

Key Points
A C181S second-site mutation suppresses myeloid transformation by a NrasG12D “knock-in” allele and perturbs hematopoiesis.
NrasG12D/G12D,C181S compound heterozygous mice develop hematologic malignancies that exhibit somatic S181C revertant mutations.

Abstract
Oncogenic RAS mutations pose substantial challenges for rational drug discovery. Sequence variations within the hypervariable region of Ras isoforms underlie differential posttranslational modification and subcellular trafficking, potentially resulting in selective vulnerabilities. Specifically, inhibiting the palmitoylation/depalmitoylation cycle is an appealing strategy for treating NRAS mutant cancers, particularly as normal tissues would retain K-Ras4b function for physiologic signaling. The role of endogenous N-RasG12D palmitoylation in signal transduction, hematopoietic differentiation, and myeloid transformation is unknown, and addressing these key questions will inform efforts to develop mechanism-based therapies. To evaluate the palmitoylation/depalmitoylation cycle as a candidate drug target in an in vivo disease-relevant model system, we introduced a C181S mutation into a conditional NrasG12D “knock-in” allele. The C181S second-site amino acid substitution abrogated myeloid transformation by NrasG12D, which was associated with mislocalization of the nonpalmitoylated N-Ras mutant protein, reduced Raf/MEK/ERK signaling, and alterations in hematopoietic stem and progenitor populations. Furthermore, hematologic malignancies arising in NrasG12D/G12D,C181S compound heterozygous mice invariably acquired revertant mutations that restored cysteine 181. Together, these studies validate the palmitoylation cycle as a promising therapeutic target in NRAS mutant cancers.
Introduction
Ras proteins promote cellular proliferation, survival, and differentiation in their active, GTP-bound conformation and terminate these signals by hydrolyzing GTP to GDP. Cancer-associated mutations encode Ras oncoproteins that accumulate in the active conformation due to impaired GTP hydrolysis, which increases proliferation and survival in many cell types.1 Somatic RAS mutations are observed in 5% to 40% of hematologic malignancies with the highest incidence in juvenile and chronic myelomonocytic leukemias1,2 and in pediatric patients with acute myeloid leukemia (AML).3 NRAS is altered ∼2.5 times more frequently than KRAS across all hematologic cancers.1 Although NRAS mutations are initiating driver events in juvenile myelomonocytic leukemias,2 these and other signaling mutations cooperate with antecedent alterations in genes that broadly regulate the epigenome, transcription factor fusions, and genes encoding for RNA splicing factors in AML pathogenesis.4 Nevertheless, RAS mutations are integral to the phenotype of uncontrolled proliferation and aberrant differentiation and are invariably lost during remission.4,5 Consistent with this idea, NRAS mutant AML cell lines and primary leukemias arising in Nras mutant mice are dependent on persistent oncogene expression ex vivo and in vivo.6 Furthermore, a recent clinical trial testing the IDH2 inhibitor AG-221 (enasidenib) in relapsed/refractory AMLs with IDH2 mutations found that cooccurring mutations in NRAS correlated with treatment failure,7 which supports cotargeting aberrant epigenetic programming and hyperactive signaling to enhance efficacy. Together, the existing data underscore the need to develop potent and selective inhibitors of N-Ras oncoproteins for use in combination with other anticancer drugs.
Blocking Ras posttranslational processing engendered intense interest in the 1990s, which resulted in the development of potent inhibitors of farnesyltransferase (FTase).8 FTase prenylates the C-terminal CAAX motif, which is essential for membrane association and biologic activity of normal and mutant Ras proteins. Unfortunately, FTase inhibitors were largely ineffective in cancer because K-Ras and N-Ras undergo lipid modification by geranylgeranyl transferase when FTase is inhibited.8 Farnesylated H-Ras, K-Ras4a, and N-Ras traffic to the Golgi where they are palmitoylated at one or more cysteine residues within the hypervariable region (HVR) to stabilize membrane association. Subsequent depalmitoylation by serine hydrolases (SHs) removes the palmitoyl moiety, which results in the orderly recycling of Ras proteins. Elegant studies recently demonstrated that most cellular N-Ras is in the cytoplasm in association with the chaperone protein VPS35 and that palmitoylated N-Ras predominantly resides at the plasma membrane.9,10 By contrast, farnesylated K-Ras4b traffics directly to the plasma membrane where its association is stabilized by electrostatic interactions through a polylysine domain within the HVR. Together, the differential processing of Ras isoforms raises the possibility that these posttranslational modifications might comprise novel and selective therapeutic vulnerabilities in RAS mutant cancers.
Cuiffo and Ren mutated the N-Ras HVR cysteine that undergoes palmitoylation (C181) in an NRASG12D oncogene and used retroviral transduction to express the double-mutant N-RasG12D,C181S protein in NIH3T3 cells.11 Despite loss of plasma membrane association and inhibition of anchorage-independent colony growth, N-RasG12D,C181S retained some oncogenic properties, which included the capacity to induce morphologic transformation and overcome density-dependent growth inhibition. These authors also transplanted lethally irradiated mice with bone marrow (BM)-derived hematopoietic stem and progenitor cells (HSPCs) transduced with the vector encoding NRASG12D,C181S. Whereas HSPCs expressing NRASG12D induced fatal chronic myelomonocytic leukemia– or AML-like myeloid disease, recipients of NRASG12D,C181S cells remained well for over 2 years. However, the mechanism underlying this phenotype was not explored. Our laboratory used a similar system to analyze the cellular and biochemical consequences of N-RasG12D,C181S expression in HSPCs ex vivo.12 Although cells expressing N-RasG12D were hyperresponsive to cytokine stimulation and exhibited cytokine-independent growth, the C181S second-site mutation reduced cytokine responses to levels below those observed with wild-type (WT) N-Ras or N-RasC181S, likely due to nonphysiologic interactions of downstream effector molecules with the retrovirally overexpressed GTP-bound N-RasG12D,C181S protein.
Although providing evidence that palmitoylation modulates the oncogenic activity of N-Ras mutant proteins, the existing data preclude drawing firm conclusions about the essentiality of palmitoylation for oncogenesis. First, N-RasG12D,C181S retained some oncogenic properties in NIH3T3 cells. Second, the activation of downstream signaling pathways and in vitro growth phenotypes of primary HPSCs transduced with NrasG12D vectors are strongly modulated by the levels of oncoprotein expression.13 Finally, overexpression systems do not accurately model the stoichiometry observed in primary leukemias, where NRAS is rarely amplified.
