Planque et al12 /1989 | 209* | 23 (1-73) | Europe, 1975-1985, retrospective | ATG, CsA, steroids | 4.1 (2.0-10.9) | Total (MDS or AML): n = 12; | MDS or AL: 15% at 7 y |
| MDS: n = 11, 4.6 (2.5-7.5) y; | |||||||
| AL: n = 5 (4 with prior MDS), 5.0 (2.8-7.6) y | |||||||
| Socié et al10 /1993† | 860 | 29‡ (0.3-83) | Europe, 1971-1991, prospective | ATG + MP ± androgens | 3.3‡ (0.1-14) | Total (MDS or AML): n = 34; | MDS: 9.6% (95% CI, 5.5% to 16.5%) at 10 y; |
| MDS: n = 28, 4.3 (0.2-10.2) y; | AL: 6.6% (95% CI, 3.6% to 11.8%) at 10 y | ||||||
| AL: n = 15 (9 with prior MDS), 3.9 (0.6-9.6) y | |||||||
| Socie et al95 /2007 | 840 | 26.8 (IQR, 15.4-46.6) | Europe, 1990-2002, retrospective | ATG + CsA, ± G-CSF | 4.7 (NR) | MDS: n = 24, 2.5 (IQR, 2-5.6) y; | MDS: 4.3% at 10 y |
| AML: n = 26, 2.2 (IQR, 0.9-5.4) y | AML: 4.6% at 10 y | ||||||
| Tichelli et al13 /1988 | 129 | 22 (1-73) | Germany, 1976-1992, prospective | ALG, norethandrolone | NR | Total (MDS or AML): n = 13; | MDS: 26% (±8%) at 15 y |
| Tichelli et al14 /1994 | MDS: n = 13, 5 (1.8-13) y; | ||||||
| AL: n = 8 (all with prior MDS) | |||||||
| Tichelli et al15 /2011 | 192 | 46 (2-81) | Germany, 2002-2008, prospective | ATG/CsA ± G-CSF | 11.7 (10.9-12.5) | Total (MDS or AML): n = 9 | 8.5% (±3%) for G-CSF group; |
| Tichelli et al139 /2019 | 8.2% (±3%) for non–G-CSF group at 15 y | ||||||
| Führer et al97 /1998 | 86 | 9 (0.9-15) | Germany/Austria, 1993-1997, prospective | ALG, CsA, ± G-CSF | NR | MDS: n = 1; | NR |
| AL: n = 4; | |||||||
| NR (0.3-1.6) | |||||||
| Huang et al98 /2015 | 100§ | 31 (5-76) | China, 2012-2014, prospective | CsA ± ATG | NR | Total (MDS or AML): n = 9; | NR |
| MDS: n = 9, 3 (0.6-7.2) y; | |||||||
| AL: n = 2 (both with prior MDS) | |||||||
| Li et al99 /2011 | 802 | 23 (3-74) | China, 1991-2007, prospective | ATG, CsA, G-CSF, androgens | 6.8 (1-28.4) | Total (MDS or AML): n = 19, 2.8 (1-20) y; | 2.5% (95% CI, 1.5% to 4.1%) at 10 y |
| MDS: n = 14; | |||||||
| AML: n = 6 (1 with prior MDS) | |||||||
| Kojima et al100 /2002 | 113 | 9 (1-18) | Japan, 1992-1997, prospective | ATG, CsA, danazol ± G-CSF | 5.3 (3.75-8.9) | Total (MDS or AML): n = 12, 4.6 (0.8-6.8) y | 13.7% (±3.9%) at 8 y |
| Ohara et al101 /1997 | |||||||
| Frickhofen et al102 /1991 | 84 | 32 (2-80) | Germany, 1986-1989, prospective | ATG, MP ± CsA | 11.3 (9.4-13.4) | Total (MDS or AML): n = 4, | 8% at 11.3 y |
| Frickhofen et al17 /2003 | NR (6.6-9.5 y) | ||||||
| Rosenfeld et al16 /2003 | 122 | 35 (NR) | USA, 1989-1998, prospective | ATG, CsA, steroids | 7.2 (NR) | MDS: n = 10||; | NR |
| AML: n = 3 | |||||||
| Paquette et al103 /1995 | 77* | 42 (16-82) | USA, 1977-1988, prospective | ATG ± androgens, steroids, cyclosporine | ∼5¶ (NR) | Total (MDS or AML): n = 5, 4.1 (2.5-10.5) y | 13% (±7%) at 10.5 y |
| Locasciulli et al104 /2001 | 87 (IST + G-CSF) | 19 (1-72) | Italy, 1978-1991, retrospective | IST + G-CSF | 3.8 (0.5-10.8) | Total (MDS or AML): n = 5, 2.1 (0.5-4.4) y; | 9% at 5 y |
| MDS: n = 3; | |||||||
| AL: n = 2 | |||||||
| Locasciulli et al104 /2001 | 57 (IST alone) | 26 (7-70) | Italy, 1978-1991, retrospective | IST (ATG, steroids, androgens, cyclosporine) | 8.6 (0.4-19) | Total (MDS or AML): n = 7, 1.0 (0.3-9.1) y; | 7% at 5 y |
| MDS: n = 5; | |||||||
| AL: n = 2 | |||||||
| Townsley et al105 /2017 | 92 | 32 (3-82) | USA, 2012-2015, prospective | ATG + CsA + eltrombopag | 1.9 (0.2-3.9) | MDS: n = 3; | N/A |
| AML: n = 1 | |||||||
| Rogers et al77 /2019 | 314 | 9.8 (1-20.3) | North America, 2002-2014, retrospective | ATG + CsA | 5 (NR) | Total (MDS or AML): n = 6 | NR |
| Doney et al106 /1997 | 227 | 25 (1-74) | USA, 1978-1986, retrospective | ATG, oxymetholone, G-CSF | NR (at least 5 y) | Total (MDS or AML): n = 20 | NR |
| Bacigalupo et al107 /2000 | 100 | 16 (1-72) | Italy, NR, prospective | 4-drug: ALG, CsA, MP, G-CSF | 3.9 (0.2-7.9) | MDS: n = 4; | NR |
| AL: n = 2 |
Rates of transformation to MDS/AML in series of AA patients treated with IST.
AL, acute leukemia; ALG, antilymphocyte globulin; ATG, antithymocyte globulin; CI, confidence interval; CsA, cyclosporine A; G-CSF, granulocyte-colony stimulating factor; IQR, interquartile range; IST, immunosuppressive therapy; MP, methylprednisolone; N/A, not applicable; NR, not reported; USA, United States of America.
Long-term survivors (>2 y) with records available.
Included some patients from the above study by de Planque et al.12
Mean value.
Patients with clinical information available.
MDS group included cytogenetic evolution.
Estimated from overall K-M curve.
Evolution to secondary MDS/AML in PNH
| Study/Year | Patients, n | Median age (range), y | Country, study period, study design | Treatment regimen | Median follow-up (range), y | No. of patients transformed to MDS/AML and, if reported, the median time to transformation | Projected incidence of transformation to MDS/AML |
|---|---|---|---|---|---|---|---|
| de Latour et al20 /2008 | 454* (Classic PNH, n = 113; AA-PNH syndrome, n = 224; Int PNH, n = 93) | 34 (24-48) | France, 1950-2005, retrospective | Steroids, IST, BMT (n = 52) | 6.8 (SE 0.5) | Total (MDS or AML), n = 26 (5.7%); | 10-y rates: Overall, 3.8% (95% CI, 0.1% to 7.5%); |
| MDS: n = 21, 1.3 (IQR 0.5-4.8) y; | Classic PNH, 3.8% (95% CI, 0.1% to 7.5%); | ||||||
| Socié et al122 /1996 | AML: n = 9 (4 with prior MDS), 1.6 (IQR 0.8-4.4) y | AA-PNH, 9% (95% CI, 4.5% to 13.6%); | |||||
| Indeterminate PNH, 3.7% (95% CI, 0.1% to 7.5%) | |||||||
| Nishimura et al19 /2004 | 176 (US); | 30 (4-80); | USA/Japan, 1966-2004, retrospective | Transfusions, androgens, prednisone, ATG/ALG, CsA, EPO, G-CSF, BMT (n = 8) | 7.6 (NR); | MDS: n = 6 (3.4%), AL: n = 1 (0.6%); | NR |
| 209 (Japan) | 45 (10-86) | 8.5 (NR) | MDS: n = 8 (3.8%), AL: n = 6 (2.9%) | ||||
| Ghosh125 /2015 | 33 | NR | India, 2010s (3-y period), prospective | NR | NR | AL, n = 1 (prior MDS) (3%); 0.5 y | NR |
| Ware et al123 /1991 | 26 | 14 (0.8-21) | USA, 1966-1991, retrospective | Prednisone, androgens, ATG | 8.5† (2.8-27) | AL, n = 1 (3.8%); 6 y | NR |
| Harris et al124 /1999 | 1760‡ | NR | Multiple, 1962-1999, retrospective | NR | NR | Total (MDS or AML), n = 38 (2.2%); | NR |
| MDS, n = 25 (1.4%); | |||||||
| AL, n = 16 (3 with prior MDS) (0.9%) |
| Study/Year | Patients, n | Median age (range), y | Country, study period, study design | Treatment regimen | Median follow-up (range), y | No. of patients transformed to MDS/AML and, if reported, the median time to transformation | Projected incidence of transformation to MDS/AML |
|---|---|---|---|---|---|---|---|
| de Latour et al20 /2008 | 454* (Classic PNH, n = 113; AA-PNH syndrome, n = 224; Int PNH, n = 93) | 34 (24-48) | France, 1950-2005, retrospective | Steroids, IST, BMT (n = 52) | 6.8 (SE 0.5) | Total (MDS or AML), n = 26 (5.7%); | 10-y rates: Overall, 3.8% (95% CI, 0.1% to 7.5%); |
| MDS: n = 21, 1.3 (IQR 0.5-4.8) y; | Classic PNH, 3.8% (95% CI, 0.1% to 7.5%); | ||||||
| Socié et al122 /1996 | AML: n = 9 (4 with prior MDS), 1.6 (IQR 0.8-4.4) y | AA-PNH, 9% (95% CI, 4.5% to 13.6%); | |||||
| Indeterminate PNH, 3.7% (95% CI, 0.1% to 7.5%) | |||||||
| Nishimura et al19 /2004 | 176 (US); | 30 (4-80); | USA/Japan, 1966-2004, retrospective | Transfusions, androgens, prednisone, ATG/ALG, CsA, EPO, G-CSF, BMT (n = 8) | 7.6 (NR); | MDS: n = 6 (3.4%), AL: n = 1 (0.6%); | NR |
| 209 (Japan) | 45 (10-86) | 8.5 (NR) | MDS: n = 8 (3.8%), AL: n = 6 (2.9%) | ||||
| Ghosh125 /2015 | 33 | NR | India, 2010s (3-y period), prospective | NR | NR | AL, n = 1 (prior MDS) (3%); 0.5 y | NR |
| Ware et al123 /1991 | 26 | 14 (0.8-21) | USA, 1966-1991, retrospective | Prednisone, androgens, ATG | 8.5† (2.8-27) | AL, n = 1 (3.8%); 6 y | NR |
| Harris et al124 /1999 | 1760‡ | NR | Multiple, 1962-1999, retrospective | NR | NR | Total (MDS or AML), n = 38 (2.2%); | NR |
| MDS, n = 25 (1.4%); | |||||||
| AL, n = 16 (3 with prior MDS) (0.9%) |
Rates of transformation to MDS/AML in series of patients with PNH.
