

Key Points
The STAR system was established to generate CAR T cell–compatible nanobodies and the respective tumor-associated antigens.
STAR-isolated CD13 nanobody and anti-TIM3 redirected the bispecific and split CAR T cells to eliminate AML in preclinical models.

Abstract
Chimeric antigen receptor (CAR) T cells have radically improved the treatment of B cell–derived malignancies by targeting CD19. The success has not yet expanded to treat acute myeloid leukemia (AML). We developed a Sequentially Tumor-Selected Antibody and Antigen Retrieval (STAR) system to rapidly isolate multiple nanobodies (Nbs) that preferentially bind AML cells and empower CAR T cells with anti-AML efficacy. STAR-isolated Nb157 specifically bound CD13, which is highly expressed in AML cells, and CD13 CAR T cells potently eliminated AML in vitro and in vivo. CAR T cells bispecific for CD13 and TIM3, which are upregulated in AML leukemia stem cells, eradicated patient-derived AML, with much reduced toxicity to human bone marrow stem cells and peripheral myeloid cells in mouse models, highlighting a promising approach for developing effective AML CAR T cell therapy.
Introduction
Cancer immunotherapy has made striking progress and changed the course of cancer therapy.1-6 Adoptive T-cell cancer therapy using chimeric antigen receptor (CAR)-expressing T cells can eradicate relapsed or refractory B-cell lymphoma or B-cell lymphocytic leukemia through targeting CD19.2,7 The CAR construct has an ectodomain, generally consisting of a single-chain variable fragment (scFv) derived from a monoclonal antibody (mAb), anchored to the cells via a transmembrane domain, followed by the intracellular costimulatory 4-1BB and/or CD28 domains, and CD3z signaling domain.8,9 Despite the remarkable achievement of CD19 CAR T cell therapy, this success has yet to be expanded to other types of cancers, such as acute myeloid leukemia (AML), which has dismal 5-year survival rate. One impediment to expanding the CAR T-cell application, among other factors, such as suppressive microenvironment,10,11 is often the lack of choices of mAbs.
The extracellular domain of a cell surface protein with a cancer-specific mutation or overexpression is ideal for targeting by CAR T-cell technology; however, the availability of mAbs suitable for developing CAR T-cell therapy against many potential targets is very limited. Moreover, many mAbs are not capable of endowing T cells with cytotoxicity, which requires the appropriate engagement between the T cell and target cell to elicit a productive immunological synapse and promote cancer cell death.12 Thus, it is imperative to generate diverse antibodies that meet these needs. Conventional antibodies cannot always bind certain antigen surfaces as a result of the large size of the antibody’s tetrameric heavy chains and light chains, coupled with the possible challenge in generating the optimal scFv.13 The camelid family of animals, which includes the llama, can produce heavy chain–only antibodies with a small (15-kDa) single variable domain (nanobodies [Nbs]) to bind various epitopes.14 Moreover, a single domain of Nbs is more effective to generate functional CAR T cells.15 The rapid identification of CAR T cell–compatible Nbs and their associated antigens would allow quick expansion of the available choices of CAR T cells by targeting previously unappreciated cell surface antigens/targets to develop potent cancer immunotherapy.
One disease in need of new therapeutic approaches is chemotherapy-resistant AML, which is highly aggressive and has a poor prognosis.16,17 CAR T cells targeting CD33, a cell surface lectin, and CD123, a subunit of the interleukin-3 receptor, were tested to suppress AML, but their application was hindered by their negative side effects on hematopoietic stem cells (HSCs) and other normal tissues.18,19 Here, we developed the Sequentially Tumor-Selected Antibody and Antigen Retrieval (STAR) system to isolate multiple Nbs that preferentially bind AML cells. In preclinical models, STAR-isolated anti-CD13, as well as an antibody against TIM3, which is upregulated in AML stem cells (LSCs), codirect CAR T cells to eradicate AML in patient-derived xenografts (PDXs), with much reduced toxicity to human HSCs.
Methods
Nb phage library construction from THP-1 cell–immunized llama
A llama was immunized with 2 × 107 THP-1 cells (Caprologics, Hardwick, MA) once a month for 3 months. Peripheral blood mononuclear cell isolation, RNA extraction, and complementary DNA (cDNA) synthesis were performed as previously described.20
Animals and in vivo models
NSG mice were conditioned with Busulfex (30 mg/kg) for 24 hours prior to tail injection with 2 × 107 patient-derived (PD) AML cells. Two weeks later, CAR or untransduced (UTD) T cells were administered into the mice. The recipient mice were euthanized at the experimental end point based on the protocol, and the long bones (femurs), spleens, and livers were collected for histological analysis by hematoxylin and eosin staining. Mice were euthanized according to protocol when moribund or upon the development of hind-limb paralysis. All experiments using mice were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Results
Generating Nbs that preferentially bind AML cells
To develop a strategy to isolate Nbs that can preferentially bind tumor cells in vitro, as well as enable the CAR T cells to induce tumor regression in vivo, we first isolated tumor-specific antibodies (Figure 1A) and then identified the matching antigens by the screening of cell surface protein cDNAs (STAR technology).

Generating Nbs that differentially bind tumor cells and empower CAR T cells to kill the tumor cells. (A) Flowchart of AML-specific CAR-compatible Nbs in vivo screening. A llama was immunized with the AML cell line THP-1. An Nb library was generated from the llama PBMCs by molecular cloning. Two rounds of conventional cell-based phage display were applied, which took the T-acute lymphoblastic leukemia cell line Jurkat and the chronic myelogenous leukemia cell line K562 as negative absorption. Thereafter, 1 round of counter-selection was applied to obtain the nanobodies with high affinity. The resultant THP-1–specific Nbs were inserted into a CAR-expressing lenti-vector to generate the Nb–sub-lib CAR (Nb-CAR) library. Human primary T cells were transduced by the Nb-CAR library and injected into NSG mice with THP-1 or K562 tumors to perform the in vivo selection. Nbs that can redirect T cells to enrich in the tumor were amplified using polymerase chain reaction and sequenced. (B) Ten million THP-1 cells or 5 million K562 cells were transplanted into NSG mice subcutaneously, followed by treatment with UTD T cells or Nb–sub-lib CAR T cells. Two weeks later, Nbs from tumor-infiltrated T cells were isolated and identified using polymerase chain reaction amplification (n = 3). (C) The 5 most frequent Nbs in the THP-1 tumor are shown. The Nb-expressing phage was directly used to test the binding to THP-1 cells, Jurkat cells, or K562 cells using a flow cytometry assay, in which the red line was flow with Nb-expressing phage, and the blue line was isotype control. bp, base pair.
Generating Nbs that differentially bind tumor cells and empower CAR T cells to kill the tumor cells. (A) Flowchart of AML-specific CAR-compatible Nbs in vivo screening. A llama was immunized with the AML cell line THP-1. An Nb library was generated from the llama PBMCs by molecular cloning. Two rounds of conventional cell-based phage display were applied, which took the T-acute lymphoblastic leukemia cell line Jurkat and the chronic myelogenous leukemia cell line K562 as negative absorption. Thereafter, 1 round of counter-selection was applied to obtain the nanobodies with high affinity. The resultant THP-1–specific Nbs were inserted into a CAR-expressing lenti-vector to generate the Nb–sub-lib CAR (Nb-CAR) library. Human primary T cells were transduced by the Nb-CAR library and injected into NSG mice with THP-1 or K562 tumors to perform the in vivo selection. Nbs that can redirect T cells to enrich in the tumor were amplified using polymerase chain reaction and sequenced. (B) Ten million THP-1 cells or 5 million K562 cells were transplanted into NSG mice subcutaneously, followed by treatment with UTD T cells or Nb–sub-lib CAR T cells. Two weeks later, Nbs from tumor-infiltrated T cells were isolated and identified using polymerase chain reaction amplification (n = 3). (C) The 5 most frequent Nbs in the THP-1 tumor are shown. The Nb-expressing phage was directly used to test the binding to THP-1 cells, Jurkat cells, or K562 cells using a flow cytometry assay, in which the red line was flow with Nb-expressing phage, and the blue line was isotype control. bp, base pair.
Using the STAR approach, we first immunized a llama with THP-1 cells, an aggressive leukemia cell line harboring the MLL-AF9 fusion protein, and then constructed an Nb-expressing phage-display library consisting of ∼109 independent members (Figure 1A).21 The library was used for panning with THP-1 cells in vitro, with negative absorption by Jurkat cells (a human acute T-cell leukemia) to exclude Nbs that recognized T cells and with negative absorption by K562 cells (a human chronic myelogenous leukemia cell line) to stringently select AML-specific Nbs. The resultant phage sublibrary (sub-lib) contained Nbs that preferentially bind the cell surface antigens of THP-1 cells with high affinity.
Identifying Nbs capable of endowing CAR T cells with anti–THP-1 tumor activity in vivo
Not all of the enriched sub-lib Nbs were capable of empowering CAR T lymphocytes with antitumor activities. Cytotoxic T-cell effector functions are triggered by antigen recognition by the T-cell receptor (TCR). Upon antigen binding, TCR activation can induce T-cell proliferation >10 000 fold, achieving clonal selection and expansion. The STAR system takes advantage of the T-cell activation system to amplify and enrich tumor-preferred CAR T cells in vivo. To identify CAR-compatible Nbs, we cloned Nbs from the phage sub-lib into a CAR construct to generate Nb-CAR T cells via lentiviral transduction, followed by CAR T-cell treatment in vivo (Figure 1B). Fourteen days after T-cell infusion, all tumor tissues were collected, and Nb sequences were decoded from the tumor-infiltrated Nb-CAR T cells. Polymerase chain reaction results indicated that Nb sequences were specifically retrieved from the Nb–sub-lib CAR T-cell–treated THP-1 tumor (Figure 1B, lane 2) but not from K562- or UTD-treated THP-1 tumor (Figure 1B). Among the 5 most enriched unique Nbs, Nb157, Nb163, Nb176, and Nb393 specifically bound THP-1 cells but not Jurkat or K562 cells (Figure 1C). Moreover, Nb157 and Nb163 also bound other AML cell lines, such as HL60, NB4, and U937 (supplemental Figure 1, available on the Blood Web site). Together, these results indicated that the STAR system is capable of enriching and isolating multiple Nbs that specifically bound AML cells.
STAR-isolated Nbs redirect CAR T cells to potently kill AML cells in vitro
To test whether STAR-isolated Nbs can guide CAR T cells to kill target tumor cells, these Nb CARs were transduced into activated primary human T cells (Figure 2A; supplemental Figure 2A-B). In the cytotoxicity assay, UTD T cells did not cause obvious cytotoxicity (Figure 2B-D); however, Nb157 CAR T cells potently and specifically killed THP-1 cells but not as many of the K562 or Jurkat cells (Figure 2B,D). Similarly, Nb163, Nb176, and Nb393 CAR T cells also specifically killed THP-1 cells (Figure 2C; supplemental Figure 3A-B). Nb157 CAR T cells also killed HL60 cells, another human AML cell line (Figure 2D).22
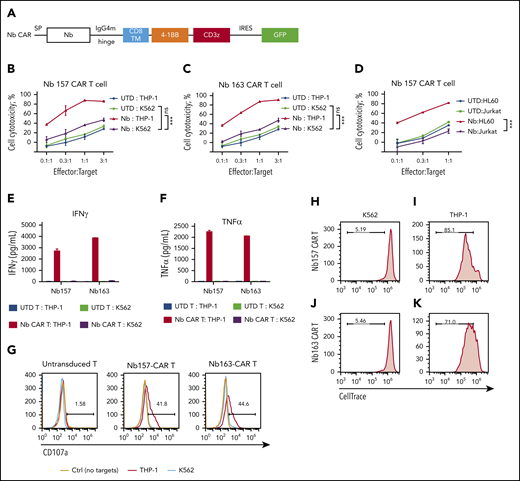
All Nbs isolated by the STAR system empower CAR T cells to potently kill AML cells in vitro. (A) Schematic diagram of Nb CAR structure, including signal peptide (SP), IgG4 mutant (IgG4m) hinge, CD8 transmembrane domain (TM), 4-1BB, and CD3z domain. (B-D) Nb CAR T cells showed potent and specific cytotoxicity against THP-1 or HL60 cells, but not K562 or Jurkat cells, in a dose-dependent manner. UTD T cells did not exert obvious killing (n = 3). THP-1 cells stimulated Nb157 or Nb163 CAR T cells, but not UTD T cells, to release cytokines, including IFN-γ (E) and TNF-α (F) (n = 3). (G) Only THP-1 cells induced Nb157 or Nb163 CAR T cells to degranulate (ie, CD107a localization to the cell membrane) after a 4-hour coculture (n = 3). CellTrace Far Red–labeled Nb157 (H-I) or Nb163 (J-K) CAR T cells were coincubated with heat-inactivated THP-1 cells (I-K) or K562 cells (H-J) for 4 days, followed by flow cytometry analysis. CAR T cells were gated on GFP+ signals (n = 3). ***P < .001, 1-way analysis of variance. ns, not significant (P > .05).