To address the palmitoylation/depalmitoylation cycle as a potential therapeutic target in a disease-relevant context, we generated mice expressing NrasG12D,C181S from the endogenous genetic locus. Here we show that abrogating N-RasG12D palmitoylation suppresses myeloid transformation in vivo by normalizing the cytokine responses of primary hematopoietic cells, while also modulating hematopoiesis. Mice coexpressing N-RasG12D and N-RasG12D,C181S proteins developed diverse hematologic disorders characterized by somatic revertant mutations that reactivate the oncogenic program. Together, these data validate targeting the palmitoylation cycle for NRAS mutant hematologic cancers.
Methods
Mouse strains
NrasG12D and Mx1-Cre mice have been described previously.13-15 The generation of NrasG12D,C181S mice is described in the supplemental Methods, available on the Blood Web site. Genotyping of all mice was performed by polymerase chain reaction using the KAPA2G Fast HotStart ReadyMix PCR Kit (Kapa Biosystems) and the primers listed in supplemental Table 2. DNA for genotyping was obtained from tail samples collected at weaning, which were lysed with 50 mM NaOH and diluted in Tris-HCl (pH 8). Kaplan-Meier curves were constructed with events defined as death or mice becoming moribund and requiring euthanasia. Statistical difference was asserted with the log-rank test using GraphPad Prism 7 (GraphPad Software). All animal studies were approved by the Committee on Animal Research at the University of California, San Francisco (UCSF).
Allelic frequency analysis
Peak signal strength for primary and minor traces was determined with Thermo Fisher Scientific Sanger Variant Analysis Tool. To evaluate the allelic frequency of the flippase recognition target (FRT) insertion, we first defined the largest region with perfect overlap between peaks derived from the nontargeted allele and peaks from FRT sequences that remained after excising the selectable marker in the gene targeting cassette (“FRT scar”). Next, we identified the positions within the overlap at which the FRT-derived and the nontargeted allele-derived sequences differed. Finally, we obtained the frequency of the FRT-derived peak at each position and calculated the overall allelic frequency of the FRT scar per mouse as average value of all positions within the overlap region.
Phospho-flow cytometry
BM cells were depleted of erythrocytes, resuspended in starvation medium (Iscove Modified Dulbecco Medium, 1% bovine serum albumin, 50 μM β-mercaptoethanol, Pen-Strep, and GlutaMAX), and plated at 2 × 107 cells per milliliter. After 1 hour of starvation, cells were stimulated for 10 minutes in medium containing recombinant murine stem cell factor (SCF; PeproTech) at the concentrations indicated in Figure 3. Cells were fixed for 10 minutes in 2% paraformaldehyde and washed twice in 2% fetal bovine serum (FBS)/Hanks Balanced Salt Solution (fluorescence-activated cell sorter [FACS] buffer). Permeabilization was performed in 95% methanol, and cells were stored at −20°C. Permeabilized cells were incubated with Pacific Blue anti-Gr-1 (RB6-8C5, Biolegend), FITC anti-Mac-1 (M1/70, BD Biosciences), phycoerythrin-Cy7 Anti-c-Kit (2B8, Invitrogen), and either rabbit anti–phosphorylated extracellular signal-regulated kinase (pERK; D13.14.4E, Cell Signaling Technology) or rabbit anti-pS6 antibodies (catalog no. 2211; Cell Signaling Technology); purified CD16/32 (UCSF Monoclonal Antibody Core) was added to block nonspecific sites. After washing in FACS buffer, cells were stained with phycoerythrin anti-rabbit secondary antibody (Jackson Immunoresearch). Cells were washed and resuspended in FACS buffer and analyzed immediately.
Proliferation curves
BM cells were first separated into Mac-1+ and Mac-1− cell fractions using the CD11b MicroBeads system (MACS Miltenyi Biotec) according to the manufacturer’s recommendations. The Mac-1− fraction was further enriched in c-Kit+ cells with CD117 MicroBeads (MACS Miltenyi Biotec). c-Kit+ cells were counted and resuspended in serum-containing medium (Iscove Modified Dulbecco Medium supplemented with 10% FBS, 50 μM β-mercaptoethanol, Pen-Strep, and GlutaMAX) with or without recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech) at the concentrations indicated in Figure 3. Cells were seeded at a density of 105 cells per milliliter and counted at 72 hours using a Vi-CELL Automated Cell Viability Analyzer (Beckman Coulter).
Ras-GTP pulldowns
Mac-1+ BM cells, isolated as stated above, were resuspended in starvation medium at a concentration of 107 cells per milliliter. Cells were incubated at 37°C for at least 2 hours and allowing for no more than 5 hours to pass between the time of death of the mouse and the end of the starvation. The stimulation sample was obtained by adding FBS and GM-CSF at a concentration of 20% and 10 ng/mL, respectively, and incubating cells for 10 minutes at 37°C. The Active Ras Pulldown and Detection Kit (Thermo Fisher Scientific) was used to lyse cells and isolate Ras-GTP. Briefly, 300 to 500 μg of protein was incubated for 1 hour with Raf binding domain–loaded beads. After bead washing, Ras-GTP was eluted in 50 μL of 2× Laemmli buffer containing 5% β-marcaptoethanol. Of the eluted fractions, 15 μL was checked by western blotting together with 30 to 50 μg of total cell proteome as input control.
Cell fractionation
Cytoplasmic and membrane-bound protein fractions from 2 × 107 BM cells were isolated with Mem-PER Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific). The manufacturer’s protocol was optimized to achieve better separation of the fractions. Specifically, 750 μL of permeabilization buffer was used to release the cytoplasmic fraction, and 150 μL of solubilization buffer was used to resuspend membrane-bound proteins. A Bicinchonic Acid Assay (Thermo Fisher Scientific) was used to ensure that, for each fraction, equal quantities of protein were loaded across different genotypes.
Results
NrasG12D,C181S homozygous and heterozygous mice do not develop hematologic neoplasms
To assess if N-Ras palmitoylation is essential for the development of hematologic malignances initiated by endogenous NrasG12D expression, embryonic stem cells containing a targeted LoxP-STOP-LoxP (LSL) inhibitory element upstream of an NrasG12D mutation within the endogenous Nras locus14 were retargeted to introduce a second-site C181S mutation in cis (supplemental Figure 1A-B). We then performed intercrosses to establish cohorts of homozygous Mx1-Cre;NrasLSL-G12D/LSL-G12D and Mx1-Cre;NrasLSL-G12D,C181S/LSL-G12D,C181S mice (hereafter referred to as NrasG12D and NrasG12D,C181S, respectively). Pups were injected with polyinosinic:polycytidylic acid at weaning to induce Cre recombinase expression from the Mx1 promoter, which resulted in efficient excision of the LSL cassette (supplemental Figure 1C). Molecular analysis confirmed the presence of the G12D and C181S mutations in cis and revealed similar levels of Nras transcripts in the BM cells of NrasG12D and NrasG12D,C181S mice (Figure 1A-B). Western blotting showed robust Ras expression in WT, NrasG12D, and NrasG12D,C181S BM cells and also demonstrated the presence of the G12D substitution in both mutant strains (Figure 1C-D). In addition, labeling experiments using the palmitate analog 17-octadecynoic acid (17-ODYA) followed by copper-catalyzed click chemistry to biotin-azide and streptavidin enrichment16 revealed palmitoylated N-RasG12D, but not N-RasG12D,C181S, in BM cells (Figure 1E). Importantly, no difference was observed probing the enriched sample with an antibody recognizing all Ras isoform, as expected from specific genetic targeting of N-Ras (Figure 1F).
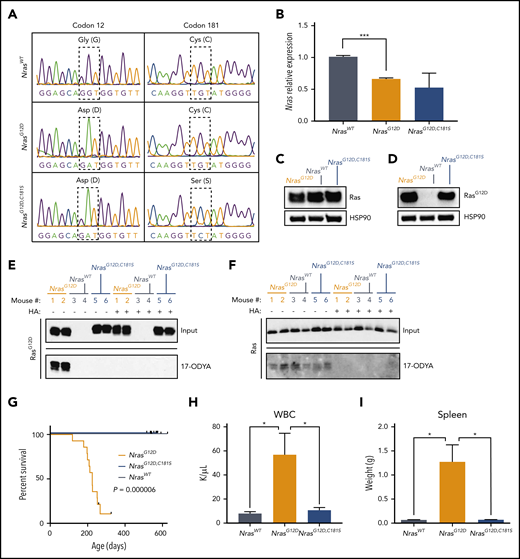
A C181S mutation prevents MPN in homozygous NrasG12Dmice. (A) BM complementary DNA sequencing indicating the positions of the G12D (left) and C181S (right) mutations (n = 3). (B) Nras transcript levels in BM cells (n = 3). (C-D) Representative western blot showing protein levels of total Ras (C) and RasG12D proteins (D) in BM cells (n = 5). Heat shock protein 90 is used as loading control. (E-F) Representative blots showing 17-ODYA labeling of palmitoylated proteins in 2 mice per genotype (out of 3 biological replicates). BM lysates (input) and 17-ODYA–labeled (palmitoylated) proteins were probed with antibodies that detect RasG12D (E) or all Ras isoforms (F). Hydroxylamine (HA) sensitivity confirms that 17-ODYA is added to Ras/RasG12D by S-palmitoylation. (G) Survival analysis of congenic NrasG12D (n = 14), NrasG12D,C181S (n = 12), and WT (n = 5) mice. (H-I) White blood cell (WBC) counts (H) and spleen sizes (I) in 6-month-old animals (n = 5). *P < .05; ***P < .001. Bar graphs show average ± standard error of the mean (SEM).
A C181S mutation prevents MPN in homozygous NrasG12Dmice. (A) BM complementary DNA sequencing indicating the positions of the G12D (left) and C181S (right) mutations (n = 3). (B) Nras transcript levels in BM cells (n = 3). (C-D) Representative western blot showing protein levels of total Ras (C) and RasG12D proteins (D) in BM cells (n = 5). Heat shock protein 90 is used as loading control. (E-F) Representative blots showing 17-ODYA labeling of palmitoylated proteins in 2 mice per genotype (out of 3 biological replicates). BM lysates (input) and 17-ODYA–labeled (palmitoylated) proteins were probed with antibodies that detect RasG12D (E) or all Ras isoforms (F). Hydroxylamine (HA) sensitivity confirms that 17-ODYA is added to Ras/RasG12D by S-palmitoylation. (G) Survival analysis of congenic NrasG12D (n = 14), NrasG12D,C181S (n = 12), and WT (n = 5) mice. (H-I) White blood cell (WBC) counts (H) and spleen sizes (I) in 6-month-old animals (n = 5). *P < .05; ***P < .001. Bar graphs show average ± standard error of the mean (SEM).
NrasG12D/WT mice on a C57BL/6 strain background exhibit myeloproliferation by 6 months of age characterized by elevated WBC counts, ineffective erythropoiesis, and splenomegaly. These mice have a median survival of 19.6 months and die of a spectrum of hematologic diseases, including myeloproliferative neoplasm (MPN), lymphoproliferative disease, histiocytic sarcoma, and myelodysplastic syndrome.15 By contrast, homozygous NrasG12D mutant mice uniformly die of an accelerated MPN and thereby comprise a robust in vivo system for testing the impact of second-site mutations.13,17 As expected, homozygous NrasG12D animals developed progressive MPN and had a median survival of 7.5 months (n = 14; Figure 1G). By contrast, 100% of WT and NrasG12D,C181S mice appeared healthy at 18 months of age. A comparative analysis of the hematopoietic compartments of 6-month-old mice revealed characteristic features of MPN in all NrasG12D mice, including markedly elevated blood leukocyte counts (mean: 58.61 k/μL; n = 6) and splenomegaly (mean: 1.72 g) (Figure 1H-I). These abnormalities were absent in NrasG12D,C181S mice, which had similar WBC counts (mean values: 10.73 k/μL and 7.94 k/μL, respectively) and spleen sizes (mean weight: 0.07 g for both groups) as WT mice. The proportion of circulating myeloid and lymphoid cells was also normal in NrasG12D,C181S mice (supplemental Figure 2). Together, these data indicate that the C181S second-site mutation abrogates the lethal MPN that results from endogenous NrasG12D expression.
We performed flow cytometry to characterize the cellular composition of the spleens and BMs of 6-month-old WT, NrasG12D, and NrasG12D,C181S mice. Consistent with a previous report,17 splenomegaly in NrasG12D mice was characterized by myeloid infiltration (mean frequencies: 25.08% Gr-1+ Mac-1+ cells and 14.71% Gr-1− Mac-1+ cells; n = 4) and increased numbers of common myeloid progenitors, granulocyte-macrophage progenitors, and megakaryocytic-erythroid progenitors (Figure 2A-B). By contrast, >90% of nucleated cells in the spleens of WT and NrasG12D,C181S mice were either B or T lymphocytes, and immature myeloerythroid progenitors were rare (Figure 2A-B). Compared with WT mice, NrasG12D,C181S animals had a significant reduction in the absolute numbers of BM Gr-1+ Mac-1+ and Gr-1− Mac-1+ cells (25.6% and 51.3% reduction, respectively) and a nonsignificant decrease in B220+ B-lineage lymphocyte counts (31% reduction; Figure 2C). Flow cytometric analysis also revealed fewer megakaryocytic-erythroid progenitors, a modest and nonsignificant reduction in common myeloid progenitors and granulocyte-macrophage progenitors, and lower counts of Lin− Sca1+ c-Kit+ (LKS) cell subpopulations in the BMs of NrasG12D,C181S mice vs WT mice (Figure 2D-E), with a modest (1.6-fold) decrease in immunophenotypically defined hematopoietic stem cells (HSCs; LKS CD48− CD150+ or LKS-SLAM) that was also not statistically significant. Cell-cycle analysis revealed an increase in the fraction of actively proliferating multipotent progenitors and HSCs in the BM of NrasG12D mice (Figure 2F-G). By contrast, the proportion of cycling multipotent progenitors and HSCs was similar in NrasG12D,C181S and WT mice. Importantly, these hematopoietic phenotypes persisted in 18- to 19-month-old NrasG12D,C181S mice, which remained well (supplemental Figure 3A-J). Thus, although abrogating N-RasG12D palmitoylation blocks myeloid transformation, homozygous NrasG12D,C181S expression has discrete effects on steady-state hematopoiesis that are not observed in Nras knock-out mice.6 These studies underscore the value of expressing mutant proteins from their endogenous genetic loci to assess functional impact in vivo.
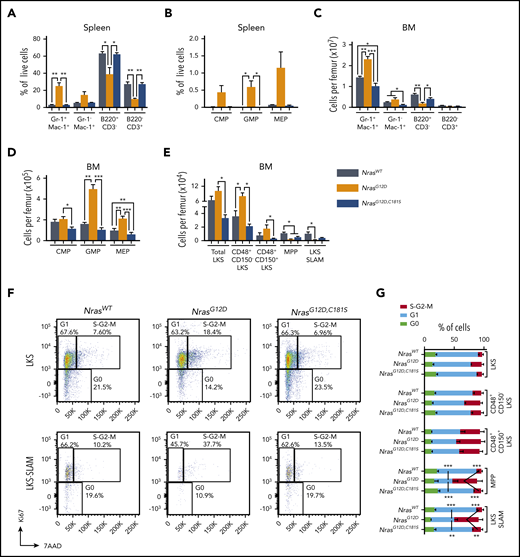
Hematopoietic populations in homozygous NrasG12D,C181Smice. (A-B) Immunophenotyping of splenocytes showing the frequencies of differentiated cells (A; n = 4) and myeloid progenitors (B; n = 5). (C-E) Immunophenotyping of BM subpopulations (n = 5) showing numbers of differentiated cells (C), myeloid progenitors (D), and immature HSPCs (E). The number of cells per femur was calculated by multiplying the frequency of each population by the total femur cell count. (F-G) Cell-cycle analysis in HSPCs (n = 3). The gating strategy for LKS and LKS-SLAM populations and the average values in LKS cells, LKS subsets, and multipotent progenitors (CD48− CD150− LKS cells), respectively. The LKS-SLAM population is composed of CD48− CD150+ LKS cells. *P < .05; **P < .1; ***P < .001. Data are average ± SEM. CMP, common myeloid progenitor; GMP, granulocyte-macrophage progenitor; MEP, megakaryocytic-erythroid progenitor.
Hematopoietic populations in homozygous NrasG12D,C181Smice. (A-B) Immunophenotyping of splenocytes showing the frequencies of differentiated cells (A; n = 4) and myeloid progenitors (B; n = 5). (C-E) Immunophenotyping of BM subpopulations (n = 5) showing numbers of differentiated cells (C), myeloid progenitors (D), and immature HSPCs (E). The number of cells per femur was calculated by multiplying the frequency of each population by the total femur cell count. (F-G) Cell-cycle analysis in HSPCs (n = 3). The gating strategy for LKS and LKS-SLAM populations and the average values in LKS cells, LKS subsets, and multipotent progenitors (CD48− CD150− LKS cells), respectively. The LKS-SLAM population is composed of CD48− CD150+ LKS cells. *P < .05; **P < .1; ***P < .001. Data are average ± SEM. CMP, common myeloid progenitor; GMP, granulocyte-macrophage progenitor; MEP, megakaryocytic-erythroid progenitor.
NRAS mutations are usually heterozygous in hematologic cancers. We therefore enumerated HSPC populations in adult mice carrying 1 NrasG12D,C181S and 1 WT Nras allele (mean age: 9 months; range 8 to 12) and compared them with age-matched control NrasG12D/WT and NrasWT/WT mice. As expected,15 NrasG12D/WT appeared well but had myeloproliferation characterized by splenomegaly (Figure 3A) and expansion of myeloid progenitors in the BM and spleen (Figure 3B-E). These phenotypes were not observed in NrasG12D,C181S/WT mice in which spleen sizes and the distribution of HSPCs and mature blood cells were normal in spleen and BM (Figure 3B-F). This normal pattern of hematopoiesis in NrasG12D,C181S/WT mice (Figure 3D-F), which contrasts with data from of NrasG12D,C181S mice (Figure 2), indicates that expression levels strongly modulate the effects of N-Ras mutant proteins.13 Furthermore, our observations indicate that loss of N-RasG12D palmitoylation impairs oncogenesis in the context of either heterozygous or homozygous NrasG12D,C181S expression.
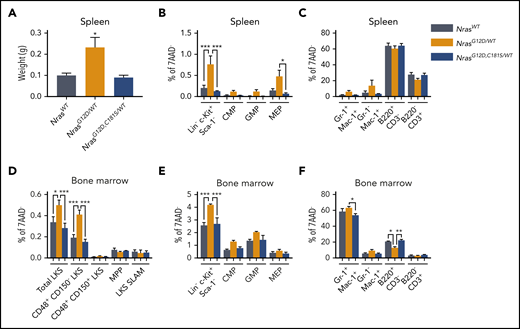
Hematopoietic populations in NrasG12D,C181S/WTmice. (A) Normal spleen size in NrasG12D,C181S/WT mice (n = 4). (B-C) Immunophenotypic analysis indicating low frequencies of myeloid progenitor cells (B) and differentiated myeloid cells (C) in the spleens of NrasG12D,C181S/WT mice (n = 4). (D-F) Flow-cytometric analysis of the BM of NrasG12D,C181S/WT mice (n = 4) showing normal frequencies of LKS cells (D), myeloid progenitors (E), and differentiated myeloid/lymphoid cells (F). Bar graphs show average values ± SEM. *P < .05; **P < .01; ***P < .001.
Hematopoietic populations in NrasG12D,C181S/WTmice. (A) Normal spleen size in NrasG12D,C181S/WT mice (n = 4). (B-C) Immunophenotypic analysis indicating low frequencies of myeloid progenitor cells (B) and differentiated myeloid cells (C) in the spleens of NrasG12D,C181S/WT mice (n = 4). (D-F) Flow-cytometric analysis of the BM of NrasG12D,C181S/WT mice (n = 4) showing normal frequencies of LKS cells (D), myeloid progenitors (E), and differentiated myeloid/lymphoid cells (F). Bar graphs show average values ± SEM. *P < .05; **P < .01; ***P < .001.
Mislocalization of N-RasG12D,C181S uncouples GTP loading from effector pathway activation
To understand how loss of N-RasG12D palmitoylation suppresses myeloid transformation and alters steady-state hematopoiesis, we analyzed the localization and activation of mutant N-Ras proteins in BM cells from homozygous mice. Using cellular fractionation and an antibody that recognizes the G12D amino acid substitution, we confirmed that endogenous N-Ras localizes to both cellular membranes and the cytoplasm9,12 and also found that the relative proportion of cytoplasmic vs membrane-associated N-Ras is markedly higher in NrasG12D,C181S vs NrasG12D cells (Figure 4A). This observation is consistent with previous data showing that palmitoylation increases the avidity of N-Ras for cellular membranes, and that its absence destabilizes this interaction. We next assessed Ras-GTP levels in myeloid (Mac-1+) BM cells from NrasG12D,C181S mice before and after cytokine stimulation (Figure 4B). Western blotting with an anti-RasG12D antibody revealed similarly elevated Ras-GTP levels in N-RasG12D and N-RasG12D,C181S myeloid cells, which increased further upon GM-CSF stimulation. Thus, the reduced amount of N-RasG12D,C181S within the membrane fraction does not alter cytokine-induced guanine nucleotide exchange.
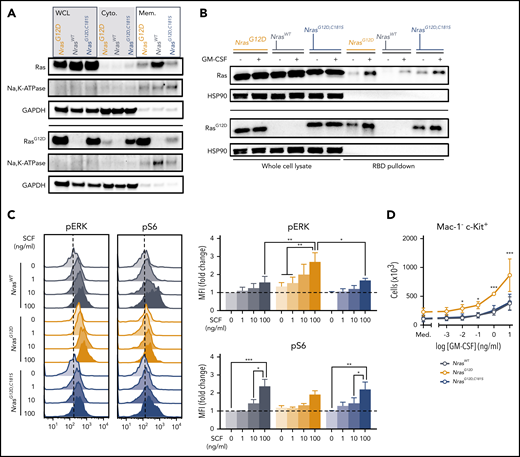
N-RasG12D,C181S mislocalization is associated with normal cytokine responses despite elevated Ras-GTP levels. (A) Levels of total Ras and RasG12D protein in BM whole-cell lysates (WCL), cytoplasmic fraction (Cyto.), and solubilized membrane fraction (Mem.) from a representative western blot (n = 3). Na, K-ATPase, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used as membrane and cytoplasm markers, respectively. (B) Raf binding domain (RBD) pulldown assays (right lanes) were performed to measure Ras-GTP levels in starved (−) or GM-CSF–stimulated (+) Mac-1+ BM cells (n = 3). Heat shock protein 90 was used as loading control. (C) Phospho-flow cytometric analysis of pERK and pS6 in Mac-1− Gr-1− c-Kit+ cells stimulated for 10 minutes with increasing doses of SCF showing representative plots (left) and median fluorescence intensity (MFI) of pERK (top right) and pS6 (bottom right) normalized to starved cells from WT mice. Bar graphs indicate mean ± SEM (n = 4). (D) Proliferation of Mac-1− c-Kit+ cells in response to increasing doses of GM-CSF. Mean ± SEM from 3 biologic replicates. Med., cytokine-free medium. *P < .05; **P < .01; ***P < .001.
N-RasG12D,C181S mislocalization is associated with normal cytokine responses despite elevated Ras-GTP levels. (A) Levels of total Ras and RasG12D protein in BM whole-cell lysates (WCL), cytoplasmic fraction (Cyto.), and solubilized membrane fraction (Mem.) from a representative western blot (n = 3). Na, K-ATPase, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used as membrane and cytoplasm markers, respectively. (B) Raf binding domain (RBD) pulldown assays (right lanes) were performed to measure Ras-GTP levels in starved (−) or GM-CSF–stimulated (+) Mac-1+ BM cells (n = 3). Heat shock protein 90 was used as loading control. (C) Phospho-flow cytometric analysis of pERK and pS6 in Mac-1− Gr-1− c-Kit+ cells stimulated for 10 minutes with increasing doses of SCF showing representative plots (left) and median fluorescence intensity (MFI) of pERK (top right) and pS6 (bottom right) normalized to starved cells from WT mice. Bar graphs indicate mean ± SEM (n = 4). (D) Proliferation of Mac-1− c-Kit+ cells in response to increasing doses of GM-CSF. Mean ± SEM from 3 biologic replicates. Med., cytokine-free medium. *P < .05; **P < .01; ***P < .001.
Having found that N-RasG12D,C181S accumulates in the GTP-bound conformation after serum withdrawal and in response to cytokine stimulation, we asked whether Ras activation is sufficient to activate downstream pathways. Unstimulated c-Kit+ cells isolated from the BMs of NrasG12D mice in serum-free medium have elevated basal levels of pERK, which increased markedly in response to submaximal concentrations of SCF (Figure 4C). By contrast, the activation profile of NrasG12D,C181S progenitors was similar to that of WT cells (Figure 4C). SCF-induced S6 phosphorylation is attenuated in Ras mutant hematopoietic cells, which is likely due to negative feedback.18 Accordingly, S6 phosphorylation was blunted in NrasG12D c-Kit+ BM cells upon SCF stimulation compared with WT progenitors, whereas NrasG12D,C181S and WT cells responded similarly (Figure 4C). We also exposed more primitive LKS cells of all 3 genotypes to a range of SCF doses and measured the phosphorylation of ERK, S6, and AKT. LKS cells were generally less responsive to cytokine stimulation with NrasG12D cells showing less activation than NrasG12D,C181S or WT cells (supplemental Figure 4A-C). Thus, although c-Kit+ and LKS BM cells exhibited distinct responses to SCF, NrasG12D,C181S cells of both populations were similar to WT cells and differed from NrasG12D cells.
We next isolated Mac-1− c-Kit+ progenitors and cultured them in medium containing a range of GM-CSF concentrations. Whereas NrasG12D progenitors proliferated in the absence of cytokines and were hyperresponsive to low doses of GM-CSF, cells from NrasG12D,C181S mice responded normally (Figure 4D). Overall, these results indicate that the GTP-loaded N-RasG12D,C181S at cellular membranes is insufficient to efficiently activate downstream signaling. Importantly, these observations in primary BM cells expressing mutant Nras from the endogenous genetic locus contrast with previous data from retroviral models in which NRASG12D,C181S overexpression suppressed Raf/MEK/ERK pathway activation and greatly attenuated GM-CSF–induced myeloid progenitor colony formation in methylcellulose medium.11,12
Extending these studies to heterozygous (NrasG12D,C181S/WT) mice confirmed that the mutant protein was mislocalized and accumulated in the cytoplasm (supplemental Figure 5A). This was associated with WT levels of phosphorylated ERK and S6 in unstimulated BM myeloid cells (supplemental Figure 5B). Consistent with our analysis of homozygous NrasG12D,C181S mice (Figure 4D), the growth of NrasG12D,C181S/WT Mac1− c-Kit+ BM cells was greatly attenuated compared with control NrasG12D/WT cells (supplemental Figure 5C). Interestingly, however, we also observed a modest, but significant, reduction in the proliferation of this population in NrasG12D,C181S/WT BM in comparison with WT controls (supplemental Figure 5C).
NrasG12D/G12D,C181S mice develop hematologic disease with revertant mutations that reinstate oncogenesis
We generated NrasG12D/G12D,C181S compound mutant mice to investigate how nonpalmitoylated N-RasG12D,C181S might affect myeloid transformation by NrasG12D allele in trans. Interestingly, these mice died prematurely with an overall median survival of 428 days (Figure 5A) that was intermediate between NrasG12D/G12D mice (225 days; Figure 1D) and heterozygous NrasG12D/WT mice on a C57Bl/6 strain background (588 days).15 Postmortem analyses of NrasG12D/G12D,C181S mice that required euthanasia for signs of systemic illness revealed splenomegaly in all 6 animals (Figure 5B; supplemental Table 1) as well as a heterogeneous spectrum of hematologic abnormalities that included anemia, polycythemia, thymic enlargement, and elevated numbers of myeloid and/or lymphoid cells (Figure 5C-F; supplemental Table 1; supplemental Figure 6). We characterized the hematologic disease in each animal to the extent the available materials permitted. This analysis included reviewing blood cell counts, blood smear morphology, histopathology of spleen and liver (and thymus of mouse 424), and performing flow cytometry. Individual diseased mice exhibited findings compatible with MPN (mice 417, 421), thymic T-lymphoblastic leukemia/lymphoma (mouse 424), and histiocytic sarcoma (mice 431, 468). We were not able to subclassify the tumor in mouse 464. The findings in this small number of animals are similar to the spectrum of fatal hematologic diseases previously seen in heterozygous NrasG12D/WT mice on a C57BL/6 strain background.15
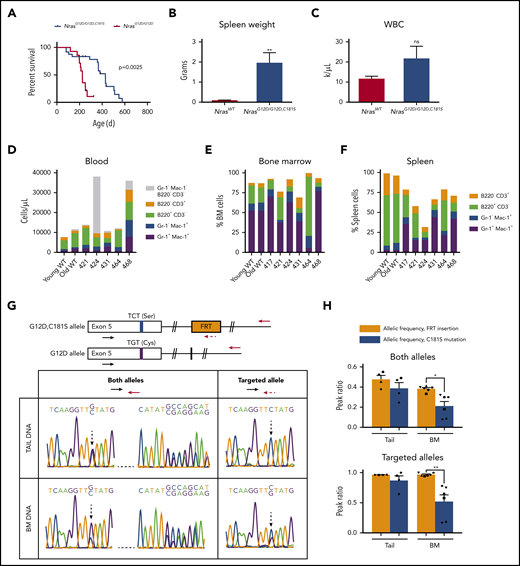
NrasG12D/G12D,C181Smice develop fatal hematologic disease characterized by S181C revertant mutations. (A) Survival of NrasG12D/G12D,C181S mice (n = 24) superimposed on that of the NrasG12D mice shown in Figure 1G. (B-C) Spleen weights (B; n = 6) and WBC counts (C; n = 5) in diseased NrasG12D/G12D,C181S mice compared with WT controls (see supplemental Figure 3). (D-F) Immunophenotypic characterization of red blood cell-lysed blood (D), BM (E), and spleen cells (F) by flow cytometry. (G) In the top panel, schematics indicating the 2 sets of primers used to amplify and sequence either both Nras alleles or the C181S-engineered allele. The bottom panel shows representative data from a diseased mouse with both reduced levels of the G>C (C181S) mutation in BM DNA and preservation of the FRT sequence. (H) Allelic frequency of the FRT sequence and the C181S mutation in the tail DNA and BM DNA amplified with primers indicated in panel G. Each symbol corresponds to an individual mouse. Bars are average ± SEM. *P < .05; **P < .1. ns, not significant.
NrasG12D/G12D,C181Smice develop fatal hematologic disease characterized by S181C revertant mutations. (A) Survival of NrasG12D/G12D,C181S mice (n = 24) superimposed on that of the NrasG12D mice shown in Figure 1G. (B-C) Spleen weights (B; n = 6) and WBC counts (C; n = 5) in diseased NrasG12D/G12D,C181S mice compared with WT controls (see supplemental Figure 3). (D-F) Immunophenotypic characterization of red blood cell-lysed blood (D), BM (E), and spleen cells (F) by flow cytometry. (G) In the top panel, schematics indicating the 2 sets of primers used to amplify and sequence either both Nras alleles or the C181S-engineered allele. The bottom panel shows representative data from a diseased mouse with both reduced levels of the G>C (C181S) mutation in BM DNA and preservation of the FRT sequence. (H) Allelic frequency of the FRT sequence and the C181S mutation in the tail DNA and BM DNA amplified with primers indicated in panel G. Each symbol corresponds to an individual mouse. Bars are average ± SEM. *P < .05; **P < .1. ns, not significant.
We transplanted BM cells from 2 diseased mice with high revertant mutation levels (417 and 424) into sublethally irradiated B6.SJL recipients (supplemental Figure 7A). Within 7 weeks, recipients of BM from mouse 417 developed splenomegaly, leukocytosis, anemia, and thrombocytopenia (supplemental Figure 7B-F). FACS analysis revealed ∼100% chimerism with expansion of immature or partially differentiated, Gr-1−/low Mac-1+ myeloid cells in the blood and BM of these mice (supplemental Figure 7G-K), indicating development of MPN. By contrast, mice transplanted with BM from mouse 424 remained well over 8 weeks of observation. We did not observe outgrowth of CD45.2+ BM cells from mouse 424, which supports either impaired homing/engraftment or poor competitiveness of this neoplasm in the stringent setting of transplantation into sublethal irradiated hosts.
The heterogeneous hematologic abnormalities in sick NrasG12D/G12D,C181S mice and the variable disease latency (supplemental Table 1) suggested that somatic genetic events contributed to leukemogenesis. As uniparental disomy with loss of the WT Nras allele is a common event in acute leukemias that develop in NrasG12D/WT mice infected with the MOL4070 retrovirus,13 we investigated diseased mice for the presence of the engineered C181S second-site mutation. Sequencing tail DNA confirmed the germline genotype of these mice and revealed the expected heterozygous WT:C181S allelic frequency (Figure 5G-H). However, the C181S substitution was present at significantly reduced frequency in the BM cells of all 6 diseased mice (range 8% to 32%). Importantly, we observed nearly heterozygous frequencies of the FRT sequence that was introduced to generate the C181S substitution of both tail DNA and BM DNA (supplemental Figure 1; supplemental Methods). We therefore used this ∼40-nucleotide sequence as a marker of the targeted allele and performed additional sequencing analysis using a primer that specifically anneals to the FRT site. This analysis identified DNA molecules with this FRT “scar” and a WT TGT nucleotide sequence at position 181 (Figure 5G-H). Together with the overall reduction of serine-encoding sequences, these data directly demonstrate the presence of revertant mutations in BM cells from diseased mice, which is indicative of strong selective pressure to reinstate oncogenic NrasG12D expression. Furthermore, the overall allelic frequency of the C181S mutation strongly correlated with that calculated from the FRT+ allele alone (supplemental Figure 8A), supporting euploidy at the Nras locus. In addition, the revertant mutation was present in BM complementary DNA from diseased mice at allelic frequencies that correlated with those obtained from sequencing genomic DNA (supplemental Figure 8B). Moreover, the frequency of the revertant mutation varied in different hematopoietic populations from the same mouse and between mice, which is fully consistent with the heterogeneous pattern of hematologic abnormalities in NrasG12D/G12D,C181S mice (supplemental Figure 8C-D). Finally, the frequency of the revertant mutation increased in secondary recipients of BM cells from mouse 417 (supplemental Figure 7L), thereby implicating the revertant mutation as key driver of both myeloid transformation and clonal outgrowth.
Discussion
This study demonstrates an essential role of palmitoylation for myeloid transformation by endogenous NrasG12D in vivo and provides genetic “proof of principle” for therapeutically targeting the palmitoylation/depalmitoylation cycle in NRAS mutant blood cancers. We show that abrogating palmitoylation impairs Raf/MEK/ERK pathway hyperactivation without reducing GTP loading of N-RasG12D,C181S. Given this underlying mechanism, we hypothesize that transformation would be similarly impaired from mutations other than G12D, as they would differently affect protein activity but not its processing or localization.19
The discrete effects of endogenous NrasG12D,C181S expression on steady-state hematopoiesis are consistent with either reduced oncogenicity or dominant negative biologic activity of the mutant protein. We previously observed potent dominant-negative effects on Raf/MEK/ERK activation and myeloid progenitor colony formation upon overexpressing NrasG12D,C181S in fetal liver cells that was likely due to effector sequestration by the mislocalized N-RasG12D,C181S protein.12 In this study, homozygous or heterozygous NrasG12D,C181S expression from the endogenous locus did not reduce basal or cytokine-induced Raf/MEK/ERK pathway activation in hematopoietic progenitors. However, the modestly impaired growth of NrasG12D,C181S/WT Mac1− c-Kit+ progenitors relative to WT cells is consistent with dominant-negative effects of the mutant protein. These observed differences between retroviral and endogenous NrasG12D,C181S expression indicate that hematopoietic cells are sensitive to N-RasG12D,C181S protein levels, as previously shown for N-RasG12D.13 Specifically, it is likely that key effector molecules are more readily available to and engaged by other Ras isoforms in hematopoietic cells expressing physiologic levels of N-RasG12D,C181S protein. Consistent with this idea, heterozygous and homozygous NrasG12D,C181S expression had differential effects on HSPC and myeloid cell numbers. The discrete, albeit modest, hematopoietic alterations in homozygous NrasG12D,C181S mice and the subtle ex vivo growth impairment of NrasG12D,C181S/WT progenitors suggest that abrogating N-RasG12D palmitoylation has negative fitness consequences. Accordingly, revertant mutations occurred in NrasG12D/G12D,C181S mice and conferred a proliferative advantage that resulted in clonal outgrowth and an increased frequency of the S181C mutation in secondary recipients of BM from a mouse with MPN. Similarly, germline mutations in FANC family genes and SAMD9/9L that result in constitutive or stress-induced HSPC deficits are associated with the acquisition of somatic revertant mutations that enhance HSPC fitness.20,21 The development of hematologic neoplasms with revertant mutations in NrasG12D/G12D,C181S mice further supports therapeutically targeting the palmitoylation machinery in NRAS mutant blood cancers.
Our observation that palmitoylation is essential for myeloid transformation by N-RasG12D is consistent with a recent CRISPR-based screen of RAS synthetic lethality in human AML cell lines that identified the noncatalytic palmitoyl transferase component GOLGA7 as a selective dependency in NRAS mutant AML.22 In addition, an analysis of the Cancer Dependency Map of the Broad Institute (https://depmap.org) supports the idea that GOLGA7 dependency extends to some nonhematological cancers with NRAS mutations (supplemental Figure 9). Of the ∼20 known mammalian palmitoyl acyl-transferases (PATs), only DHHC9 has been directly shown to catalyze N-Ras palmitoylation in conjunction with GOLGA7.23 N-Ras palmitoylation is partially reduced in hematopoietic cells from ZDHHC9−/−NrasG12D mice, and these animals develop MPN, albeit at an older age than NrasG12D mice.24 The absence of MPN in NrasG12D,C18S mice and the observation that GOLGA7, but not ZDHHC9, is essential for the proliferation of NRAS-mutant AML cell lines22 support the idea that one or more additional PATs can palmitoylate N-Ras in hematopoietic cells. Characterizing the full complement of N-Ras PATs in mammalian cells is a key question with clear therapeutic implications, particularly as we found that a 50% reduction in endogenous N-Ras palmitoylation markedly extended the survival of NrasG12D/G12D,C181S mice compared with NrasG12D animals, and also imposed selective pressure favoring the outgrowth of clones with revertant mutations.
One potential alternative to directly inhibit PATs involves targeting SHs that depalmitoylate N-Ras. Inhibiting N-Ras depalmitoylation results in protein mislocalization due to entropy-driven diffusion.25 “First-generation” SH inhibitors both reduced Raf/MEK/ERK signaling in cancer cell lines and impaired the growth of Nras mutant myeloid cells.12 N-Ras is a substrate for ABHD17 enzymes,26 and these and other potential depalmitoylating SHs are alternative therapeutic targets in NRAS mutant cancers, particularly as N-Ras is one of the few palmitoylated proteins that is dynamically regulated.16 Irrespective of whether either PATs or SHs prove to be better drug targets in NRAS mutant cancers, interfering with the palmitoylation/depalmitoylation cycle, may enhance the efficacy of existing inhibitors of Ras effectors through cooperative “synthetic lethality,” an approach recently explored in KRAS mutant cancer cells treated with Raf/MEK inhibitor combination therapies.27
Clinical experience with tyrosine kinase inhibitors indicates that advanced cancers almost invariably relapse, which is frequently due to acquired “on-target” resistance mutations that abrogate drug binding. We therefore expect that inhibiting N-Ras palmitoylation in NRAS mutant cancers will create substantial pressure for the acquisition of resistance mutations. Interestingly, however, as N-Ras palmitoylation requires C181, amino acid substitutions at this position are an unlikely mechanism of resistance to PAT inhibitors. This general idea also raises the intriguing idea of deploying the thiol group as a covalent handle for developing small molecules that directly block access to C181. The existence of a pocket for drug binding has allowed the development of such “irreversible inhibitors” of K-RasG12C, some of which can induce objective tumor shrinkage.28,29 Similarly, characterizing the crystal structure of the farnesylated N-Ras HVR may reveal opportunities for inhibiting oncogenic signaling through direct nucleophilic attack on C181.
All data are available in the main text or the supplemental materials.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Benjamin Braun for valuable insights on interrogating the Broad Institute DepMap for GOLGA7 dependency in cancer cells.
Research reported in this publication was supported by the National Institutes of Health, National Cancer Institute under award numbers R01CA193994 and R37CA72614, the Leukemia and Lymphoma Society LLS Fellowship 5465-18 (N.A.Z.), and the Damon Runyon Cancer Research Foundation Fellowship DRG-2149-13 (A.J.F).
Authorship
Contribution: N.A.Z., A.J.F., and K.S. conceptualized the study; N.A.Z., A.J.F., J.R.R., J.C.W., R.M.S., and B.F.C. formed the methodology; B.J.H. created the software; N.A.Z. and J.R.R. were responsible for validation; N.A.Z. and B.J.H. performed the formal analysis; N.A.Z., A.J.F., J.R.R., J.C.W., A.M.L., M.P., R.M.S., A.I., and S.C.K. performed the investigation; K.M.H., B.F.C., and K.S. were responsible for resources; N.A.Z. and K.S. wrote the original draft; N.A.Z., A.J.F., J.R.R., B.J.H., A.M.L., S.C.K., K.M.H., and K.S. reviewed and edited the paper; N.A.Z., A.J.F., and J.R.R. were responsible for visualization; K.S. supervised; and N.A.Z. and K.S. handled the project administration and funding.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict-of-interest disclosure: A.J.F. is currently employed at Calico LLC. N.A.Z. is currently employed at Amgen Inc. The remaining authors declare no competing financial interests.
Correspondence: Kevin Shannon, University of California, San Francisco, Helen Diller Family Cancer Research Building, 1450 3rd St, Room 263, San Francisco, CA 94158; e-mail: kevin.shannon@ucsf.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal