

In this issue of Blood, have identified multiple novel somatic mutations in BCL-2 occurring concurrently with the recently reported Gly101Val BCL-2 resistance mutation in patients with chronic lymphocytic leukemia (CLL) receiving venetoclax.1 Their study demonstrates that, in addition to functional resistance mechanisms such as aberrant expression of other antiapoptotic proteins, multiple acquired resistance mutations in BCL-2 can occur in different CLL cells in a single patient.
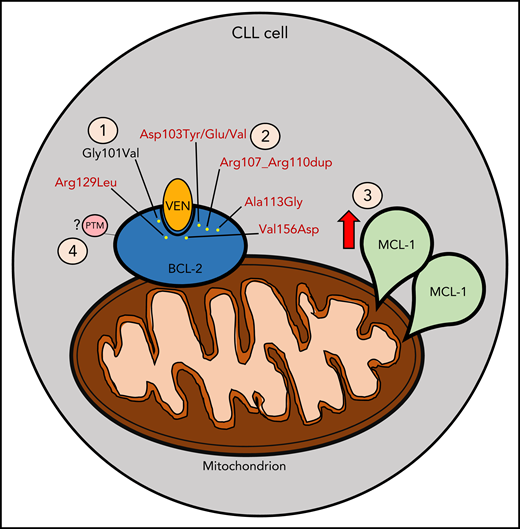
Mechanisms contributing to venetoclax (VEN) resistance. (1) Gly101Val mutation (in black) acquired following venetoclax treatment as previously described. (2) New mutations (in red) identified concomitantly with Gly101Val, as described by Blombery and colleagues. (3) MCL-1 overexpression following VEN treatment. (4) Potential posttranslational modification (PTM) that may contribute to VEN resistance (eg, phosphorylation events).
Mechanisms contributing to venetoclax (VEN) resistance. (1) Gly101Val mutation (in black) acquired following venetoclax treatment as previously described. (2) New mutations (in red) identified concomitantly with Gly101Val, as described by Blombery and colleagues. (3) MCL-1 overexpression following VEN treatment. (4) Potential posttranslational modification (PTM) that may contribute to VEN resistance (eg, phosphorylation events).
Venetoclax is a BH3-mimetic drug that binds specifically to the hydrophobic groove of BCL-2, thereby displacing proapoptotic proteins and rapidly inducing apoptosis in cells that rely on BCL-2 for survival.2 Recently, venetoclax has been found to be highly efficacious in both CLL and acute myeloid leukemia (AML), leading to approval in both diseases. In relapsed or refractory CLL, continuous venetoclax monotherapy induced response in ∼80% of patients, and 20% achieved complete remission (CR), including in patients with high-risk TP53 aberrant disease.3 When given in combination with CD20 monoclonal antibodies, time-limited venetoclax therapy can provide durable benefit for patients with relapsed/refractory CLL.4 The combination of venetoclax with hypomethylating agents in elderly patients with AML led to achievement of CR in about two-thirds of patients, with a median overall survival of ∼18 months.5
Despite the favorable outcomes with continuous venetoclax monotherapy in relapsed or refractory CLL, recent work by Blombery et al reported CLL disease progression in 21 of 67 responders, with 7 of these patients harboring a critical Gly101Val mutation at the binding groove of BCL-2 that was absent prior to initiating venetoclax treatment.2 This mutation, which was confirmed by another group,6 was shown to markedly reduce the affinity of venetoclax for BCL-2, thereby conferring acquired resistance.2 This discovery was the first clear mechanistic insight into resistance to BCL-2–selective inhibition in CLL.
In their follow-up report, Blombery and colleagues have opened a Pandora’s Box by demonstrating the existence of numerous other acquired BCL-2 mutations in patients with CLL treated with venetoclax monotherapy. By utilizing more sensitive and specific next-generation sequencing techniques, they found that 91% (10/11) of the patients studied harbored other mutations in different CLL cells in addition to Gly101Val. These mutations include Asp103Tyr/Glu/Val, situated in the P4 pocket responsible for hydrogen binding to the azaindole moiety of venetoclax, Val156Asp, situated at the base of the P2 pocket in proximity to the chlorophenyl moiety of venetoclax, in-frame insertion Arg107_Arg110dup, as well as 2 other mutations Ala113Gly and Arg129Leu previously observed in other B-cell lymphomas. Importantly, these mutations were not detected prior to venetoclax exposure, suggesting acquisition during the course of treatment. BCL-2 Asp103 is of particular note, because it is comparable to a BCL-xL Glu residue, which is a critical determinant for the selective binding of venetoclax to BCL-2 in preference to BCL-xL. The authors further elucidated the relevance of the Asp103Glu BCL-2 mutant by demonstrating a reduced binding affinity of Asp103Glu BCL-2 mutant to venetoclax, while in contrast, this mutation enhanced sensitivity to the dual BCL-2/BCL-xL inhibitor navitoclax.
Although the identification of these new BCL-2 resistance mutations provides valuable insight into one important mechanism of venetoclax resistance, these mutations are not universally found in resistant patients, and even when identified, often are only present in a minority of subclones. Thus, additional genomic and functional mechanisms likely also contribute to venetoclax resistance (see figure). For example, functional resistance to venetoclax may also arise through increased expression of other antiapoptotic proteins, such as MCL-1, or by altered cellular energy metabolism.7 Indeed, both the current and the prior reports by this group have also demonstrated MCL-1 locus focal copy number gain and BCL-xL overexpression, respectively.2 Such changes could potentially shift the survival dependency of resistant cells away from BCL-2, rendering venetoclax less effective. Another possible resistance mechanism suggested by preclinical models is posttranslational modification of BCL-2 by events such as phosphorylation.8
Despite the important insights gained from Blombery and colleagues, many crucial questions remain unanswered. Will similar resistance mutations as what has been observed here with venetoclax monotherapy also be found in CLL patients who develop resistance to time-limited venetoclax-based combination therapies, and if so, will the mutational profile be different? Will these combination therapies decrease the risk of developing resistance mutations? Will the same mutations be found in frontline CLL patients treated with venetoclax combinations? Are other venetoclax combination partners, such as inhibitors of Bruton tyrosine kinase, less likely to lead to such mutations as compared with combination with CD20 monoclonal antibodies? Beyond CLL, will these same resistance mutations also be identified in patients with other hematologic malignancies, such as AML, who are treated with venetoclax? Is it possible to develop novel BCL-2 inhibitors that remain active even against tumor cells harboring these BCL-2 mutations?
As the field moves forward, it will be important to determine the optimal approaches to preventing or overcoming such BCL-2 resistance mutations and to develop strategies to counter functional resistance mechanisms. One approach worthy of further investigation is the idea of adding or switching specific BH3-mimetic drugs based on serial assessment of genomic and functional resistance mechanisms. Next-generation sequencing testing continues to become less expensive and more widely used and may someday be able to be used in practice to monitor for genomic resistance. Functional resistance can be assessed with techniques such as BH3 profiling, which identifies cellular dependency on different antiapoptotic BCL-2 proteins for survival, and could be used to adapt therapy based on changes in antiapoptotic dependencies.9
As venetoclax continues to be explored in other hematologic malignancies such as non-Hodgkin lymphoma10 and beyond, a more comprehensive understanding of venetoclax resistance mechanisms will be increasingly important to optimize the care of patients treated with this powerful new drug.
Conflict-of-interest disclosure: S.J.F.C. has no relevant conflicts of interest. M.S.D. is on Consultancy/Advisory Boards at AbbVie, Adaptive, Ascentage, Beigene, Celgene, Genentech, Pharmacyclics, Janssen, Astra-Zeneca, MEI Pharma, Verastem, Syros, and Sunesis and has received research funding from Ascentage, Genentech, Pharmacyclics, TG Therapeutics, Surface Oncology, MEI Pharma, Verastem, and Astra-Zeneca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal