

Key Points
Limited concordance and differing trajectories of clonal hematopoiesis in identical twins emphasize nonheritable factor importance.
Identification of elderly monozygotic twins with identical driver mutations suggests a common cellular origin in utero.

Abstract
Although acquisition of leukemia-associated somatic mutations by 1 or more hematopoietic stem cells is inevitable with advancing age, its consequences are highly variable, ranging from clinically silent clonal hematopoiesis (CH) to leukemic progression. To investigate the influence of heritable factors on CH, we performed deep targeted sequencing of blood DNA from 52 monozygotic (MZ) and 27 dizygotic (DZ) twin pairs (aged 70-99 years). Using this highly sensitive approach, we identified CH (variant allele frequency ≥0.5%) in 62% of individuals. We did not observe higher concordance for CH within MZ twin pairs as compared with that within DZ twin pairs, or to that expected by chance. However, we did identify 2 MZ pairs in which both twins harbored identical rare somatic mutations, suggesting a shared cell of origin. Finally, in 3 MZ twin pairs harboring mutations in the same driver genes, serial blood samples taken 4 to 5 years apart showed substantial twin-to-twin variability in clonal trajectories. Our findings propose that the inherited genome does not exert a dominant influence on the behavior of adult CH and provide evidence that CH mutations may be acquired in utero.
Introduction
Clonal hematopoiesis (CH), the disproportionate expansion of blood cell clones harboring leukemia-associated somatic mutations, becomes more prevalent with advancing age and is the precursor of many hematological malignancies.1-5 Acquisition of such mutations in 1 or more hematopoietic stem cell (HSC) is inevitable by the age of 50 to 60 years,6 yet the consequences of mutation acquisition are highly variable between individuals. A number of small studies tracking clonal size longitudinally suggest that clones in different individuals with similar or even identical mutations behave differently over time.5-7 Indeed, this could also be inferred by the fact that clinically silent CH is common, whereas hematological cancers are rare.5 Importantly, acquisition of additional driver mutations is not always necessary for malignant progression; for example, the JAK2-V617F mutation8 is the sole identifiable driver in many cases of myeloproliferative neoplasms, and SF3B1 mutations9,10 are often the only driver in myelodysplastic syndromes. The factors that allow mutant clones to expand in some individuals, and those restraining them in others, are not understood.
There is some evidence that the inherited genome might play a role in this process. For example, recent studies reported (i) heritable genetic variants associated with increased risk of developing MPNs,11,12 (ii) familial clustering of CH driven by TET2 mutations,13 and (iii) increased prevalence of CH among relatives of individuals with myeloid, but not lymphoid, malignancies.14 Moreover, a number of germline variants have emerged as important determinants of hematological phenotypes in the general population and it is plausible that these exert epistatic effects on CH evolution.15 To investigate whether the inherited genome influences CH development, we performed deep targeted sequencing on blood DNA from 52 monozygotic (MZ) and 27 dizygotic (DZ) twin pairs, and analyzed patterns of twin-to- twin concordance for CH.
Study design
We studied blood DNA from 158 individuals from the TwinsUK cohort, comprising 52 MZ and 27 DZ twin pairs with no history of hematological malignancy, aged 70 to 99 years (150 women, 8 men).16 Samples were obtained with informed consent and appropriate ethics committee approval (REC reference EC04/015). Target enrichment for 41 genes implicated in CH and myeloid malignancies (Agilent SureSelect; ELID 0735431; supplemental Table 1, available on the Blood Web site) was performed successfully for 154 samples. Libraries were sequenced on Illumina HiSeq 2000 and variant calling was performed as we described previously.5 Briefly, somatic single-nucleotide variants and small indels were called using Shearwater (v.1.21.5), an algorithm designed to detect subclonal mutations in deep sequencing experiments.17 Two additional variant-calling algorithms were applied to complement this approach: CaVEMan (v.1.11.2) for single-nucleotide variants, and Pindel (v.2.2) for indels. Finally, allele counts at recurrent mutation hotspots were verified using an in-house script (github.com/cancerit/allelecount). Driver mutations were defined according to evidence for functional relevance in CH and hematological malignancy (supplemental Table 2). Methodological validation of our approach is outlined in supplemental Figure 1 and supplemental Tables 3 and 4.
Statistical analyses were performed in R (version 3.4.0). The Fisher exact test was used to assess twin concordance for CH. Null distributions of CH within the MZ and DZ groups were generated using random sample permutation (1000 iterations). The openMX R package was used for maximum likelihood modeling of genetic and environmental contributions to CH.18
Results and discussion
Mutational landscape in the cohort
Using deep sequencing (mean, 1650X) and sensitive variant calling, we identified CH (variant allele frequency [VAF] ≥0.5%) in 62% of individuals (95 of 154; Figure 1A), with larger clones (VAF ≥2%) present in 41 individuals (Figure 1B). Somatic driver mutations were identified in 16 of the 41 genes sequenced, with mutations in the epigenetic regulators DNMT3A and TET2 predominant (Figure 1C-E; supplemental Table 5). Almost one-third of individuals (48 of 154) harbored multiple distinct mutations (Figure 1F), often in the same gene and at different VAFs (supplemental Figure 2), suggesting the presence of multiple clones or subclones.

CH in individuals. (A) Red bars represent individuals in MZ twin pairs; blue bars represent those in DZ pairs. Dark red/blue (lower stacks) depict the proportions with CH; pale red/blue (upper stacks) depict those without CH. Absolute numbers of individuals in each proportion are shown within each bar. There were no DZ individuals above the age of 80 years. (B) Distribution of the maximum VAF per individual among those with CH. VAFs are divided into 0.5% bins. (C) In the main grid, each column represents 1 individual, and each row 1 gene. If a grid square is colored, a mutation was detected, and the specific color indicates the mutation type (see key). The plot on the right shows the proportion of the cohort harboring a mutation in each gene. (D-E) Somatic variants identified in DNMT3A (D) and TET2 (E). Conserved/functional protein domains are colored red, and intervening domains gray. Each circle connected to the protein cartoon represents a mutation. Missense mutations are represented above and truncating mutations below the protein, with the color of the circle indicating specific mutation type (as per the key in panel C). (F) Distribution of the total number of mutations per individual. fs, frameshift.
CH in individuals. (A) Red bars represent individuals in MZ twin pairs; blue bars represent those in DZ pairs. Dark red/blue (lower stacks) depict the proportions with CH; pale red/blue (upper stacks) depict those without CH. Absolute numbers of individuals in each proportion are shown within each bar. There were no DZ individuals above the age of 80 years. (B) Distribution of the maximum VAF per individual among those with CH. VAFs are divided into 0.5% bins. (C) In the main grid, each column represents 1 individual, and each row 1 gene. If a grid square is colored, a mutation was detected, and the specific color indicates the mutation type (see key). The plot on the right shows the proportion of the cohort harboring a mutation in each gene. (D-E) Somatic variants identified in DNMT3A (D) and TET2 (E). Conserved/functional protein domains are colored red, and intervening domains gray. Each circle connected to the protein cartoon represents a mutation. Missense mutations are represented above and truncating mutations below the protein, with the color of the circle indicating specific mutation type (as per the key in panel C). (F) Distribution of the total number of mutations per individual. fs, frameshift.
CH in twin pairs
Comparing age-matched samples from MZ and DZ groups (age range, 70-80 years; n = 33 MZ pairs and 24 DZ pairs), the overall prevalence of CH was very similar (59% and 54%, respectively; P = .70). We did not observe significantly higher concordance for CH within MZ twin pairs as compared with DZ pairs (P = .59; Figure 2A). Furthermore, using random permutation to model the null distribution, we found no difference in the observed distributions of CH among either MZ or DZ twins as compared with those expected by chance (P = 1 for MZ; P = .86 for DZ; Figure 2A). Excess twin concordance was also not observed when CH positivity was defined by (i) mutation in DNMT3A, (ii) mutation in TET2, and (iii) mutation in any gene with VAF >2% (supplemental Figure 3).
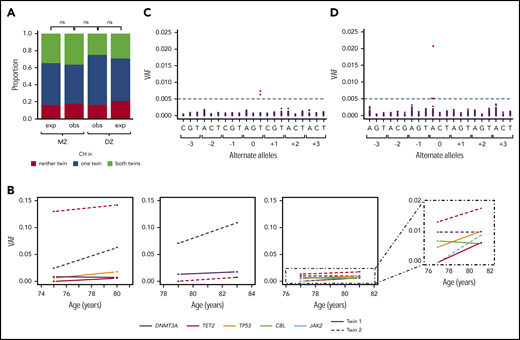
CH in twin pairs. (A) Concordance for CH status in age-matched MZ (n = 33) and DZ (n = 24) twin pairs. Observed (obs) proportions are those identified in the cohort, and expected (exp) are those generated by random sample permutation. There was no significant difference when comparing (i) the observed distributions between MZ and DZ twins (P = .59) and (ii) the observed vs expected distributions within the MZ (P = 1) or DZ (P = .86) twin groups (Fisher exact test). (B) Change in VAF over time in the 3 twin pairs in which both individuals had CH and serial samples were available. Each box surrounded by a solid line represents a twin pair. The fourth box surrounded by a dashed line is data from the third twin pair with the y-scale magnified to the lower VAFs. In each box, change in VAF over time is represented by a solid line for 1 twin, and a dotted line for the other, with line color indicating which gene was mutated. (C-D) The variants identified in 2 MZ twin pairs in which both twins harbored identical nonsense mutations: KDM6A (NM_021140:c.C2074T:p.Q692X) (C) and DNMT3A (NM_175629:c.C1792T:p.R598X) (D). Each “triplet” represents the 3 nonreference bases at each genomic position, centered on the identified variant position (denoted as 0). For each alternate allele, the VAF of each of the 154 individuals in the cohort is plotted. The horizontal dashed line represents the lower limit of sensitivity of variant calling, with calls below this VAF considered error. MZ twins with identical mutations are plotted in red, all other individuals in black. ns, not significant.
CH in twin pairs. (A) Concordance for CH status in age-matched MZ (n = 33) and DZ (n = 24) twin pairs. Observed (obs) proportions are those identified in the cohort, and expected (exp) are those generated by random sample permutation. There was no significant difference when comparing (i) the observed distributions between MZ and DZ twins (P = .59) and (ii) the observed vs expected distributions within the MZ (P = 1) or DZ (P = .86) twin groups (Fisher exact test). (B) Change in VAF over time in the 3 twin pairs in which both individuals had CH and serial samples were available. Each box surrounded by a solid line represents a twin pair. The fourth box surrounded by a dashed line is data from the third twin pair with the y-scale magnified to the lower VAFs. In each box, change in VAF over time is represented by a solid line for 1 twin, and a dotted line for the other, with line color indicating which gene was mutated. (C-D) The variants identified in 2 MZ twin pairs in which both twins harbored identical nonsense mutations: KDM6A (NM_021140:c.C2074T:p.Q692X) (C) and DNMT3A (NM_175629:c.C1792T:p.R598X) (D). Each “triplet” represents the 3 nonreference bases at each genomic position, centered on the identified variant position (denoted as 0). For each alternate allele, the VAF of each of the 154 individuals in the cohort is plotted. The horizontal dashed line represents the lower limit of sensitivity of variant calling, with calls below this VAF considered error. MZ twins with identical mutations are plotted in red, all other individuals in black. ns, not significant.
In 8 individuals (4 MZ twin pairs), serial blood samples were taken 4 to 5 years apart. CH clones were identified in both twins in 3 of 4 of these pairs, and intertwin variability in clonal size and trajectory was seen in all 3 (Figure 2B). This was the case even for clones harboring mutations in the same gene.
In summary, we find no evidence of high concordance for CH in elderly MZ twins. In addition, we observe disparity in clonal size and trajectory over time, even between MZ twins harboring mutations in the same driver genes. Although our cohort size is too small to precisely quantify genetic vs environmental contributions, maximum likelihood modeling provides support for a substantial influence of noninherited factors upon CH emergence and behavior (supplemental Table 6). Overwhelming evidence shows that mutation acquisition is widespread and inevitable in the aging hematopoietic system,6 as is the case in other healthy tissues studied to date,19,20 indicating that this is not the rate-limiting step in CH development. By suggesting that the inherited genome does not play a dominant role, our study frames nongenetic events as important factors in CH emergence. Altered interactions of the HSC with its environment, associated with processes such as aging, senescence, inflammation, and infection, are plausible operators, some of which are supported by evidence from experimental models.21
Despite the overall lack of concordance for CH, we did identify 2 MZ pairs in which both twins harbored identical nonsense mutations, namely KDM6A Q692X in 1 pair and DNMT3A R598X in the other (Figure 2C-D; supplemental Table 5). KDM6A (= UTX) is a histone H3 lysine 27 demethylase that acts as a tumor suppressor in a number of different cancers, including 2% to 3% of myeloid malignancies.22 There are no somatic mutation hotspots in KDM6A and the substitution identified here is not reported in either the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (cancer.sanger.ac.uk/cosmic) or in several large, albeit less sensitive, CH studies (total number of participants >30 000).1,2,4 Although mutations in DNMT3A are generally more prevalent, the particular mutation detected here is not common. In this light, the likelihood that each member of these 2 twin pairs acquired the same mutation independently and by chance is extremely small. A more plausible explanation is that the somatic mutation occurred just once during embryogenesis, either prior to twinning or in an HSC whose progeny reached both twins through shared circulation in utero. Although monozygotic twin sharing of somatic mutations has been demonstrated in other settings, including pediatric leukemia, this is the first description of possible acquisition of adult-type CH driver mutations in utero.23-25
In conclusion, the lack of strong concordance for CH and the variable clonal trajectories between MZ twins indicate that the inherited genome does not exert a profound influence on the emergence and behavior of CH in older adults. In addition, sharing of rare somatic mutations by MZ twins raises the possibility that mutations driving adult CH may sometimes be acquired in utero.
The sequencing data reported in this article have been deposited in the European Genome-phenome Archive (accession number EGAD00001005055).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the participants of the TwinsUK Registry. The authors also thank Kirsten Grønbæk, Kaare Christensen, Jacob Werner Hansen, and Hannes Ponstingl for helpful discussions.
This work was supported by Wellcome Trust (WT098051). G.S.V. was supported by a Cancer Research UK Senior Cancer Fellowship (C22324/A23015). Work in the laboratory of G.S.V. was also supported by the European Research Council, the Kay Kendall Leukaemia Fund, Bloodwise, the Leukemia & Lymphoma Society, and the Rising Tide Foundation for Clinical Cancer Research. M.A.F. was supported by a Wellcome Clinical Fellowship.
Authorship
Contribution: G.S.V., T.M., and P.D. conceived and designed the study; G.S.V. supervised the study; M.A.F., T.M., and M.Z. performed the bulk of the bioinformatic and statistical analyses with help from M.S.V.; N.P. performed sequencing validation experiments; P.M.W. performed heritability modeling; K.S., R.R., and C.J.S. guided the choice of twin samples and advised on analysis; and G.S.V., M.A.F., T.M., and M.Z. wrote the manuscript with input from all coauthors.
Conflict-of-interest disclosure: G.S.V. is a consultant for Kymab Ltd and OxStem Ltd, and receives a research grant from Celgene. The remaining authors declare no competing financial interests.
Correspondence: George S. Vassiliou, University of Cambridge and Wellcome Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SA, United Kingdom; e-mail: gsv20@sanger.ac.uk.