BMT, bone marrow transplantation; EPO, erythropoietin; Int PNH, intermediate PNH; SE, standard error. See Table 1 for expansion of other abbreviations.
Patients with documented follow-up.
Mean value.
Compiled 15 series of ≥20 PNH patients from 1965 through 1995; see references within.
The relationship between CH and hematologic malignancies: wrongly accused or guilty as charged?
CH is a phenomenon in which an HSPC gains a growth or a survival advantage, most commonly due to an acquired genetic change, leading to a competitive outgrowth of the mutant HSPC’s progeny. The process through which this occurs is called clonal evolution. Although all hematologic malignancies arise from an abnormal hematopoietic cell and, thus, by definition, are clonal, CH is distinct from malignancy. CH is a nonneoplastic condition that can be associated with diverse genetic alterations, some of which improve cell fitness whereas others are neutral “passengers.”
Whether the presence of mutations predisposes to malignancy, as opposed to being a general biomarker of aging and marrow stress, continues to be a subject of active debate.21 When ultrasensitive, error-corrected next-generation sequencing is used to detect somatic mutations, nearly all middle-aged adults can be found to have minute mutant clones with mutations in DNMT3A and TET2, which remain stable over a decade of follow-up.22 Using whole-genome sequencing (WGS), CH can be identified in 12.5% of individuals overall and over one-half of the people aged 85 years and older.23 The high prevalence of CH indicates that the mere presence of mutations is insufficient for malignant transformation because most individuals do not develop hematologic malignancies. Conversely, the majority of patients who develop hematologic malignancy do so without antecedent CH detectable by standard techniques, further calling into question the direct relationship between CH and malignancy.24,25 Instead, it has been hypothesized that some forms of age-related CH may actually be adaptive, sustaining blood production despite hematopoietic aging and HSPC senescence, or may occur by neutral drift without apparent driver mutations.26-28
Despite these uncertainties, it is also clear that individuals with CH do have a significantly increased risk of hematologic malignancies (hazard ratio, 2.4-12.9 across studies) and higher all-cause mortality (hazard ratio, 1.3-1.4).23-25,29 The association with hematologic malignancies was observed regardless of whether a putative driver mutation was identified,23-25 suggesting that CH may be a biomarker of more general stem cell damage (eg, stem cell loss and telomere attrition) that drives evolution to malignancy. Importantly, emerging data indicate that mutations have different consequences depending on the function of the mutated gene: for example, mutations in TP53 and U2AF1 carry a high risk of leukemic transformation, whereas mutant DNMT3A and TET2 are associated with a more benign clinical course.29 In patients with idiopathic cytopenia, somatic mutations, particularly in spliceosome genes, alone or in combination with mutations in TET2, DNMT3A, or ASXL1, were found to predict development of MDS,30 with cytopenias presumably indicative of HSPC compromise. The risk of malignancy was higher with a higher mutant allelic fraction and a greater number of mutations,23,24,29,30 suggesting that, similar to murine data, certain mutations may cooperate to increase leukemic susceptibility.31,32
CH in AA: general concepts
Among the few nonneoplastic hematologic diseases in which somatic mutations have been reported,33-36 AA and PNH are the 2 most strongly associated with CH, with CH detected in over two-thirds of AA patients.37,38 The reasons for the nearly ubiquitous CH in the recovering marrow of AA patients are severalfold. In the typical AA disease course, an autoimmune attack leads to bone marrow aplasia with massive contraction of the HSPC pool (Figure 1). After immunosuppressive therapy, AA patients have a prolonged recovery period, with up to one-third of patients experiencing a relapsing/remitting course. In this context, any cell that can evade the autoimmune attack or attenuate lymphocyte-mediated cytotoxicity will have a competitive advantage. Similarly, mutations that help to preserve the stem cell pool, improve cell survival, or enhance proliferation are favored (Figure 1). Additionally, as neutral, stochastic mutations increase with older age,39 preexisting background mutations can be “swept up” during HSPC clonal expansion.
What causes clonal expansion of PNH cells?
Although a significant number of PNH patients present with classical PNH without a prior history of BMF, the primary risk factor for PNH is a history of AA.40-42 Indeed, somatic loss of PIGA, which causes loss of the GPI-APs and the PNH cell phenotype, is the most common manifestation of CH in AA, with nearly 40% of AA patients having a detectable clone of PNH cells during their disease course.43 Ten percent to 30% of AA patients will have clonal expansion of PNH cells over time, leading to the clinical syndrome of hemolytic PNH disease.12-17,44 Although other BMF states with an immune component (eg, hypoplastic MDS) have been associated with PNH,42,45 some of these may actually represent moderate AA, as evidenced by their stable disease course and responsiveness to immunosuppressive therapy.
The mechanisms that give rise to PIGA-mutant clones in an AA bone marrow environment have been the subject of active investigation for decades.8,41 Various experimental models demonstrated that PNH cells have no intrinsic growth advantage compared with PIGA wild-type cells.46-48 Although somatic mutations may lead to enhanced growth by cooperating with PIGA loss (as hypothesized for JAK2 mutations36 and dysregulation of HMGA249,50 ), such cooperative mutations occur only in a small minority of PNH patients.36,51 Similarly, the hypothesis that PIGA-mutant cells arise as a result of genetic instability52-54 has been disproven.55,56
Instead, current evidence points to a selective advantage of GPI-AP–deficient cells within an autoimmune milieu of AA. The potential mechanisms of immune evasion by PNH cells include the resistance of PNH cells to natural killer cell and T-cell activation through the NKG2D receptor due to lack of GPI-linked NKG2D ligands,57,58 evasion of CD1d-restricted autoimmune T-cell attack against the GPI anchor moiety,59 and reduced rates of apoptosis in GPI− blood cells in the presence of mononuclear cells60 (Figure 3A-B). Similar to AA, an association of PNH with certain HLA class I and II alleles has been reported, pointing to the potential role of an HLA-mediated immune mechanism in PNH clone expansion.61,62
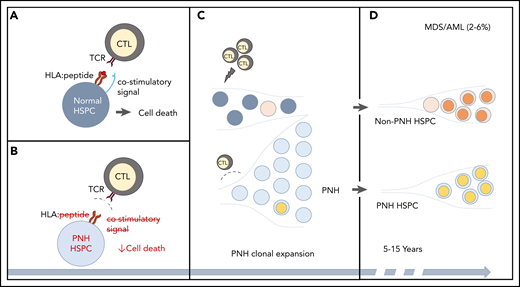
CH in PNH. (A-B) A schematic of the relationship between immune-mediated BMF and the development of PNH. (A) In immune-mediated BMF, such as AA, HSPCs (dark gray circles) undergo immune-mediated attack by cytotoxic T lymphocytes (CTLs), which recognize AA autoantigen presented in the context of an HLA molecule (red lines, HLA:peptide) via a T-cell receptor (TCR). Effective CTL activation requires the presence of a costimulatory signal (blue line). (B) PNH HSPCs (light gray circles) are believed to evade autoimmune T-cell recognition by 1 or more mechanisms, which include lack of a GPI-anchored autoantigen, lack of a costimulatory interaction that may involve GPI-anchored proteins, or relative resistance to T-cell–mediated cell death. (C) The evasion of the CTL-mediated immune attack leads to clonal expansion by the PNH HSPCs. Ongoing autoimmune selective pressure may lead to the emergence of additional somatic mutations (depicted by colored circles) both in the ancestral, previously normal HSPCs and in the PNH HSPCs. (D) With long-term follow-up, stepwise accumulation of additional genetic events, either in the ancestral HSPCs or in the PNH clone, can lead to secondary MDS-AML in 3% to 6% of PNH patients.
CH in PNH. (A-B) A schematic of the relationship between immune-mediated BMF and the development of PNH. (A) In immune-mediated BMF, such as AA, HSPCs (dark gray circles) undergo immune-mediated attack by cytotoxic T lymphocytes (CTLs), which recognize AA autoantigen presented in the context of an HLA molecule (red lines, HLA:peptide) via a T-cell receptor (TCR). Effective CTL activation requires the presence of a costimulatory signal (blue line). (B) PNH HSPCs (light gray circles) are believed to evade autoimmune T-cell recognition by 1 or more mechanisms, which include lack of a GPI-anchored autoantigen, lack of a costimulatory interaction that may involve GPI-anchored proteins, or relative resistance to T-cell–mediated cell death. (C) The evasion of the CTL-mediated immune attack leads to clonal expansion by the PNH HSPCs. Ongoing autoimmune selective pressure may lead to the emergence of additional somatic mutations (depicted by colored circles) both in the ancestral, previously normal HSPCs and in the PNH HSPCs. (D) With long-term follow-up, stepwise accumulation of additional genetic events, either in the ancestral HSPCs or in the PNH clone, can lead to secondary MDS-AML in 3% to 6% of PNH patients.
For PNH patients who present with classical PNH disease without a history of BMF, the reasons for PNH clonal expansion are unknown. One possibility is that PNH developed following a clinically occult episode of moderate AA. One notable exception with an alternative mechanism is the recently described rare variant of PNH, phosphatidylinositol glycan anchor biosynthesis class T (PIGT)-PNH, which emerges due to the loss of PIGT, an enzyme responsible for the attachment of the GPI anchor to proteins.63,64 Unlike PIGA, which is X-linked and requires 1 inactivating mutation to produce a PNH phenotype, PIGT is autosomal, requiring biallelic inactivation for GPI-AP loss. Known cases of PIGT-PNH occurred in the absence of BMF.63,64 Instead, PIGT-PNH patients inherited 1 loss-of-function mutation in PIGT and subsequently developed a deletion of the myeloid common deleted region on chromosome 20q, containing PIGT and associated with clonal expansion in myeloid malignancies.63,64
Common somatic alterations in AA
CH occurs in over 70% to 80% of AA patients, and over 60% of patients of pediatric age37,38 (Figure 4). Mutations typically develop within 6 months of immunosuppressive therapy, but can already be present at diagnosis.37,65 The profile of somatic mutations in AA differs from age-associated CH and MDS, with the 2 most common somatic changes in AA, mutational inactivation of PIGA and the loss of HLA class I alleles, being pathognomonic of immune-mediated marrow failure. HLA loss can occur due to a loss-of-function mutation in 1 of the HLA class I genes or through the loss of 1 of the parental HLA haplotypes.37,38,65-67 The latter can be detected on single-nucleotide polymorphism array as copy number–neutral loss of heterozygosity of chromosome arm 6p at the site of the major histocompatibility locus.67,68 Copy number–neutral loss of heterozygosity of chromosome arm 6p is relatively specific to AA, occurring in <1% of patients with MDS or normal aging,69,70 but found in 11% to 13% of AA patients. Interestingly, PIGA and HLA mutations have not been found within the same hematopoietic clone in AA patients,65,71,72 suggesting a lack of additive advantage from having these 2 mutations together and pointing to a similar mechanism of clonal selection by evasion of HLA-restricted autoimmune attack.

The landscape of CH in AA. (A) Left, When all available modalities to assess for genetic alterations are used, CH can be detected in over 70% to 80% of AA patients (pie chart). Right, Bar chart showing the relative proportions of recurrent mutations. The 2 most commonly mutated genes in AA are PIGA and HLA (blue bars); inactivating mutations in these genes are pathognomonic of immune-mediated BMF. Mutations in genes associated with MDS (red bar) are found in ∼25% to 30% of AA patients. (B) Table showing available information on the prognostic significance of recurrent genetic alterations in AA, which is discussed in "Common somatic alterations in AA" and "Evolution to post-AA secondary MDS/AML." §Based on Negoro et al.51 #Based on Yoshizato et al.37
The landscape of CH in AA. (A) Left, When all available modalities to assess for genetic alterations are used, CH can be detected in over 70% to 80% of AA patients (pie chart). Right, Bar chart showing the relative proportions of recurrent mutations. The 2 most commonly mutated genes in AA are PIGA and HLA (blue bars); inactivating mutations in these genes are pathognomonic of immune-mediated BMF. Mutations in genes associated with MDS (red bar) are found in ∼25% to 30% of AA patients. (B) Table showing available information on the prognostic significance of recurrent genetic alterations in AA, which is discussed in "Common somatic alterations in AA" and "Evolution to post-AA secondary MDS/AML." §Based on Negoro et al.51 #Based on Yoshizato et al.37
Other mutations found frequently in AA patients include epigenetic modifier genes BCOR and BCORL1, DNMT3A, and ASXL1.37,51,73 Overall, mutations in genes associated with aging and hematologic malignancies occur in ∼25% to 30% of AA patients74 (Figure 4). In pediatric AA, malignancy-associated mutations are found less frequently, likely due to the lower rate of age-related CH.37,74-77
In addition to somatic mutations, ∼10% of AA patients develop cytogenetic abnormalities (CAs), ranging in frequency from 3% to 26%.78 Recurrent CAs in AA include whole or partial loss of chromosome 7, trisomy 8, and deletion 13q, and, less frequently, trisomy 6, trisomy 15, and trisomy 21. CAs can be detected at diagnosis in ∼1% to 4% of AA patients78 and may introduce diagnostic uncertainty in distinguishing AA from hypoplastic MDS. Because metaphase cytogenetics are often inadequate in AA because of low cellularity, low aspirate quality, and poor cell growth, it is important to use fluorescence in situ hybridization or single-nucleotide polymorphism array in conjunction with a traditional karyotype.
There are several distinctions between the typical CAs in AA and primary MDS. Although deletion 13q continues to be included on the 2016 list of MDS-defining CAs by the World Health Organization (WHO),79 it carries a favorable prognosis in AA, where it is associated with an improved response to immunosuppression, lack of morphologic dysplasia, and a disease course much more consistent with AA than MDS.80-82 Interestingly, in AA, isolated deletion 13q frequently occurs in conjunction with a PNH clone, but derives from PIGA wild-type, GPI-AP+ cells, suggesting that it may represent an alternative pathway of immune evasion through an as-yet-unknown mechanism.80 Similar to deletion 13q, trisomy 8 carries a favorable prognosis and a high rate of response to immunosuppression when present in association with a PNH clone.83,84 In contrast, monosomy 7 connotes a poor prognosis84 and is considered a harbinger of malignant transformation. Unlike in primary MDS, where monosomy 7 commonly occurs in conjunction with complex karyotype and loss of TP53, monosomy 7 in AA typically presents as an isolated abnormality and may occur in the absence of somatic mutations.51,85
Common somatic alterations in PNH
Because PNH is caused by a clonal expansion of PIGA-mutant cells, all PNH patients, by definition, have CH. However, similar to AA, PNH patients can acquire additional genetic abnormalities in their hematopoietic cells.36 When whole exome sequencing was used to characterize somatic mutations in classical PNH, 10 of 12 PNH patients (83%) had somatic mutations other than PIGA, with an average of 2 additional mutations (range, 0-6) per patient.36 Unlike in AA, where somatic HLA loss is common, to date, HLA mutations have not been identified in patients with classical PNH,65 suggesting that HLA loss likely confers no additional advantage in a PNH cell. In contrast, malignancy-associated mutations were found in 27% to 44% of PNH patients.36,51 The most commonly mutated genes were TET2 and JAK2, followed by ASXL1, BCOR, MECOM, and RIT1.36,51 Clonal architecture analysis revealed that accessory non-PIGA somatic mutations can occur either as a subclone of the PNH cell or can arise independently in the ancestral, PIGA wild-type HSPC36 (Figure 3).
Although PNH is generally considered a disease of normal karyotype, PNH patients can also develop CAs.86-89 Most common CAs in PNH are similar to AA and include trisomy 8, deletion 13q, monosomy 7, and trisomy 6,86,89 where deletion 13q is similarly associated with a relatively benign clinical course.86,88,89 Rearrangements of chromosome 12q12-12q14, leading to dysregulated expression of HMGA2, have also been reported.90 For other CAs, the clinical significance is less clear. In a retrospective analysis of 46 PNH patients, CAs were seen in 11 patients (24%), but none developed excess blasts or leukemia.86 Clonal architecture analysis of 9 PNH patients with CA found that in all but 1 patient, CAs arose in a non-PNH stem cell, suggesting that CAs arise due to external selective pressure, as opposed to an intrinsic, PNH cell-specific chromosomal instability.89
A well-known curiosity of PNH is spontaneous remission,91,92 which can occur in up to 15% of patients.56 Recent studies of PNH spontaneous remission suggested that the diminution of the PNH clone does not imply the restoration of healthy hematopoiesis. Instead, PNH remission may represent clonal replacement or evolution, which, in some cases, can occur without an apparent clinical consequence, but in others may herald malignant transformation.93,94 This phenomenon is similar to other types of clonal evolution in AA/PNH and reflects ongoing clonal selection over time.
Evolution to post-AA secondary MDS/AML
Although CH develops in most AA patients, only a minority of patients develop secondary MDS/AML10,12-17,95-107 (Table 1). The true rate of post-AA malignant transformation is likely lower than the ∼15% to 20% reported by historical studies because of the potential inclusion of patients with hypoplastic MDS and inherited BMF, focus on severe AA, and less effective therapies in older series. The discrepancy between nearly ubiquitous CH in AA and the severalfold lower frequency of secondary MDS/AML underscores that in AA, just like in age-related CH, clonality by itself is not a predictor of malignant transformation. Instead, the nearly ubiquitous CH reflects the unique marrow environment of AA, defined by HSPC-directed autoimmunity and a severely depleted stem cell pool (Figure 1).
The risk of secondary MDS/AML goes up with disease duration without reaching a plateau,10,17,99 occurring in 4% to 8% of patients by 5 to 6 years of follow-up and in 9% to 26% by 10 years (Table 1). Importantly, multiple factors, not captured by aggregated statistics, likely contribute to a given patient’s risk of malignancy, including age at AA diagnosis, constitutional genetic differences, somatic mutations at onset of AA, and telomere lengths (TLs). Disease-specific factors, such as AA severity, time to therapy, and treatment response, additionally contribute to the risk of malignancy by determining HSPC depletion and the selective pressure on the remaining cells.
Mutations present in the HSPCs at the time of AA onset act as a substrate for clonal selection when the autoimmune attack and the potential for clonal sweeps are the strongest. Accordingly, AA patients with MDS-associated somatic mutations detected early in the AA course had the highest rate of subsequent MDS/AML progression.51,73 Conversely, with less telomere attrition and fewer aging/malignancy-associated mutations in childhood, a pediatric diagnosis of AA may be protective against malignant transformation compared with AA diagnosis later in life. In agreement with this, pediatric AA patients developed fewer clones with aging- and MDS-associated somatic mutations compared with adult patients.38,65,74,75 The low rate of malignant transformation in pediatric patients was confirmed in a recent analysis of AA patients in the North American Pediatric Aplastic Anemia Consortium, which found a 2% frequency of MDS/AML after a median of 5 years of follow-up.77 The lower MDS/AML frequency compared with historical studies of pediatric AA97,100 may be in part due to improved diagnostic precision of AA, with older studies more likely to include patients with occult inherited BMF syndromes.
Although available data do not answer this question definitively, it stands to reason that prolonged or refractory disease course may increase the risk of post-AA malignancy and that factors associated with a more severe disease course (eg, inheriting an AA HLA-risk allele65,66 ) may potentiate clonal selection. Conversely, early remission would be predicted to protect the remaining stem cell pool, removing the autoimmune pressure that causes expansion of existing clones and selects for additional mutations.51,73 Best illustrated in the case of PNH, AA/PNH patients had less PNH clone expansion when they received standard immunosuppression therapy compared with less effective cyclosporine monotherapy or no treatment at all.108 Similarly, spontaneous remissions of CAs, reported in 5% to 10% of AA patients, have also been noted in conjunction with immunosuppressive therapy and seem to correlate with treatment response.65,107,109-113 Other immune-responsive lesions (eg, HLA loss, del 13q, BCOR/BCORL1 mutations) may be similarly stabilized by AA remission37,86,88,89 ; however, whether this is true for other genetic alterations is not clear.
Of somatic abnormalities in AA, monosomy 7 is most strongly associated with post-AA malignant transformation.51,84,85,114 Monosomy 7 can develop in the absence of somatic mutations and has been linked to accelerated telomere attrition.85 Telomere attrition may be a product of the exhausted stem cell pool, contributing to senescence, replicative stress, and chromosomal instability.115 The relationship between leukocyte TLs in AA and clonal evolution may be more complicated, as short TLs within the lymphocyte compartment reflect lymphocyte activation and replicative history116 and may be a marker of greater AA severity. Indeed, shorter TLs at diagnosis were associated with a more severe disease course, worse overall survival, and an increased risk of CA, including monosomy 7.117 It is important to distinguish between telomere attrition in immune-mediated AA and telomere shortening caused by inherited telomere maintenance defects in the BMF syndrome dyskeratosis congenita.118 Although AA cohorts may inadvertently include rare patients with occult dyskeratosis congenita,105,119 these probably represent a minority of analyzed patients, whereas the bulk of the TL effect in AA seems to be related to acquired TL defects related to cell replicative history.
The role of somatic mutations in post-AA leukemogenesis is less clear. In contrast to studies of CH in the healthy aged population that included thousands of individuals,22,24,25,29,120,121 far fewer AA patients have been analyzed, and the available data mostly come from retrospective studies at tertiary referral centers, with short clinical follow-up and the potential for referral and ascertainment biases.37,51,73,85,98-100,114 These limitations restrict our ability to tease out the role of specific mutations in malignant transformation in AA. Nonetheless, some insights can be gleaned from the available data. Patients who progressed to post-AA MDS/AML were more likely to have clonal expansions of cells bearing somatic mutations detected early in their AA course, at a higher mutant allelic fraction, and had a longer duration of disease with mutations as compared with nonprogressors.51,73 A multi-institutional study of 439 patients with AA reported an association of MDS/AML progression with an “unfavorable group” of mutations including DNMT3A, ASXL1, RUNX1, JAK2, and JAK3, whereas alterations in a “favorable” group of BCOR, BCORL1, and PIGA genes correlated with a reduced risk of transformation and improved survival.37 An analysis comparing 23 patients with post-AA/PNH secondary MDS to 133 patients with AA alone found that mutations in ASXL1, RUNX1, splicing factors, and CBL were significantly more frequent in patients with secondary MDS.51 Two additional retrospective studies also identified mutations in ASXL1 as a possible risk factor for MDS transformation.73,98 The contribution of mutations to the risk of malignancy in a given patient is less clear, and has to be considered within the context of other patient- and disease-specific factors, as well as the function of the mutated gene in hematopoiesis, aging, and autoimmunity.
Evolution to post-PNH secondary MDS/AML
Similar to AA, PNH patients can develop secondary hematologic malignancies, most commonly MDS and AML.19,20,122-128 The frequency of post-PNH secondary MDS/AML is threefold to sevenfold less frequent compared with AA, estimated to occur in 2.3% to 6.4% of PNH patients by 7 to 10 years of follow-up. Patients with AA/PNH syndrome have an intermediate rate of secondary malignancies (9%; 95% confidence interval, 4.5-13.6)20 (Table 2). The lower rate of secondary MDS/AML in PNH is likely due to the reduced autoimmune selection on the PNH cell, which has evolved to evade the autoimmune attack in AA. The lower selective pressure on the PNH HSPC likely explains why, in most (although not all124,129-134 ) cases, post-PNH malignancy originates from the PIGA wild-type ancestral HSPC93,135-137 and is accompanied by PNH remission at the time of leukemia onset.93,135-137 Data on the role of specific mutations in post-PNH secondary MDS/AML are very limited. A single-center analysis of genetic alterations in 49 patients with PNH compared with 23 patients with post-AA and post-PNH secondary MDS showed a higher rate of mutations in ASXL1, RUNX1, and SETBP1 in the patients with secondary MDS.51
Effects of AA and PNH therapy on clonal evolution
At present, therapeutic decisions in AA patients are not driven by the detection of somatic mutations. However, with the improving ability to characterize genetic alterations in AA patients in the clinic, there has been increasing interest in the role of AA therapies on long-term clonal complications in AA.
Earlier retrospective analyses of AA patients treated with granulocyte colony-stimulating factor (G-CSF) compared with immunosuppression alone seemed to show a higher rate of emergence of monosomy 7 and development of MDS in G-CSF–treated patients.100,101 However, when analyzed prospectively in a randomized manner, the use of G-CSF was not associated with increased clonal evolution, malignant transformation, or mortality outcomes,15,104,138 with the earlier observations now attributed to confounding by indication, with more severely affected patients being more likely to have been prescribed G-CSF.139,140
The introduction of small molecule thrombopoietin agonist eltrombopag into the treatment of AA both in the upfront105 and the refractory105,141,142 settings introduced concerns about the promotion of clonal evolution by eltrombopag. In the National Institutes of Health (NIH) cohort, 8 of 43 refractory AA patients treated with eltrombopag developed clonal cytogenetic abnormalities that included loss or partial deletion of chromosome 7. A follow-up analysis using an extended 24-week course of eltrombopag in patients with refractory AA showed early evolution to an abnormal karyotype (most commonly monosomy 7) in 18% of patients, with no change in the allelic fraction of somatic mutations.143 The unusually high frequency of early CAs temporally following eltrombopag therapy suggests a potential causative relationship between eltrombopag and the detection of CAs. This could occur through an eltrombopag-mediated expansion of preexisting cytogenetically abnormal cells or due to eltrombopag-mediated enhancement of damaged HSPC growth beyond normal limits of replicative senescence. A recent study found that irradiated CD34+ cells treated with eltrombopag had a higher rate of nonhomologous end-joining and greater survival of genetically damaged irradiated HSPCs compared with cells not treated with eltrombopag.144 Long-term follow-up and close surveillance of eltrombopag-treated patients will be essential in clarifying the role of eltrombopag therapy, if any, on the development of late hematologic malignancies in this population. The ongoing European Society for Blood and Marrow Transplantation (EBMT) randomized controlled trial of immunosuppression with and without eltrombopag in an upfront setting (RACE)145 will be particularly informative for treatment-naive patients, who are anticipated to have a lower rate of clonal complications compared with patients with relapsed/refractory AA.
An unresolved issue of relevance to AA patients with a subclinical PNH clone is whether eltrombopag has a role in promoting PNH clonal expansion. In a study of 8 relapsed/refractory AA patients with a detectable PNH clone treated with eltrombopag, 4 patients had >20% PNH clone expansion on eltrombopag therapy; the other 4 had stable or minimally increased PNH clone size.143 In treatment-naive AA patients receiving eltrombopag together with standard immunosuppression, no specific trend of PNH clone size was observed.105
Finally, in PNH, the role of C5 complement inhibition on clonal evolution has been evaluated both in prospective and retrospective studies.146-149 Treatment with the C5 inhibitor eculizumab did not affect the PNH granulocyte clone size and did not alter the rates of MDS/AML in eculizumab-treated compared with untreated patients. Interestingly, for the exceptional patients with the rare subtype of PIGT-mutated PNH, eculizumab may play a role in PIGT-PNH granulocyte clone expansion. The reasons for this are not entirely clear, but may be due to the increased intensity of peripheral complement activation leading to complement-mediated destruction of PIGT-PNH granulocytes in addition to PNH erythrocytes.63
Conclusions
AA and PNH are 2 closely related BMF disorders of autoimmune etiology. The majority of AA and PNH patients experience clonal evolution with the development of somatic alterations over the course of their disease, and ∼15% to 20% of AA patients and 2% to 6% of PNH patients develop secondary MDS/AML by 10 years of follow-up. As in other types of secondary leukemias, somatic mutations associated with post-AA and post-PNH secondary MDS/AML confer poor prognosis.37,51,73,98,150 With the growing appreciation of CH and somatic mutations in AA and PNH, advanced diagnostics, including next-generation sequencing panels, have entered the clinical care of AA patients. However, our ability to predict an individual’s risk of secondary MDS/AML based on their somatic mutation profile remains limited. Factors linked to an increased risk of post-AA and post-PNH secondary MDS/AML include disease duration, older age, relapsed/refractory disease, accelerated telomere attrition, and certain genetic alterations. Patients with high-risk features should undergo closer follow-up with periodic blood count surveillance and be counseled regarding the potential for secondary MDS/AML. Consideration should be given to a preemptive assessment of transplant donor options. Areas of active investigation include the effect of thrombopoietin mimetic therapy on clonal evolution in AA, as well as the optimal choice and sequencing of immunosuppression-based and transplant approaches and posttreatment surveillance.
Acknowledgments
The authors thank the patients and families for participating in the AA and PNH studies. The authors also thank the 3 anonymous reviewers whose thoughtful comments helped to improve this manuscript.
This work was supported by National Institutes of Health grants K08 HL132101 from the National Heart, Lung, and Blood Institute (D.V.B.) and T32 CA009679-28 from the National Cancer Institute (L.S.).
Authorship
Contribution: L.S. and D.V.B. performed literature searches and data analysis, and wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daria V. Babushok, Division of Hematology-Oncology, Department of Medicine, University of Pennsylvania, BRB II/III Room 808, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: daria.babushok@pennmedicine.upenn.edu.