All Nbs isolated by the STAR system empower CAR T cells to potently kill AML cells in vitro. (A) Schematic diagram of Nb CAR structure, including signal peptide (SP), IgG4 mutant (IgG4m) hinge, CD8 transmembrane domain (TM), 4-1BB, and CD3z domain. (B-D) Nb CAR T cells showed potent and specific cytotoxicity against THP-1 or HL60 cells, but not K562 or Jurkat cells, in a dose-dependent manner. UTD T cells did not exert obvious killing (n = 3). THP-1 cells stimulated Nb157 or Nb163 CAR T cells, but not UTD T cells, to release cytokines, including IFN-γ (E) and TNF-α (F) (n = 3). (G) Only THP-1 cells induced Nb157 or Nb163 CAR T cells to degranulate (ie, CD107a localization to the cell membrane) after a 4-hour coculture (n = 3). CellTrace Far Red–labeled Nb157 (H-I) or Nb163 (J-K) CAR T cells were coincubated with heat-inactivated THP-1 cells (I-K) or K562 cells (H-J) for 4 days, followed by flow cytometry analysis. CAR T cells were gated on GFP+ signals (n = 3). ***P < .001, 1-way analysis of variance. ns, not significant (P > .05).
We also found that THP-1 cells, but not K562 cells, specifically stimulated Nb157 and Nb163 CAR T cells to release cytokines, including tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) (Figure 2E-F). A similar increase in cytokine release was detected with the coincubation of HL60 cells and Nb157 CAR T cells (supplemental Figure 3C). Moreover, THP-1 cells specifically induced CAR T-cell degranulation, as reflected by an increase in cell surface CD107a (Figure 2G), and specifically induced proliferation of Nb157 and Nb163 CAR T cells (Figure 2H-K). Together, these findings demonstrated that Nb157 and Nb163 CAR T cells were specifically activated by AML cells, leading to enhanced proliferation, cytokine release, and degranulation to kill the AML cells.
Nb CAR T cells potently induce AML tumor regression in vivo
To determine whether Nb CAR T cells suppressed AML tumors in vivo, THP-1 cells were subcutaneously transplanted into NSG mice, followed by treatment with Nb157 CAR T cells, Nb163 CAR T cells, or UTD T cells. The tumors from UTD T cell–treated mice grew exponentially (Figure 3A-B). Notably, the tumors in Nb157 or Nb163 CAR T cell–treated mice failed to grow substantially and eventually regressed (Figure 3A-B). Histological studies indicated that, although tumors in the UTD T-cell–treated group contained abundant tumor cells (Figure 3C; UTD), the Nb CAR T cells eradicated the cancer cells, leaving fibrotic tissues at the tumor site (Figure 3C). Nb157 CAR T cells also demonstrated significant antitumor efficacy against HL60 tumors in vivo (Figure 3D). Nb157 and Nb163 CAR T cells failed to induce regression of the nontarget K562 tumors (Figure 3E; supplemental Figure 3D-E), supporting the specificity of CAR T cells’ anti-AML efficacy. Furthermore, a low dose of Nb157 or Nb163 CAR T cells (1.5 million cells per mouse) led to complete remission of the THP-1 tumor, without recurrence, throughout a prolonged maintenance period of 52 days (Figure 3F), indicating the ability of Nb CAR T cells to eradicate the tumors in mice.
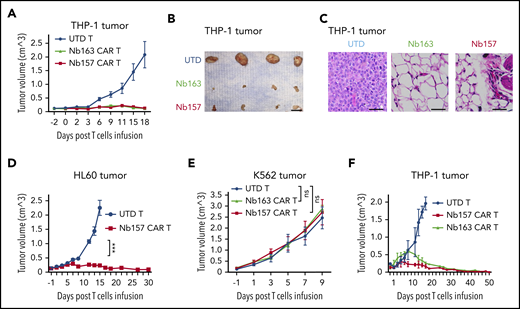
Nb-redirected CAR T cells potently eradicate AML tumor in vivo. (A-B) Ten million THP-1 cells were transplanted into NSG mice subcutaneously. The tumor reached 150 mm3 after ∼14 days. Three million Nb157 T cells, Nb163 CAR T cells, or UTD T cells were injected IV into the mice separately (n = 4). Tumor engraftment was monitored every other day. Scale bar, 10 mm. (C) Hematoxylin and eosin–stained THP-1 xenografts after treatment with UTD T cells, Nb163 CAR T cells, or Nb157 CAR T cells. Scale bars, 100 μm. (D) Three million Nb157 CAR T cells or UTD T cells were injected IV separately into NSG mice bearing HL60 tumors. Tumor engraftment was monitored every other day (n = 4). (E) Five million K562 cells were transplanted into NSG mice subcutaneously. The tumor reached 150 mm3 after ∼10 days. Three million Nb157 CAR T cells, Nb163 CAR T cells, or UTD T cells were injected into NSG mice separately. Tumor engraftment was monitored every other day (n = 4). (F) A total of 1.5 million Nb157 CAR T cells, Nb163 CAR T cells, or UTD T cells was injected IV separately into NSG mice bearing THP-1 tumor. Tumor engraftment was monitored every other day until the tumors were gone completely (n = 4). ***P < .001, 1-way analysis of variant. ns, not significant.
Nb-redirected CAR T cells potently eradicate AML tumor in vivo. (A-B) Ten million THP-1 cells were transplanted into NSG mice subcutaneously. The tumor reached 150 mm3 after ∼14 days. Three million Nb157 T cells, Nb163 CAR T cells, or UTD T cells were injected IV into the mice separately (n = 4). Tumor engraftment was monitored every other day. Scale bar, 10 mm. (C) Hematoxylin and eosin–stained THP-1 xenografts after treatment with UTD T cells, Nb163 CAR T cells, or Nb157 CAR T cells. Scale bars, 100 μm. (D) Three million Nb157 CAR T cells or UTD T cells were injected IV separately into NSG mice bearing HL60 tumors. Tumor engraftment was monitored every other day (n = 4). (E) Five million K562 cells were transplanted into NSG mice subcutaneously. The tumor reached 150 mm3 after ∼10 days. Three million Nb157 CAR T cells, Nb163 CAR T cells, or UTD T cells were injected into NSG mice separately. Tumor engraftment was monitored every other day (n = 4). (F) A total of 1.5 million Nb157 CAR T cells, Nb163 CAR T cells, or UTD T cells was injected IV separately into NSG mice bearing THP-1 tumor. Tumor engraftment was monitored every other day until the tumors were gone completely (n = 4). ***P < .001, 1-way analysis of variant. ns, not significant.
Moreover, other STAR-isolated Nbs (Nb173 and Nb393) also endowed CAR T cells with anti-AML activity in vivo (supplemental Figure 3F-G), which was confirmed by histological studies (supplemental Figure 3H). Collectively, these results demonstrate that all 4 of the STAR-isolated Nbs were capable of empowering cognate CAR T cells to potently kill AML cells, highlighting an effective approach to isolate CAR-compatible antibodies for developing novel CAR T-cell therapy.
Identification of CD13 as a CAR T-cell target to kill AML cells
To identify the antigens of the isolated Nbs, ∼3000 human membrane protein cDNAs were individually overexpressed in HEK293T cells and screened by flow cytometry (Figure 4A). Nb157 and Nb163 bound the cells transfected with human aminopeptidase N (CD13) (Figure 4B).23 CD13 cDNA expression was also confirmed by western blot (Figure 4C). CD13 is preferentially expressed in acute myeloid blast cells.24 To further confirm that Nb157 or Nb163 CAR T cells killed AML cells by targeting CD13, 3 CD13-knockout THP-1 cell lines were constructed using a guide RNA (gRNA)/Cas9 system, followed by verification via western blot (Figure 4D). Consistently, flow cytometry analysis showed that CD13 knockout abrogated the binding of Nb157 and Nb163 (Figure 4E) and abolished the killing of target cells by Nb157 CAR T cells (Figure 4F). CD13 knockout also diminished the killing of the targets by Nb163 CAR T cells significantly, but not completely, suggesting that possible off-CD13 antigen is recognized by Nb163 (Figure 4F). Together, these results demonstrate that Nb157 CAR T cells potently kill AML cells by specifically targeting CD13.
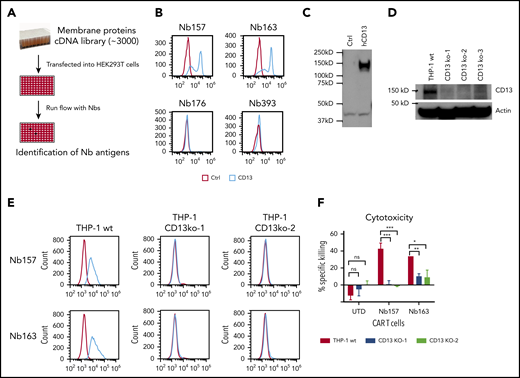
Identification of CD13 as a target to kill AML cells by CAR T cells. (A) Experimental schema. About 3000 cell membrane protein cDNAs were purified and transfected into HEK293T cells separately, followed by flow analysis with Nbs expressing phage and FITC-labeled secondary antibody against phage M13 protein. (B) Flow analysis of Nbs binding to HEK293T cells with CD13 overexpression. (C) Confirmation of CD13 cDNA expression in HEK293T cells by western blot. (D) Western blot was performed to confirm the gRNA/CRISPR-guided CD13-knockout effect in THP-1 cells. Three independent gRNAs were transduced into THP-1 separately, followed by puromycin selection and single individual clone expansion. (E) Flow analysis of Nb157- or Nb163-binding CD13-knockout THP-1 cells. (F) Cytotoxicity assay of CAR/UTD T cells to wild-type (wt) THP-1 or 2 CD13-knockout THP-1 cell lines (CD13 KO) (n = 4). *P < .05, **P <.01, d***P < .001, Student t test. ns. not significant.
Identification of CD13 as a target to kill AML cells by CAR T cells. (A) Experimental schema. About 3000 cell membrane protein cDNAs were purified and transfected into HEK293T cells separately, followed by flow analysis with Nbs expressing phage and FITC-labeled secondary antibody against phage M13 protein. (B) Flow analysis of Nbs binding to HEK293T cells with CD13 overexpression. (C) Confirmation of CD13 cDNA expression in HEK293T cells by western blot. (D) Western blot was performed to confirm the gRNA/CRISPR-guided CD13-knockout effect in THP-1 cells. Three independent gRNAs were transduced into THP-1 separately, followed by puromycin selection and single individual clone expansion. (E) Flow analysis of Nb157- or Nb163-binding CD13-knockout THP-1 cells. (F) Cytotoxicity assay of CAR/UTD T cells to wild-type (wt) THP-1 or 2 CD13-knockout THP-1 cell lines (CD13 KO) (n = 4). *P < .05, **P <.01, d***P < .001, Student t test. ns. not significant.
Nb157 CAR T cells eradicate PD AML cells in NSG mouse models
To determine whether Nb CAR T cells can also kill PD AML cells, we found that Nb157 and Nb163 can bind PD AML cells by recognizing CD13 (Figure 5A-B). PD AML cells were also potently killed by Nb157 and Nb163 CAR T cells in the in vitro cytotoxicity assays (Figure 5C-D). Thereafter, we sought to examine the therapeutic effect of the CAR T cells against PD AML in vivo. To this end, NSG mice were transplanted with PD AML cells, followed by treatment with UTD or Nb157 CAR T cells 2 weeks later. The Kaplan-Maier curve showed that all control mice had died by 45 days after PD AML cell infusion; however, Nb157 CAR T-cell infusion significantly prolonged the life of the treated mice to >90 days, without obvious toxicity or weight loss (Figure 5E; supplemental Figure 4A).

Nb157 CAR T cells display antitumor activity in patient-derived AML cells in an NSG mouse model. Nb157 (A) and Nb163 (B) recognized PD AML cells by flow analysis. Nb157 (C) and Nb163 (D) CAR T cells specifically killed PD AML cells in vitro in a dose-dependent manner (n = 4). (E) Nb157 CAR T cells efficiently prolonged survival of NSG mice bearing PD AML. In brief, 20 million PD AML cells were injected into NSG mice, followed by treatment with 3 million Nb157 CAR T cells or UTD T cells, and survival of mice was monitored (n = 10, each group). (F-I) PD AML in NSG bone marrow and spleen was monitored after Nb157 CAR T cell treatment by staining with anti-human CD45/CD3/CD33, followed by flow cytometry analysis (n = 3). (J-K) At the end points of each group of experiments, mice spleens were harvested and fixed with paraformaldehyde, followed by immunofluorescence staining of anti-human CD3(red)/CD33(green) and DAPI (blue; nuclear).
Nb157 CAR T cells display antitumor activity in patient-derived AML cells in an NSG mouse model. Nb157 (A) and Nb163 (B) recognized PD AML cells by flow analysis. Nb157 (C) and Nb163 (D) CAR T cells specifically killed PD AML cells in vitro in a dose-dependent manner (n = 4). (E) Nb157 CAR T cells efficiently prolonged survival of NSG mice bearing PD AML. In brief, 20 million PD AML cells were injected into NSG mice, followed by treatment with 3 million Nb157 CAR T cells or UTD T cells, and survival of mice was monitored (n = 10, each group). (F-I) PD AML in NSG bone marrow and spleen was monitored after Nb157 CAR T cell treatment by staining with anti-human CD45/CD3/CD33, followed by flow cytometry analysis (n = 3). (J-K) At the end points of each group of experiments, mice spleens were harvested and fixed with paraformaldehyde, followed by immunofluorescence staining of anti-human CD3(red)/CD33(green) and DAPI (blue; nuclear).
To investigate the dynamics of CAR T-cell treatment in vivo, we monitored the change in AML and UTD/CAR T cells in the bone marrow, spleen, and peripheral blood. In the first week after T-cell treatment, few CD33+ AML cells, but a large percentage of CD3+ T cells (>90% of human CD45+), were detectable in the bone marrow in both groups (Figure 5F-G). Notably, in the second week after T-cell treatment, compared with UTD T cells, Nb157 CAR T cells markedly reduced the accumulation of CD33+ AML cells in bone marrow (Figure 5H-I). Consistently, CD33+ AML cells decreased substantially following Nb157 CAR T-cell treatment (supplemental Figure 4B), and more T cells were detected in the peripheral blood (supplemental Figure 4C). However, CAR T cells in peripheral blood decreased by the third week after CAR T-cell infusion (supplemental Figure 4C), likely reflecting tumor regression and a decrease in the antitumor response.
Furthermore, immunofluorescent staining of the mouse spleens revealed a large number of CD33+ AML cells in the UTD group (Figure 5J). However, all CD33+ AML cells were eradicated, with a large number of CD3+ T cells in the Nb157 CAR T-cell group (Figure 5K). Consistently, flow cytometry also showed enrichments for CAR T cells in the bone marrow and spleen were from 30% (day 0) to 70% (day 14 post T cell infusion) (supplemental Figures 2B and 4D). CD8+ T-cell percentage was also higher in the Nb157 CAR T-cell group (59%) than in the UTD T-cell group (33%) (supplemental Figure 4E). Meanwhile, Nb157 CAR can induce persistent memory T-cell phenotypes, including central memory and effector memory populations (supplemental Figure 4F), which were correlated with complete remissions in CAR T-cell clinical therapy.25,26 Together, these results indicate that Nb157 CAR T cells effectively eliminate PD AML cells in the bone marrow and spleen of recipient mice and significantly prolong their survival.
Combinatory bispecific and split CAR T cells targeting CD13 and TIM3 redirect T cells to eradicate AML xenografts and AML PDXs in vivo
Because CAR T-cell therapy may cause on-target/off-tumor side effects,27,28 it is ideal to reduce the toxicity by increasing the specificity with multiple tumor markers. In this regard, novel bispecific CAR T cells were developed to synergistically kill the experimental tumor models by targeting >1 tumor-associated antigen (TAA).29,30
One other potential TAA, TIM3, an immune-suppressing receptor, is highly expressed in the majority of human AML LSCs31,32 but not in HSCs. A combinatory bispecific and split CAR (BissCAR) T-cell system was developed to effectively kill CD13+TIM3+ LSCs, while maintaining a reduced impact on normal cells that only express CD13 (Figure 6A). TIM3 expression was extremely low in normal donor bone marrow but high in the LSC subset (CD34+CD38−CD90−) (Figure 6B, upper panels). In contrast, a high percentage of TIM3 and CD13 double-positive cells was detected in the LSC-enriched population (CD34+CD38−) from PD AML cells but not normal donor bone marrow (Figure 6B), indicating the high coexpression of CD13 and TIM3 in LSCs, which was consistent with previous reports.31,32

In vivo, combinatorial bispecific and split CD13 and TIM3 CAR T cells eradicate tumor expressing CD13 and TIM3, but not tumor expressing only CD13. (A) Schematic diagram of combinatorial bispecific and split CD13 and TIM3. Nb157 linked with CD3z recognized CD13 on normal HSCs or LSCs. Anti-TIM3 linked with CD28 and 4-1BB recognized TIM3 only on LSCs. Such Biss CAR T cells can be fully activated only by LSCs but not by HSCs. (B) Flow cytometry showing the expression of TIM3, CD90, CD13 on normal donor bone marrow cells (ND-BM) or PD AML cells, which were gated from CD45+Lin−CD34+CD38− subsets. Ten million NB4 (C) or NB4-TIM3 (D) cells were transplanted subcutaneously into NSG mice to form 100-mm3 tumors. Three million combinatorial BissCAR T cells, conventional Nb157 CAR T cells, or UTD T cells were injected IV into each NSG mouse with the tumors. The engraftment volume was monitored by measuring the length and width of the tumor every other day (n = 4). (E) Three weeks after mice with NB4 or NB4-TIM3 tumors were treated with BissCAR T cells or UTD T cells, human T-cell (CD3+) numbers in mouse peripheral blood were analyzed by flow cytometry and quantified using CountBright counting beads (n = 3). *P < .05, Student t test.
In vivo, combinatorial bispecific and split CD13 and TIM3 CAR T cells eradicate tumor expressing CD13 and TIM3, but not tumor expressing only CD13. (A) Schematic diagram of combinatorial bispecific and split CD13 and TIM3. Nb157 linked with CD3z recognized CD13 on normal HSCs or LSCs. Anti-TIM3 linked with CD28 and 4-1BB recognized TIM3 only on LSCs. Such Biss CAR T cells can be fully activated only by LSCs but not by HSCs. (B) Flow cytometry showing the expression of TIM3, CD90, CD13 on normal donor bone marrow cells (ND-BM) or PD AML cells, which were gated from CD45+Lin−CD34+CD38− subsets. Ten million NB4 (C) or NB4-TIM3 (D) cells were transplanted subcutaneously into NSG mice to form 100-mm3 tumors. Three million combinatorial BissCAR T cells, conventional Nb157 CAR T cells, or UTD T cells were injected IV into each NSG mouse with the tumors. The engraftment volume was monitored by measuring the length and width of the tumor every other day (n = 4). (E) Three weeks after mice with NB4 or NB4-TIM3 tumors were treated with BissCAR T cells or UTD T cells, human T-cell (CD3+) numbers in mouse peripheral blood were analyzed by flow cytometry and quantified using CountBright counting beads (n = 3). *P < .05, Student t test.
NB4(CD13+TIM3−) and NB4-TIM3(CD13+TIM3+) cell lines were generated to mimic the HSC and LSC models (supplemental Figure 5A-B). Next, a conventional TIM3-BBz CAR was generated (supplemental Figure 5C), which guided the T cells to kill NB4-TIM3 cells potently and specifically in vitro and suppressed NB4-TIM3 tumor growth in vivo (supplemental Figure 5D-E).
We then constructed the BissCAR, in which Nb157 recognizing CD13 was linked to CD3z and anti-TIM3 scFv recognizing TIM3 was linked to CD28 and 4-1BB costimulatory domains (Figure 6A; supplemental Figure 5F). The resulting BissCAR expression on the T cells was verified by flow cytometry (supplemental Figure 5G). An in vitro killing assay showed that BissCAR T cells killed NB4 and NB4-TIM3 cells, because the CD13 recognition elicited CD3z signaling to induce target death in vitro (supplemental Figure 5H).29 Moreover, compared with NB4 cells, NB4-TIM3 cells increased the secretion of IFN-γ and TNF-α from BissCAR T cells (supplemental Figure 5I-J). The enhanced cytokine secretion was also dependent on NB157-CD3z signaling, because K562-TIM3(CD13−TIM3+) cells failed to induce the BissCAR T cells to release the cytokines (supplemental Figure 5I-J).
In the NB4 xenograft models, BissCAR T cells only moderately suppressed tumor growth compared with complete elimination when using Nb157 CAR T-cell treatment (Figure 6C). However, BissCAR T cells could eradicate the NB4-TIM3 tumor as potently as Nb157 CAR T cells (Figure 6D). These results indicate that BissCAR T cells are capable of completely shrinking the tumor expressing CD13 and TIM3, but they spared the cells expressing only CD13. Consistently, BissCAR T-cell number in peripheral blood in NB4-TIM3 tumor-bearing mice was significantly higher than in NB4 tumor-bearing mice (Figure 6E).
We further explored whether BissCAR T cells can suppress PD AML cells. To this end, PD AML cells were transplanted into NSG mice to induce leukemia, followed by treatment with BissCAR or UTD T cells 2 weeks later (Figure 7A). The appearance of CD33+ AML cells or CD3+ T cells in peripheral blood was monitored weekly (Figure 7B-C). The results indicated that, following the first week of injection, peripheral blood AML cells gradually decreased in the BissCAR T-cell group (Figure 7B), consistent with heavy leukemic infiltration in the spleen in the later stage (Figure 5H; supplemental Figure 4B). Notably, treatment with BissCAR T cells, but not with UTD T cells, increased peripheral T-cell number 1 week after the T-cell injection, reflecting the quick activation and proliferation of CAR T cells to kill AML cells (Figure 7C). Consistently, BissCAR T-cell treatment significantly prolonged survival of the mice compared with the UTD T-cell group (Figure 7D). It has been reported that various immune-suppressing factors weaken the immunotherapy for AML, such as the PD-1, TIM3 immune checkpoint molecules, and regulatory T cells (Tregs).33-35 Compared with UTD T cells, BissCAR T cells have lower PD-1 and TIM3 expression in the bone marrow (supplemental Figure 6A-F). BissCAR T cells and UTD T cells have similar low PD-1 and TIM3 expression in the mouse spleen; however, the PD-1/TIM3 levels were not correlated with resistance to CAR T cells, because CAR T cells eradicated AML in our xenograft and PDX models. Low percentages of Tregs were consistently detected in the bone marrow and spleen of mice injected with BissCAR T cells or UTD T cells (supplemental Figure 6G-K). We did not observe any T-cell suppression from Tregs, because of the robust elimination of the leukemia. Therefore, the results demonstrate that BissCAR T cells can effectively eradicate the double-positive PD AML cells in this clinically relevant model.
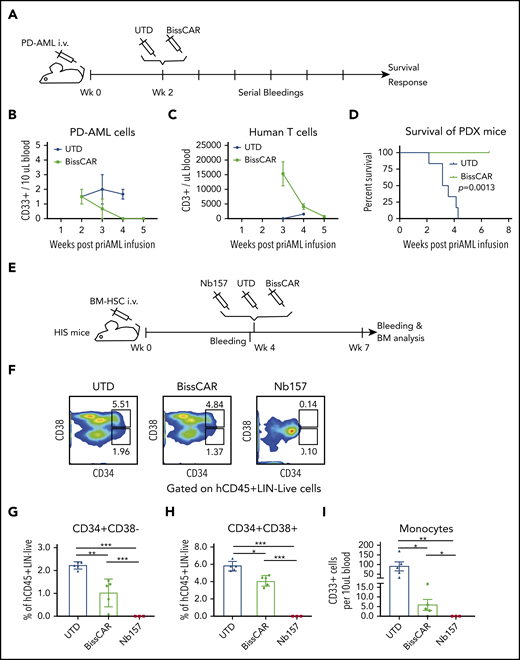
BissCAR T cells targeting CD13 and TIM3 eradicate AML PDXs, but with reduced toxicity to human HSCs in vivo. (A) Schematic diagram of AML PDX mice treated with control or BissCAR T cells. Twenty million PD AML cells were injected into NSG mice, followed by injection of 5 million BissCAR T cells or UTD T cells 2 weeks later. Next, human peripheral blood CD3+ cells were analyzed by serial bleeding weekly. (B-C) PD AML cells or T cells in mouse peripheral blood were monitored weekly by flow staining with anti-human CD33 or anti-human CD3 antibodies. Blood volume was normalized and quantified using CountBright counting beads (n = 3). (D) Mice survival was monitored and recorded (n = 6 per group). (E) Schematic diagram of HIS mice for evaluation of human HSC toxicity. A total of 1.5 million normal donor bone marrow (BM) CD34+ cells was injected into each NSG mouse. Four weeks later, 3 million Nb157 anti-TIM3 BissCAR T cells, conventional Nb157 CAR T cells, or UTD T cells were injected IV, followed by flow cytometry analysis of peripheral blood and bone marrow (n = 5 per group for BissCAR and UTD T cells; n = 3 per group for Nb157 T cells). (F) Bone marrow of HIS mice, which were treated with T cells for 3 weeks, was analyzed by flow cytometry after staining with CD45/Lin/CD34/CD38/7-AAD. Representative fluorescence-activate cell sorting plots were used to identify HSC (CD34+CD38−) and myeloid progenitors (CD34+CD38+). (G) HSCs (CD45+Lin−CD34+CD38−) in the bone marrow of HIS mice were analyzed by flow cytometry 3 weeks after the initial treatment. (H) Myeloid progenitors (CD45+Lin−CD34+CD38−) in the bone marrow of HIS mice were analyzed by flow cytometry 3 weeks after the initial treatment. (I) Monocytes (human CD45+CD33+) from peripheral blood of HIS mice were analyzed by flow cytometry 3 weeks after the initial treatment; cell number and blood volume were quantified using CountBright counting beads. In (G-I), n = 5 per group for BissCAR T cells and UTD T cells, n = 3 per group for Nb157 T cells. *P < .05, **P < .01, ***P < .001, Student t test.
BissCAR T cells targeting CD13 and TIM3 eradicate AML PDXs, but with reduced toxicity to human HSCs in vivo. (A) Schematic diagram of AML PDX mice treated with control or BissCAR T cells. Twenty million PD AML cells were injected into NSG mice, followed by injection of 5 million BissCAR T cells or UTD T cells 2 weeks later. Next, human peripheral blood CD3+ cells were analyzed by serial bleeding weekly. (B-C) PD AML cells or T cells in mouse peripheral blood were monitored weekly by flow staining with anti-human CD33 or anti-human CD3 antibodies. Blood volume was normalized and quantified using CountBright counting beads (n = 3). (D) Mice survival was monitored and recorded (n = 6 per group). (E) Schematic diagram of HIS mice for evaluation of human HSC toxicity. A total of 1.5 million normal donor bone marrow (BM) CD34+ cells was injected into each NSG mouse. Four weeks later, 3 million Nb157 anti-TIM3 BissCAR T cells, conventional Nb157 CAR T cells, or UTD T cells were injected IV, followed by flow cytometry analysis of peripheral blood and bone marrow (n = 5 per group for BissCAR and UTD T cells; n = 3 per group for Nb157 T cells). (F) Bone marrow of HIS mice, which were treated with T cells for 3 weeks, was analyzed by flow cytometry after staining with CD45/Lin/CD34/CD38/7-AAD. Representative fluorescence-activate cell sorting plots were used to identify HSC (CD34+CD38−) and myeloid progenitors (CD34+CD38+). (G) HSCs (CD45+Lin−CD34+CD38−) in the bone marrow of HIS mice were analyzed by flow cytometry 3 weeks after the initial treatment. (H) Myeloid progenitors (CD45+Lin−CD34+CD38−) in the bone marrow of HIS mice were analyzed by flow cytometry 3 weeks after the initial treatment. (I) Monocytes (human CD45+CD33+) from peripheral blood of HIS mice were analyzed by flow cytometry 3 weeks after the initial treatment; cell number and blood volume were quantified using CountBright counting beads. In (G-I), n = 5 per group for BissCAR T cells and UTD T cells, n = 3 per group for Nb157 T cells. *P < .05, **P < .01, ***P < .001, Student t test.
Combinatory BissCAR T cells targeting CD13 and TIM3 have reduced toxicity to HSCs in vivo
We also investigated the impact of BissCAR T cells on normal human HSCs. Humanized immune system (HIS) mice were used to assess hematopoietic toxicity of BissCAR T cells (Figure 7E). NSG mice were conditioned with busulfan and engrafted with bone marrow CD34+ cells from a normal adult donor, followed by treatment with BissCAR, Nb157 CAR, or UTD T cells 4 weeks later. Bone marrow from these mice was collected for analysis 3 weeks after treatment. Nb157 CAR T cells almost completely depleted CD34+CD38− HSCs, CD34+CD38+ myeloid progenitors, and peripheral monocytes (Figure 7F-I). Notably, BissCAR T cells significantly reduced the toxicity to HSCs, retaining ∼50% of the human HSC-enriched population and the myeloid progenitors of normal control mice (Figure 7F-H). Moreover, BissCAR T cells significantly reduced the monocytes in peripheral blood and allowed the protection of part of the monocytes in peripheral blood in BissCAR T-cell–injected mice compared with Nb157 CAR T-cell–injected mice (Figure 7I). Together, these results indicate that BissCAR T cells effectively eradicate PD AML cells (Figure 7B-D) and have much reduced toxicity to sensitive human HSCs (Figure 7F-I), suggesting BissCAR T cells as a valuable approach to treat human AML with reduced and tolerable hematopoietic toxicity.
Discussion
Remarkable success in CAR T-cell therapy for B-cell malignancies highlights an important and promising direction to improve cancer immunotherapy. Here, we developed the STAR system to generate antibodies to evaluate CAR T-cell efficacies in vitro and in vivo, accelerating the pace of development of effective immunotherapy. The STAR system can isolate numerous specific CAR-compatible antibodies against various TAAs. The STAR approach may miss some low-expressed or low-immunogenic TAAs. Human CD13 is overexpressed in AML. However, in our system, we isolated 2 Nbs recognizing human CD13, which has >80% homology with llama CD13, indicating that the STAR approach has the power to generate Nbs for fairly homologous antigens. Notably, all of the retrieved Nbs, when assembled into CAR T cells, were capable of potently killing AML in vitro and in vivo. Together, these findings highlight the fact that the STAR system can uncover new mAbs and their targets for developing novel and effective immunotherapy for AML and other types of cancers.
As a proof of concept, we used subcutaneously formed tumor from AML cells to isolate tumor-preferred CAR-compatible Nbs. Nb157 CAR T cells showed a marked ability to eradicate AML cell xenografts and PDXs, the subcutaneous tumor model and blood disseminated model, respectively. The target CD13 is preferentially expressed on AML cells and LSCs24 and is moderately expressed on monocytic leukemia cells.36,37 Anti-CD13–based bispecific CAR T-cell engagers inhibited AML cell colony formation in culture,38 and the anti-CD13 mAbs could inhibit the growth of AML cells.37,39 However, no further preclinical evaluation was reported for the relevant CAR T-cell system. There are several reasons for this. First, it is well known that suitable antibodies are crucial for immunotherapy, and the rarity of available mAbs against CD13 to be tested may account, in part, for the current status. Second, compared with Nbs, it is harder for regular antibodies to penetrate tumor sites. Third, the possible toxicity was caused by targeting only CD13; in this regard, BissCAR T cells may increase the chance of success. Our findings demonstrate that targeting CD13 by CAR T cells is able to potently kill AML cells.
CD13 is also moderately expressed in a few nonleukemia cells, such as colon epithelial cells and kidney tubular epithelial cells.36,40 TIM3 has a limited expression profile in exhausted T cells and certain antigen-presenting cells.41-43 Targeting CD13 alone could lead to CAR T-cell–mediated on-target/off-tumor toxicity toward human HSCs and other normal cells. It appears that ∼75% of the PD-AML has CD13+/TIM3+ LSCs and 50% of the PD-AML has CD13+/TIM3+ bulk blasts based on the cell surface protein expression, regardless of the cytogenetic or molecular subset.37,44 BissCAR, a dual CAR T-cell system, selectively kills CD13 and TIM3–expressing leukemia cells. Because no known life-essential cells express both CD13 and TIM3, it is likely that BissCAR T cells are sufficient to eliminate the AML, with minimal or tolerable damage to other normal tissues expressing only CD13.
Although the current TIM3 CAR T cells were unable to regress the NB4-TIM3 tumor in vivo (supplemental Figure 5E), the TIM3-28BB split CAR, which carried the costimulatory domains, was sufficient to assist the BissCAR T cells to increase cytokine release in vitro (supplemental Figure 5I-J) and eradicate the NB4-TIM3 tumor in vivo (Figures 6D and 7D). Although CD13 CAR T cells led to elimination of HSCs, BissCAR T cells had reduced toxicity to HSCs and progenitors, retaining ∼50% of the cells (Figure 7F-H). Nevertheless, the reduced, yet remaining, toxicity of BissCAR T cells to HSCs makes it attractive to generate more potent TIM3 antibodies and/or modify the combinations of the antibodies in the BissCAR to enhance the synergistic killing of the tumor cells while further reducing the toxicity.
In this respect, the BissCAR T cells that we developed showed greater specificity for eradicating CD13 and TIM3 double-positive cancer cells (Figure 6C-D). Moreover, BissCAR T cells are also potent in eliminating AML PDXs in vivo, and they reduce the killing of human HSCs, retaining ∼50% of human HSCs and the progenitor population in vivo compared with CD13 CAR T cells (Figure 7). BissCAR exhibited an antileukemia efficacy equivalent to conventional Nb157 CAR (Figures 3A, 5E, 6D, and D7D) but with reduced toxicity. Together, these findings indicate that BissCAR T cells specifically killed CD13+ TIM3+ double positive tumors, but with reduced and likely acceptable toxicity to HSCs and other normal cells with only CD13 expression. Thus, BissCAR T cells may be valuable assets for the development of other treatments for human AML. To expedite the process of getting CAR T cells into AML patients, it is conceivable to produce them, using an off-the-shelf CAR T-cell approach, from allogeneic T cells with modified TCRs or MHCs.45
In summary, the current study establishes a technical platform, the STAR system, which simultaneously isolates multiple tumor-select and CAR-compatible Nbs, and demonstrates that the isolated Nb redirected BissCAR T cells specifically to eradicate human AML expressing CD13 and TIM3, with reduced toxicity to HSCs, thus expediting the development of effective CAR T-cell–mediated immunotherapy.
For original data, please e-mail Xianxin Hua (huax@pennmedicine.upenn.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ting Pan and Bingfeng Liu (Sun Yat-Sen University) and John Scholler (University of Pennsylvania) for methodology assistance with antibody and CAR T-cell functional assays.
This work was supported, in part, by the National Institutes of Health, National Cancer Institute grant R01-CA-178856 and National Institute of Diabetes and Digestive and Kidney Diseases grant R01-DK097555, the American Association for Cancer Research–Neuroendocrine Tumor Research Foundation, a Harrington Discovery Institute Takeda Rare Disease Scholar award, and a Clinical and Translational Research Award–Institute for Translational Medicine and Therapeutics pilot grant at the University of Pennsylvania.
Authorship
Contribution: X. He and X. Hua designed and generated Nb CAR constructs, analyzed data, and prepared the manuscript; X. He and Z.F. performed most of the experiments and created the figures; J.M., S.L, Y.C., B.G., Y.W., B.W.K. and K.P.O. performed some of the experiments; D.L.S. designed pComb3X-compatible camelid VH primers and provided phage display protocols and guidance; C.H.J. analyzed and interpreted data and provided reagents; and X. Hua conceived and supervised the project. All authors commented on and revised the manuscript and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xianxin Hua, Department of Cancer Biology, Abramson Family Cancer Research Institute, University of Pennsylvania, 412 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104-6160; e-mail: huax@pennmedicine.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal