

Key Points
Risk of CMV in HCT recipients is poorly captured by assessment of quantitative defects in the T-cell pool or production of interferon-γ alone.
Virus-specific CD8+ T-cell functional profiles predict risk of clinically significant CMV, independent of steroids or T-cell depletion.

Abstract
Cytomegalovirus (CMV) is the most common viral infection in hematopoietic cell transplantation (HCT) recipients. We performed deep phenotyping of CMV-specific T cells to predict CMV outcomes following allogeneic HCT. By using 13-color flow cytometry, we studied ex vivo CD8+ T-cell cytokine production in response to CMV-pp65 peptides in 3 clinically distinct subgroups of CMV-seropositive HCT patients: (1) Elite Controllers (n = 19): did not have evidence of CMV DNAemia on surveillance testing; (2) Spontaneous Controllers (n = 16): spontaneously resolved low-grade CMV DNAemia without antiviral therapy; and (3) Noncontrollers (NC; n = 21): experienced clinically significant CMV. Two CMV-specific CD8+ T-cell functional subsets were strongly associated with risk of CMV: (i) the nonprotective signature (NPS; IL-2−IFN-γ+TNF-α−MIP-1β+), found at increased levels among NC; and (ii) the protective signature (PS; IL-2+IFN-γ+TNF-α+MIP-1β+) found at low levels among NC. High levels of the NPS and low levels of PS were associated with an increased 100-day cumulative incidence of clinically significant CMV infection (35% vs 5%; P = .02; and 40% vs 12%; P = .05, respectively). The highest predictive value was observed when these signatures were combined into a composite biomarker consisting of low levels of the PS and high levels of the NPS (67% vs 10%; P < .001). After adjusting for steroid use or donor type, this composite biomarker remained associated with a fivefold increase in the risk of clinically significant CMV infection. CMV-specific CD8+ T-cell cytokine signatures with robust predictive value for risk of CMV reactivation should prove useful in guiding clinical decision making in HCT recipients.
Introduction
Cytomegalovirus (CMV) is the most important opportunistic infection in allogeneic hematopoietic cell transplantation (HCT) recipients.1 More than 70% of CMV-seropositive HCT recipients exhibit detectable CMV DNA levels within the first 100 days posttransplant.2,3 CMV reactivation can result in tissue-invasive disease1,4-6 with mortality rates as high as 60% in those with CMV pneumonitis5,6 that have decreased only slightly in the modern era.7 CMV infection has also been associated with increased risk of secondary bacterial and fungal infections,3,8,9 and with an increased risk of graft-versus-host disease (GVHD)10,11 and nonrelapse mortality (NRM).2,3,12-15 Data from a seminal Center for International Blood and Marrow Transplant Research study of 9469 patients transplanted between 2003 and 2010 confirmed that early CMV reactivation remains significantly associated with increased NRM in the contemporary era.14 Even at relatively low viral load levels (250 IU/mL), CMV reactivation remains associated with increased risk of mortality in the posttransplant period.2,16
Recently, letermovir was approved by the Food and Drug Administration for CMV prophylaxis in CMV-seropositive HCT recipients. However, not all CMV-seropositive HCT recipients develop CMV reactivation even in the absence of prophylaxis. More than half of the patients in the placebo arm of the pivotal letermovir trial (#NCT02137772) did not develop clinically significant CMV infection by week 24.17 We have previously reported that 28% of allogeneic HCT patients who develop detectable CMV DNAemia experience spontaneous clearance.3 Thus, a significant proportion of CMV-seropositive HCT recipients might not develop DNAemia or might experience self-resolved episodes, and such individuals are not likely to benefit of prophylaxis or preemptive antiviral therapy, respectively. Therefore, improved strategies for identification of patients at risk of CMV reactivation requiring therapy are needed.
Control of CMV reactivation is highly dependent on CMV-specific T-cell responses.18-22 In the face of severe lymphopenia following HCT, the reconstituting immune system controls viral replication in a subset of recipients.3 Despite dramatic technical advances over the past 2 decades, the clinical utility of monofunctional assays of virus-specific T cells to predict CMV reactivation following HCT remains equivocal. We hypothesized that deep functional phenotyping of CMV-specific T cells may better predict CMV outcomes following allogeneic HCT. Confirming our hypothesis, we identified 2 novel CMV-specific CD8+ T-cell cytokine signatures with robust independent and combined predictive value for risk of clinically significant CMV infection.
Methods
Study subjects
We used a previously described cohort of 174 consecutive allogeneic HCT performed at the Sylvester Comprehensive Cancer Center at the University of Miami between August 2012 and April 20163 and identified 56 CMV-seropositive patients who fell into 1 of the following clinical categories (Figure 1A): (1) Elite Controllers (EC; n = 19): CMV-seropositive recipients who despite being at risk did not have evidence of CMV DNAemia on surveillance testing; (2) Spontaneous Controllers (SC; n = 16): CMV-seropositive recipients who spontaneously resolved low-grade CMV DNAemia (median peak CMV DNA level in this group was <200 IU/mL) without antiviral therapy; and (3) Noncontrollers (NC; n = 21): CMV-seropositive recipients who experienced clinically significant CMV defined as CMV disease23 or CMV DNAemia leading to preemptive treatment. All the individuals in the NC group experienced high-grade CMV DNAemia (peak >1000 IU/mL) lasting >7 days. Schematic viral kinetics of patient groups with different CMV outcomes is shown in Figure 1B. Fourteen severely lymphopenic patients with insufficient number of events in the CMV-specific CD8+ T-cell gate in flow cytometry analyses were censored (supplemental Figure 1, available on the Blood Web site). The study was approved by our Institutional Review Board, consistent with principles in the Declaration of Helsinki.
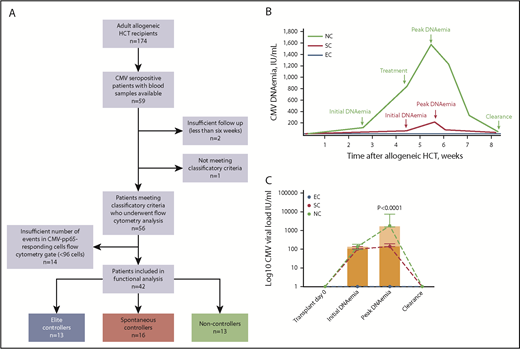
NCs exhibit higher CMV DNA levels than SCs. (A) Study flow. (B) Schematic representation of CMV DNAemia kinetics in 3 patient groups by CMV clinical phenotypes during the first 100 days posttransplant. CMV DNAemia values and dates were retrieved from electronic medical records for CMV-seropositive allogeneic HCT recipients. CMV DNA levels were monitored twice a week while inpatient and weekly thereafter until day 100. EC (blue) had no evidence of CMV reactivation posttransplant. SCs (purple) had detectable CMV DNAemia but at low DNA levels (typically <200 IU/mL) and experienced self-resolved episodes of DNAemia (ie, without need for antiviral therapy). NCs (black) experienced CMV reactivation at higher DNA levels (>1000 IU/mL) lasting >7 days and received preemptive antiviral therapy. See text and Table 1 for specific time to onset, duration, and CMV DNA levels by patient groups. (C) CMV DNAemia kinetics for the study cohort by patient group. Actual values for initial and peak CMV DNA levels are shown. None of the patients underwent transplant with detectable DNAemia. Initial DNAemia refers to the first detectable DNAemia after transplant. Peak DNAemia refers to the highest CMV DNA value (IU/mL) reached during the first episode of CMV reactivation. Bars correspond to median with IQR for each group. Y-axis corresponds to log10 scale. P value for comparison of peak DNAemia between SC and NC by using Mann-Whitney U test is shown. Seven patients from the NC group who failed to achieve clearance of DNAemia in response to antiviral therapy are not shown here. EC (n = 19); SC (n = 16); NC (n = 14).
NCs exhibit higher CMV DNA levels than SCs. (A) Study flow. (B) Schematic representation of CMV DNAemia kinetics in 3 patient groups by CMV clinical phenotypes during the first 100 days posttransplant. CMV DNAemia values and dates were retrieved from electronic medical records for CMV-seropositive allogeneic HCT recipients. CMV DNA levels were monitored twice a week while inpatient and weekly thereafter until day 100. EC (blue) had no evidence of CMV reactivation posttransplant. SCs (purple) had detectable CMV DNAemia but at low DNA levels (typically <200 IU/mL) and experienced self-resolved episodes of DNAemia (ie, without need for antiviral therapy). NCs (black) experienced CMV reactivation at higher DNA levels (>1000 IU/mL) lasting >7 days and received preemptive antiviral therapy. See text and Table 1 for specific time to onset, duration, and CMV DNA levels by patient groups. (C) CMV DNAemia kinetics for the study cohort by patient group. Actual values for initial and peak CMV DNA levels are shown. None of the patients underwent transplant with detectable DNAemia. Initial DNAemia refers to the first detectable DNAemia after transplant. Peak DNAemia refers to the highest CMV DNA value (IU/mL) reached during the first episode of CMV reactivation. Bars correspond to median with IQR for each group. Y-axis corresponds to log10 scale. P value for comparison of peak DNAemia between SC and NC by using Mann-Whitney U test is shown. Seven patients from the NC group who failed to achieve clearance of DNAemia in response to antiviral therapy are not shown here. EC (n = 19); SC (n = 16); NC (n = 14).
Antiviral prophylaxis and CMV preemptive therapy
All of the patients received antiviral prophylaxis with acyclovir 800 mg orally twice daily; CMV monitoring and preemptive therapy were performed per local protocol.3 CMV DNAemia lasted >7 days in all treated patients, with a median CMV viral load of >850 IU/mL preceding initiation of preemptive therapy.
Mononuclear cell collection and processing
Blood samples were collected prospectively on post-HCT day 30. Peripheral blood mononuclear cells were prepared by Ficoll centrifugation and cryopreserved in freezing media (90% fetal bovine serum/10% dimethyl sulfoxide) at 5 to 10 × 106/mL. Viability % postthaw was consistently >90%.
Cytokine flow cytometry
CMV-specific T-cell stimulation was performed as previously described.24 After staining, cells were analyzed by 13-color 15-parameter flow cytometry using an LSR-II cytometer (BD Biosciences) using FlowJo software (Tree Star).
Statistical modeling
Demographic, medical, and treatment characteristics were summarized using descriptive statistics. Mann-Whitney U test, Kruskal-Wallis test, Fisher’s exact test, and log-rank test were used where appropriate. Univariable analyses were performed using Cox proportional hazards regression model to assess associations between CMV outcomes and individual biomarkers. Optimal biomarker cutoffs were identified by using minimum P value approach.25 In functional analyses involving 15 possible signatures, we conducted Monte Carlo simulation (1000 repeats) to identify the minimum number of cytokine-positive events with acceptable error rate under 5% (supplemental Figure 1). A multivariable model was created using variables with a P value <.05 in the univariable model, and those were considered to be clinically relevant. All tests were 2-sided and P < .05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism Software, Inc, version 7.03, and statistical software R version 3.1.1.
Results
Patient characteristics
Characteristics of the 42 study subjects included in functional analyses are shown in Table 1. All patients were CMV seropositive, and all episodes of CMV reactivation occurred within first 100 days post-HCT (ie, early CMV). The NC group was enriched for recipients of unrelated donors treated with ATG as part of the conditioning regimen, and patients in this group received lower median doses of CD34+ cells (Table 1). Otherwise, there were no significant differences in the baseline characteristics among the 3 patient groups (Table 1). Median donor chimerism after HCT was 99.3% (IQR, 97.5% to 99.9%; n = 26). Median time from transplant to donor chimerism assessment was 92 days (IQR, 45-191). Invasive fungal and bacterial infections were greater than threefold higher among NC compared with EC/SC (38 vs 10%, respectively; P = .08; Table 1). Although the magnitude of first detectable CMV DNAemia was similar between groups (102 vs 137 IU/mL for SC vs NC, respectively; P = .21) compared with SC, the median peak CMV DNAemia was significantly higher among NC: 140 (IQR, 137-185) vs 1785 (IQR, 1557-5802) IU/mL, respectively (P < .0001; Figure 1C), and the median duration of CMV DNAemia was longer: 22 (IQR, 5-32) vs 40 (IQR, 30-60) days, respectively (P < .001). CMV DNAemia occurred earlier in NC compared with the SC group: 18 (IQR, 5-33) vs 34 (15-57) days, respectively (P = .03). Thus, the NC group exhibited different viral kinetics relative to the SC group, with higher CMV DNAemia that occurred earlier and lasted longer (Table 1; Figure 1B-C).
Characteristics of study subjects
| Characteristic | All patients | EC (n = 13) | SCs (n = 16) | NCs (n = 13) | P* | P† |
|---|---|---|---|---|---|---|
| Age, median (IQR), y | 57 (48-63) | 57 (46-61) | 57 (51-62) | 58 (46-65) | .86 | .91 |
| Male sex | 26 (62) | 7 (54) | 10 (63) | 9 (69) | .79 | >.99 |
| Follow-up, median (IQR), post-HCT days | 511 (227-778) | 373 (210-922) | 602 (453-822) | 529 (76-772) | .48 | .37 |
| Time to CMV reactivation, median (IQR), posttransplant days | 30 (12-35) | NA | 34 (15-57) | 18 (5-33) | n/a | .03 |
| CMV reactivation within 30 d posttransplant | 12 (29) | NA | 6 (38) | 6 (46) | n/a | .72 |
| Initial CMV DNAemia, median (IQR), IU/mL | 106 (96-137) | NA | 102 (96-137) | 106 (96-181) | n/a | .62 |
| Peak CMV DNAemia, median (IQR), IU/mL | 377 (137-1800) | NA | 140 (137-192) | 1,832 (1600-9189) | n/a | <.0001 |
| CMV DNAemia preceding therapy, median (IQR), IU/mL | 873 (700-1500) | NA | n/a | 873 (700-1500) | n/a | n/a |
| Duration of CMV DNAemia, median (IQR), d | 30 (22-40) | NA | 22 (5-32) | 40 (30-60) | n/a | .0008 |
| CMV DNAemia lasting >7 d | 24 (57) | NA | 11 (69) | 13 (100) | n/a | .048 |
| Recurrent CMV DNAemia (ie, ≥2 episodes of viremia)‡ | 10 (26) | NA | 4 (25) | 6 (60) | n/a | .11 |
| CMV disease | 4 (9.5) | 0 | 1 (6.3) | 3 (23) | .18 | .29 |
| Underlying diagnosis | ||||||
| Leukemia | 24 (57) | 10 (77) | 8 (50) | 6 (46) | .25 | >.99 |
| Lymphoma | 6 (14) | 1 (8) | 1 (6) | 4 (31) | .22 | .14 |
| MDS/MPN | 12 (29) | 2 (15) | 7 (44) | 3 (23) | .25 | .43 |
| Conditioning regimen | ||||||
| Myeloablative§ | 11 (26) | 3 (23) | 2 (13) | 6 (46) | .2 | .1 |
| Stem cell source | ||||||
| Peripheral blood | 41 (98) | 13 (100) | 16 (100) | 12 (92) | .62 | .44 |
| Bone marrow | 1 (2) | 0 | 0 | 1 (8) | ||
| Type of donor | ||||||
| Unrelated‖ | 20 (48) | 4 (31) | 5 (31) | 11 (85) | .006 | .008 |
| HLA-matched related | 22 (52) | 9 (69) | 11 (69) | 2 (15) | ||
| Malignancy status at time of transplant | ||||||
| Complete remission | 26 (62) | 9 (69) | 9 (56) | 8 (62) | .92 | >.99 |
| Time to engraftment, median (IQR), d | 10 (11-13) | 11 (9-18) | 11 (10-13) | 12 (11-19) | .49 | .28 |
| aGVHD (grade 2-4)¶ | 13 (31) | 3 (23) | 5 (31) | 5 (38) | .65 | .71 |
| Steroids (>0.5mg/kg), day 30 | 6 (14) | 2 (15) | 1 (6) | 3 (23) | .47 | .29 |
| (D)onor/(R)ecipient CMV serostatus | ||||||
| D− R+ | 12 (29) | 3 (23) | 6 (38) | 3 (23) | .63 | .45 |
| D+ R+ | 30 (71) | 10 (77) | 10 (62) | 10 (77) | ||
| CD34+ cells infused (1 × 106), median (IQR) | 7.4 (5.8-9.4) | 7.8 (6.7-12) | 7.6 (6.5-10) | 6.0 (3.5-7.3) | .04 | .045 |
| Charlson Comorbidity Score, median (IQR) | 3 (2-5) | 3 (2-4) | 3 (2-5) | 3 (2-7) | .68 | .49 |
| Lymphoid malignancy | 11 (26) | 5 (38) | 1 (6) | 5 (38) | .06 | .06 |
| Absolute lymphocyte count, day 30, cells/µL | 800 (480-1380) | 800 (350-1390) | 950 (725-1623) | 700 (250-1300) | .35 | .17 |
| Time to “day 30” sample collection, median (range), d | 29 (27-31) | 28 (25-31) | 30 (28-32) | 29 (27-31) | .11 | .15 |
| 1-y bacterial or fungal invasive infections | 8 (19) | 2 (15) | 1 (6) | 5 (38) | .13 | .06 |
| 100-d all-cause mortality | 5 (12) | 1 (8) | 0 | 4 (31) | .03 | .03 |
| 1-y all-cause mortality | 9 (21) | 2 (15) | 2 (13) | 5 (38) | .25 | .19 |
| 3-y all-cause mortality | 10 (24) | 2 (15) | 3 (19) | 5 (38) | .39 | .41 |
| 100-d NRM | 3 (7) | 1 (8) | 0 | 2 (15) | .28 | .19 |
| 1-y NRM | 5 (12) | 1 (8) | 1 (6) | 3 (23) | .49 | .29 |
| 3-y NRM | 5 (12) | 1 (8) | 1 (6) | 3 (23) | .49 | .29 |
| Characteristic | All patients | EC (n = 13) | SCs (n = 16) | NCs (n = 13) | P* | P† |
|---|---|---|---|---|---|---|
| Age, median (IQR), y | 57 (48-63) | 57 (46-61) | 57 (51-62) | 58 (46-65) | .86 | .91 |
| Male sex | 26 (62) | 7 (54) | 10 (63) | 9 (69) | .79 | >.99 |
| Follow-up, median (IQR), post-HCT days | 511 (227-778) | 373 (210-922) | 602 (453-822) | 529 (76-772) | .48 | .37 |
| Time to CMV reactivation, median (IQR), posttransplant days | 30 (12-35) | NA | 34 (15-57) | 18 (5-33) | n/a | .03 |
| CMV reactivation within 30 d posttransplant | 12 (29) | NA | 6 (38) | 6 (46) | n/a | .72 |
| Initial CMV DNAemia, median (IQR), IU/mL | 106 (96-137) | NA | 102 (96-137) | 106 (96-181) | n/a | .62 |
| Peak CMV DNAemia, median (IQR), IU/mL | 377 (137-1800) | NA | 140 (137-192) | 1,832 (1600-9189) | n/a | <.0001 |
| CMV DNAemia preceding therapy, median (IQR), IU/mL | 873 (700-1500) | NA | n/a | 873 (700-1500) | n/a | n/a |
| Duration of CMV DNAemia, median (IQR), d | 30 (22-40) | NA | 22 (5-32) | 40 (30-60) | n/a | .0008 |
| CMV DNAemia lasting >7 d | 24 (57) | NA | 11 (69) | 13 (100) | n/a | .048 |
| Recurrent CMV DNAemia (ie, ≥2 episodes of viremia)‡ | 10 (26) | NA | 4 (25) | 6 (60) | n/a | .11 |
| CMV disease | 4 (9.5) | 0 | 1 (6.3) | 3 (23) | .18 | .29 |
| Underlying diagnosis | ||||||
| Leukemia | 24 (57) | 10 (77) | 8 (50) | 6 (46) | .25 | >.99 |
| Lymphoma | 6 (14) | 1 (8) | 1 (6) | 4 (31) | .22 | .14 |
| MDS/MPN | 12 (29) | 2 (15) | 7 (44) | 3 (23) | .25 | .43 |
| Conditioning regimen | ||||||
| Myeloablative§ | 11 (26) | 3 (23) | 2 (13) | 6 (46) | .2 | .1 |
| Stem cell source | ||||||
| Peripheral blood | 41 (98) | 13 (100) | 16 (100) | 12 (92) | .62 | .44 |
| Bone marrow | 1 (2) | 0 | 0 | 1 (8) | ||
| Type of donor | ||||||
| Unrelated‖ | 20 (48) | 4 (31) | 5 (31) | 11 (85) | .006 | .008 |
| HLA-matched related | 22 (52) | 9 (69) | 11 (69) | 2 (15) | ||
| Malignancy status at time of transplant | ||||||
| Complete remission | 26 (62) | 9 (69) | 9 (56) | 8 (62) | .92 | >.99 |
| Time to engraftment, median (IQR), d | 10 (11-13) | 11 (9-18) | 11 (10-13) | 12 (11-19) | .49 | .28 |
| aGVHD (grade 2-4)¶ | 13 (31) | 3 (23) | 5 (31) | 5 (38) | .65 | .71 |
| Steroids (>0.5mg/kg), day 30 | 6 (14) | 2 (15) | 1 (6) | 3 (23) | .47 | .29 |
| (D)onor/(R)ecipient CMV serostatus | ||||||
| D− R+ | 12 (29) | 3 (23) | 6 (38) | 3 (23) | .63 | .45 |
| D+ R+ | 30 (71) | 10 (77) | 10 (62) | 10 (77) | ||
| CD34+ cells infused (1 × 106), median (IQR) | 7.4 (5.8-9.4) | 7.8 (6.7-12) | 7.6 (6.5-10) | 6.0 (3.5-7.3) | .04 | .045 |
| Charlson Comorbidity Score, median (IQR) | 3 (2-5) | 3 (2-4) | 3 (2-5) | 3 (2-7) | .68 | .49 |
| Lymphoid malignancy | 11 (26) | 5 (38) | 1 (6) | 5 (38) | .06 | .06 |
| Absolute lymphocyte count, day 30, cells/µL | 800 (480-1380) | 800 (350-1390) | 950 (725-1623) | 700 (250-1300) | .35 | .17 |
| Time to “day 30” sample collection, median (range), d | 29 (27-31) | 28 (25-31) | 30 (28-32) | 29 (27-31) | .11 | .15 |
| 1-y bacterial or fungal invasive infections | 8 (19) | 2 (15) | 1 (6) | 5 (38) | .13 | .06 |
| 100-d all-cause mortality | 5 (12) | 1 (8) | 0 | 4 (31) | .03 | .03 |
| 1-y all-cause mortality | 9 (21) | 2 (15) | 2 (13) | 5 (38) | .25 | .19 |
| 3-y all-cause mortality | 10 (24) | 2 (15) | 3 (19) | 5 (38) | .39 | .41 |
| 100-d NRM | 3 (7) | 1 (8) | 0 | 2 (15) | .28 | .19 |
| 1-y NRM | 5 (12) | 1 (8) | 1 (6) | 3 (23) | .49 | .29 |
| 3-y NRM | 5 (12) | 1 (8) | 1 (6) | 3 (23) | .49 | .29 |
Data are presented as absolute number (percentage), unless specified otherwise. Statistically significant P values (P < .05) are indicated in bold.
aGVHD, acute graft-versus-host disease; ATG, antithymocyte globulin; IQR, interquartile range; MDS/MPN, myelodysplastic syndrome/ myeloproliferative neoplasm.
P value for comparison between the 3 groups by using Kruskal-Wallis or Fisher’s exact test.
P value for comparison between SCs and NCs groups by using Mann-Whitney U or Fisher’s exact test.
Excluding 3 patients from the NCs group who never achieved viral clearance.
Other patients received reduced intensity conditioning. Missing data on 1 patient from SCs group.
Includes 4 mismatched unrelated donors and 16 matched unrelated donors. All unrelated donor recipients at our center undergo T-cell depletion with ATG during conditioning regimen. Typical dose of ATG at our center is 4 mg/kg total.
Median time from transplant to aGVHD was 108 d (IQR, 43-123).
Risk of CMV infection is not associated with T-cell quantitative defects
There were no differences in the absolute counts of CMV-specific CD4+ and CD8+ T cells (ie, producing any cytokine in response to in vitro stimulation with CMV-pp65) between individuals experiencing reactivation vs those who did not (Figure 2A and data not shown). The absolute count of CMV-specific CD8+ T cells, measured on posttransplant day 30, was not predictive of the risk of CMV reactivation (any DNAemia; Figure 2B) or clinically significant CMV (supplemental Figure 2). We also stratified the cohort by absolute lymphocyte count above or below 300 cells per microliter on day 30 and found no statistical difference in the cumulative incidence of CMV reactivation (any DNAemia) or clinically significant CMV (ie, DNAemia triggering preemptive therapy) in the first 100 days posttransplant between groups (supplemental Figure 2). The proportion of CMV(pp65)-responsive cells (ie, cells that recognized and responded to CMV-pp65 peptides in vitro by producing at least 1 of the 4 tested cytokines) within the total CD8+ T-cell pool was 4.3% (range, 0.2% to 24%). This frequency of CMV responsive cells within the CD8+ T-cell compartment was similar between EC, SC, and NC (Figure 2C).
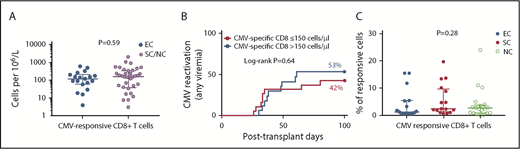
Virus-specific CD8+ lymphocyte count does not discriminate between HCT patients with different CMV outcomes. CMV(pp65)-specific CD8+ T-cell counts are shown. (A) CMV-specific CD8+ T cells as a function of absolute lymphocyte count, for patients with and without CMV reactivation. Y-axis corresponds to log10 scale. (B) The 100-day cumulative incidence of CMV reactivation (any DNAemia) by CMV-specific CD8+ T-cell count above (n = 17) or below (n = 19) the median (150 cells per microliter). (C) The proportion of CMV-responding cells (by producing any cytokine in response to in vitro stimulation with CMV-pp65) within the CD8+ T-cell compartment. T-cell counts were measured on day +30. Median with IQR is shown for each group. P value in panel A corresponds to comparison between groups by using Mann-Whitney U test. P value in panel B corresponds to comparison between groups by using Log-rank test. P value for the Kruskal-Wallis test is shown in panel C; similarly, no statistical differences were observed between groups when using Mann-Whitney U test (not shown). EC (n = 19); SC (n = 16); and NC (n = 21).
Virus-specific CD8+ lymphocyte count does not discriminate between HCT patients with different CMV outcomes. CMV(pp65)-specific CD8+ T-cell counts are shown. (A) CMV-specific CD8+ T cells as a function of absolute lymphocyte count, for patients with and without CMV reactivation. Y-axis corresponds to log10 scale. (B) The 100-day cumulative incidence of CMV reactivation (any DNAemia) by CMV-specific CD8+ T-cell count above (n = 17) or below (n = 19) the median (150 cells per microliter). (C) The proportion of CMV-responding cells (by producing any cytokine in response to in vitro stimulation with CMV-pp65) within the CD8+ T-cell compartment. T-cell counts were measured on day +30. Median with IQR is shown for each group. P value in panel A corresponds to comparison between groups by using Mann-Whitney U test. P value in panel B corresponds to comparison between groups by using Log-rank test. P value for the Kruskal-Wallis test is shown in panel C; similarly, no statistical differences were observed between groups when using Mann-Whitney U test (not shown). EC (n = 19); SC (n = 16); and NC (n = 21).
Functionality of CMV-specific CD8+ T cells
Interferon-γ (IFN-γ) was the most common cytokine produced in response to CMV-pp65 and was produced by 81% of responding CD8+ T cells; this was followed in decreasing order by macrophage inflammatory protein 1β (MIP-1β; 67%), tumor necrosis factor-α (TNF-α; 24%), and interleukin-2 (IL-2; 5%), respectively. Production of TNF-α was reduced in NC, whereas production of IFN-γ, MIP-1β, or IL-2 by virus-specific CD8+ T cells did not discriminate between patient groups (supplemental Figure 3). The numbers of T cells producing IFN-γ were comparable between patients with clinically significant CMV infection and those who experienced self-resolved episodes of DNAemia or did not reactivate CMV (Figure 3A). In time to event analyses, the frequencies of CD8+ T cells producing IFN-γ failed to predict the 100-day risk of CMV reactivation (any DNAemia) and clinically significant reactivation (Figure 3B-C).

IFN-γ production does not discriminate between HCT patients with different CMV outcomes. (A) Proportion of IFN-γ–producing cells within the CMV(pp65)-responsive CD8+ T-cell pool across groups. T-cell responses were measured on day +30. Medians (81%, 82%, and 77% for EC, SC, and NC, respectively) with IQR are shown for each group. P value corresponds to comparison between groups by using Kruskal-Wallis test. Similarly, no statistical differences between groups by using Mann-Whitney U test (not shown). EC (n = 13); SC (n = 16); NC (n = 13). (B) The 100-day cumulative incidence of CMV reactivation (any DNAemia) by levels of IFN-γ using 69% as cutoff. Number of subjects in log-rank analysis was 9 and 27 for IFN-γ low and high, respectively. (C) The 100-day cumulative incidence of clinically significant CMV by levels of IFN-γ using 72% as cutoff. Number of subjects in log-rank analysis was 13 and 33 for IFN-γ low and high, respectively. Specific cutoffs were identified using the most significant P value approach.
IFN-γ production does not discriminate between HCT patients with different CMV outcomes. (A) Proportion of IFN-γ–producing cells within the CMV(pp65)-responsive CD8+ T-cell pool across groups. T-cell responses were measured on day +30. Medians (81%, 82%, and 77% for EC, SC, and NC, respectively) with IQR are shown for each group. P value corresponds to comparison between groups by using Kruskal-Wallis test. Similarly, no statistical differences between groups by using Mann-Whitney U test (not shown). EC (n = 13); SC (n = 16); NC (n = 13). (B) The 100-day cumulative incidence of CMV reactivation (any DNAemia) by levels of IFN-γ using 69% as cutoff. Number of subjects in log-rank analysis was 9 and 27 for IFN-γ low and high, respectively. (C) The 100-day cumulative incidence of clinically significant CMV by levels of IFN-γ using 72% as cutoff. Number of subjects in log-rank analysis was 13 and 33 for IFN-γ low and high, respectively. Specific cutoffs were identified using the most significant P value approach.
We next estimated the proportion of polyfunctional CMV-specific T cells (ie, producing 2 or more cytokines simultaneously in response to CMV peptides) within the CMV-specific CD8+ T-cell compartment. The functional capacity of CMV-specific T cells was assessed by higher-order flow cytometry measuring the coproduction of intracellular IFN-γ, MIP-1α, TNF-α, and/or IL-2 via Boolean gating. When compared with EC and SC, CMV-specific CD8+ T cells in the NC group had a reduced polyfunctional capacity exhibiting the lowest proportion of cells expressing 3 to 4 cytokines simultaneously in response to CMV-pp65 stimulation: 26% and 35% vs 18%, respectively (P = .02). The proportion of quadruple producer CMV-specific CD8+ T cells was 2 times higher in EC and SC compared with NC: 2.7% in EC and 2.4 in SC vs 1.2% in NC (P = .001). In contrast, the proportion of quadruple producers in the CMV-pp65–specific CD4+ T-cell compartment was identical (3%) for all patient groups (data not shown). These results suggest that failure to control CMV reactivation following allogeneic HCT is associated with loss of polyfunctionality within the CMV-specific CD8+ T-cell compartment, in the presence of preserved absolute numbers of lymphocytes, and absolute numbers of total and monofunctional CMV-specific T cells.
Functional signatures associated with risk of CMV reactivation
Deep immunophenotyping using IFN-γ, ΜIP-1β, TNF-α, and IL-2 allowed us to delineate 15 functional populations of CD8+ T cells that recognized and responded to CMV peptides (Figure 4): 1 functional subset was a quadruple producer; 4 subsets were triple producers; 6 subsets were double producers; and 4 subsets were single producers.

Distribution of deep functional signatures across CMV clinical groups. Deep immunophenotyping based on expression of IFN-γ, ΜIP-1β, TNF-α, and/or IL-2 was used to delineate fifteen functional populations within the CMV(pp65)-specific CD8+ T-cell compartment (n = 42): 1 functional subset was a quadruple producer; 4 subsets were triple producers; 6 subsets were double producers; and 4 subsets were single producers. Mean and standard error of the mean for each signature in the 3 patient groups are shown. Dotted boxes show the NPS (IL-2−IFN-γ+TNF-α−ΜIP-1β+) and the PS (IL-2+IFN-γ+TNF-α+ΜIP-1β+) were found to be associated with increased and reduced risk of CMV reactivation, respectively, in analyses shown in Figures 5 and 6 and Table 2. EC (n = 13); SC (n = 16); NC (n = 13).
Distribution of deep functional signatures across CMV clinical groups. Deep immunophenotyping based on expression of IFN-γ, ΜIP-1β, TNF-α, and/or IL-2 was used to delineate fifteen functional populations within the CMV(pp65)-specific CD8+ T-cell compartment (n = 42): 1 functional subset was a quadruple producer; 4 subsets were triple producers; 6 subsets were double producers; and 4 subsets were single producers. Mean and standard error of the mean for each signature in the 3 patient groups are shown. Dotted boxes show the NPS (IL-2−IFN-γ+TNF-α−ΜIP-1β+) and the PS (IL-2+IFN-γ+TNF-α+ΜIP-1β+) were found to be associated with increased and reduced risk of CMV reactivation, respectively, in analyses shown in Figures 5 and 6 and Table 2. EC (n = 13); SC (n = 16); NC (n = 13).
Among 15 cytokine signatures measured at day +30, we identified 1 CMV-specific CD8+ T-cell functional subset that was strongly associated with increased risk of CMV reactivation. These cells were characterized by production of IFN-γ and MIP-1β but not IL-2 or TNF-α (IL-2−IFN-γ+TNF-α−MIP-1β+); this signature was found at significantly higher levels within the CMV-specific CD8+ T-cell population among patients who experienced CMV reactivation compared with EC (median [IQR]: 22 [10-32] vs 5.7 [2.8-18], respectively; P = .002); the highest (median, IQR) levels of this signature were observed among patients who failed to control viral replication and were eventually treated with antivirals: 5.7 (2.8-18), 15.4 (6.5-30), and 29.1 (19-35) for EC, SC, and NC, respectively (P = .04 for EC vs SC; P = .0003 for EC vs NC; Figure 5A). Because this CD8+ T-cell cytokine functional signature was found at higher levels among patients who experienced CMV reactivation, we will refer to this subset as the nonprotective signature (NPS) throughout the rest of the article. In a time-to-event analysis, the cumulative incidence of CMV reactivation (any DNAemia) during the first 100 days posttransplant was significantly higher among individuals with high levels of the NPS (71% vs 11%; log-rank P = .006; Figure 5B).
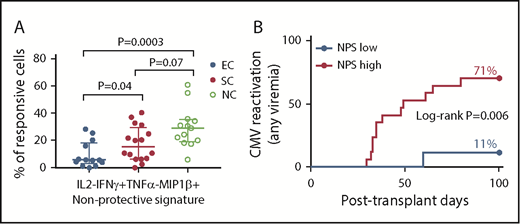
Skewed distribution of the NPS in patients with CMV reactivation (any DNAemia). (A) The skewed distribution of the NPS (IL-2−IFN-γ+TNF-α−ΜIP-1β+ cells within the CMV(pp65)-specific CD8+ T-cell compartment) in patients with CMV reactivation, with the highest levels among those individuals with clinically significant CMV. P value for comparision between groups using Mann-Whitney U test. EC (n = 13); SC (n = 16); NC (n = 13). (B) The 100-day cumulative incidence of CMV reactivation (any DNAemia) by levels of NPS, using 5.7% as cutoff. Specific cutoff was identified using the most significant P value approach. Number of subjects in log-rank analysis was 9 and 17 for NPS low and high, respectively.
Skewed distribution of the NPS in patients with CMV reactivation (any DNAemia). (A) The skewed distribution of the NPS (IL-2−IFN-γ+TNF-α−ΜIP-1β+ cells within the CMV(pp65)-specific CD8+ T-cell compartment) in patients with CMV reactivation, with the highest levels among those individuals with clinically significant CMV. P value for comparision between groups using Mann-Whitney U test. EC (n = 13); SC (n = 16); NC (n = 13). (B) The 100-day cumulative incidence of CMV reactivation (any DNAemia) by levels of NPS, using 5.7% as cutoff. Specific cutoff was identified using the most significant P value approach. Number of subjects in log-rank analysis was 9 and 17 for NPS low and high, respectively.
We next assessed associations between CMV-specific T-cell signatures and the risk of CMV reactivation requiring therapy. None of the 15 CD4+ T-cell signatures was associated with risk of clinically significant CMV (data not shown). Compared with patients without clinically significant CMV, the NC group exhibited a higher proportion of CMV-specific CD8+ T cells expressing the NPS (9.8 [IQR, 4.9-23] vs 29 [IQR, 19-35], respectively, P = .003; Figure 6A), and a trend toward a lower proportion of quadruple producer (IL-2+IFN-γ+TNF-α+MIP-1β+) CMV-specific CD8+ T cells (0.5 [0.1-1.8] vs 1.8 [0.6-4], respectively, P = .07; Figure 6B). Because these cells were depleted among patients with clinically significant CMV, we termed this signature the protective signature (PS). In time to event analysis, high levels of the NPS within the overall CMV-specific CD8+ T-cell population were associated with an increased 100-day cumulative incidence of clinically significant CMV infection of 35% compared with 5% incidence observed among individuals with low levels of the NPS (P = .02; Figure 6C). Lower levels of the PS were also associated with higher cumulative incidence of clinically significant CMV (40% vs 12%; P = .054; Figure 6D). The highest predictive value was observed when these 2 signatures were combined into a composite biomarker consisting of low levels of the PS and high levels of the NPS. Compared with patients with other signatures, patients identified by this composite biomarker had a significantly increased cumulative incidence of clinically significant CMV (10% vs 67%; P < .001; Figure 6E) during the first 100 days after HCT.

Functional signatures are associated with risk of clinically significant CMV. (A) The skewed distribution of the NPS (IL-2−IFN-γ+TNF-α-ΜIP-1β+ cells within the CMV(pp65)-specific CD8+ T-cell compartment) in patients with clinically significant CMV. (B) A trend toward reduced levels of the PS (IL-2+IFN-γ+TNF-α+MIP-1β+ cells) in patients with clinically significant CMV. (A-B) Immunophenotyping was performed on day +30. Medians with IQR are shown for each group. Differences between groups by using Mann-Whitney U test are shown. EC (n = 13); SC (n = 16); NC (n = 13). (C-E) Cumulative incidence of clinically significant CMV by levels of NPS using 16% as cutoff (C); by levels of PS using 0.45 as cutoff (D); and by a composite biomarker consisting of high levels of NPS and low levels of PS using above cutoffs (E). Specific cutoffs were identified using the most significant P value approach. Number of subjects in log-rank analysis shown in panel C was 19 and 17 for low and high NPS groups, respectively. Number of subjects in log-rank analysis shown in panel D was 10 and 26 for low and high PS groups, respectively. Number of subjects in log-rank analysis in panel E was 6 and 30 for composite biomarker and other signatures, respectively. Note increased cumulative incidence of clinically significant CMV infection in patients with high levels of the NPS, low levels of PS, and those with the composite biomarker.
Functional signatures are associated with risk of clinically significant CMV. (A) The skewed distribution of the NPS (IL-2−IFN-γ+TNF-α-ΜIP-1β+ cells within the CMV(pp65)-specific CD8+ T-cell compartment) in patients with clinically significant CMV. (B) A trend toward reduced levels of the PS (IL-2+IFN-γ+TNF-α+MIP-1β+ cells) in patients with clinically significant CMV. (A-B) Immunophenotyping was performed on day +30. Medians with IQR are shown for each group. Differences between groups by using Mann-Whitney U test are shown. EC (n = 13); SC (n = 16); NC (n = 13). (C-E) Cumulative incidence of clinically significant CMV by levels of NPS using 16% as cutoff (C); by levels of PS using 0.45 as cutoff (D); and by a composite biomarker consisting of high levels of NPS and low levels of PS using above cutoffs (E). Specific cutoffs were identified using the most significant P value approach. Number of subjects in log-rank analysis shown in panel C was 19 and 17 for low and high NPS groups, respectively. Number of subjects in log-rank analysis shown in panel D was 10 and 26 for low and high PS groups, respectively. Number of subjects in log-rank analysis in panel E was 6 and 30 for composite biomarker and other signatures, respectively. Note increased cumulative incidence of clinically significant CMV infection in patients with high levels of the NPS, low levels of PS, and those with the composite biomarker.
We next examined whether the predictive value of functional signatures identified here added diagnostic information not captured by clinical judgment alone. Administration of ATG to recipients from unrelated donors was the only clinical factor found to be strongly associated with the risk of clinical significant CMV in this cohort (Table 2; Figure 7A). The incidence of clinically significant CMV infection was significantly higher among those who underwent T-cell depletion compared with those who did not (56% vs 9%; P < .001). However, among ATG-treated patients, the risk of clinically significant CMV was further stratified by the NPS: ATG-treated patients with low levels of the NPS had similar risk of clinically significant CMV to that of patients who did not receive ATG, whereas ATG-treated patients with high levels of NPS exhibited the highest incidence of clinically significant reactivation (14 vs 63%; P = .05; Figure 7B). The highest incidence of CMV reactivation (any DNAemia) and clinically significant CMV were observed among ATG-treated patients with high levels of the NPS and those with the composite biomarker, respectively (supplemental Figure 4).
Probability of CMV infection
| CMV reactivation (any DNAemia) | Clinically significant CMV | |||||||
|---|---|---|---|---|---|---|---|---|
| Variable | HR | Lower | Upper | P | HR | Lower | Upper | P |
| Univariate analysis | ||||||||
| Unrelated donor* | 2.16 | 0.72 | 6.47 | .17 | 10.1 | 1.21 | 83.6 | .03 |
| Lymphoid malignancy | 0.22 | 0.03 | 1.72 | .15 | 1.5 | 0.29 | 7.66 | .64 |
| CMV seronegative donor | 0.56 | 0.18 | 1.73 | .32 | 2.4 | 0.29 | 19.9 | .42 |
| Steroids† | 0.58 | 0.05 | 6.88 | .67 | 2.94 | 0.4 | 21.8 | .29 |
| IL-2+IFN-γ+TNF-α+MIP-1β+ CD8+ T cells‡ (PS) | 1.89 | 0.58 | 6.16 | .29 | 0.26 | 0.06 | 1.14 | .07 |
| IL-2−IFN-γ+TNF-α−MIP-1β+ CD8+ T cells§ (NPS) | 8.15 | 1.1 | 62.9 | .04 | 7.16 | 0.86 | 59.5 | .07 |
| Composite biomarker‖ | 1.34 | 0.41 | 4.37 | .63 | 6.92 | 1.44 | 31.1 | .01 |
| CMV reactivation (any DNAemia) | Clinically significant CMV | |||||||
|---|---|---|---|---|---|---|---|---|
| Variable | HR | Lower | Upper | P | HR | Lower | Upper | P |
| Univariate analysis | ||||||||
| Unrelated donor* | 2.16 | 0.72 | 6.47 | .17 | 10.1 | 1.21 | 83.6 | .03 |
| Lymphoid malignancy | 0.22 | 0.03 | 1.72 | .15 | 1.5 | 0.29 | 7.66 | .64 |
| CMV seronegative donor | 0.56 | 0.18 | 1.73 | .32 | 2.4 | 0.29 | 19.9 | .42 |
| Steroids† | 0.58 | 0.05 | 6.88 | .67 | 2.94 | 0.4 | 21.8 | .29 |
| IL-2+IFN-γ+TNF-α+MIP-1β+ CD8+ T cells‡ (PS) | 1.89 | 0.58 | 6.16 | .29 | 0.26 | 0.06 | 1.14 | .07 |
| IL-2−IFN-γ+TNF-α−MIP-1β+ CD8+ T cells§ (NPS) | 8.15 | 1.1 | 62.9 | .04 | 7.16 | 0.86 | 59.5 | .07 |
| Composite biomarker‖ | 1.34 | 0.41 | 4.37 | .63 | 6.92 | 1.44 | 31.1 | .01 |
| Variable | aHR | Lower | Upper | P | aHR | Lower | Upper | P |
|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | ||||||||
| Unrelated donor* | 3.3 | 1.04 | 10.3 | .04 | 7.9 | 0.93 | 66.6 | .06 |
| IL-2−IFN-γ+TNF-α−MIP-1β+ CD8+ T cells§ (NPS) | 10.9 | 1.4 | 87.5 | .02 | ||||
| Composite biomarker‖ | 5.1 | 1.12 | 23.1 | .04 | ||||
| Multivariate analysis 2 | ||||||||
| Steroids† | 0.39 | 0.03 | 4.41 | .45 | 3.61 | 0.41 | 31.7 | .25 |
| IL-2−IFN-γ+TNF-α−MIP-1β+ CD8+ T cells§ (NPS) | 8.97 | 1.15 | 69.7 | .04 | ||||
| Composite biomarker‖ | 5.42 | 1.18 | 24.8 | .03 |
| Variable | aHR | Lower | Upper | P | aHR | Lower | Upper | P |
|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | ||||||||
| Unrelated donor* | 3.3 | 1.04 | 10.3 | .04 | 7.9 | 0.93 | 66.6 | .06 |
| IL-2−IFN-γ+TNF-α−MIP-1β+ CD8+ T cells§ (NPS) | 10.9 | 1.4 | 87.5 | .02 | ||||
| Composite biomarker‖ | 5.1 | 1.12 | 23.1 | .04 | ||||
| Multivariate analysis 2 | ||||||||
| Steroids† | 0.39 | 0.03 | 4.41 | .45 | 3.61 | 0.41 | 31.7 | .25 |
| IL-2−IFN-γ+TNF-α−MIP-1β+ CD8+ T cells§ (NPS) | 8.97 | 1.15 | 69.7 | .04 | ||||
| Composite biomarker‖ | 5.42 | 1.18 | 24.8 | .03 |
aGVHD grade 2 to 4 was not included as a predictor in this analysis because CMV DNAemia preceded the onset of aGVHD in all cases. Statistically significant P values (P < .05) are indicated in bold.
aHR, adjusted hazard ratio; HR, hazard ratio.
All unrelated donor recipients at our center undergo T-cell depletion with ATG during the conditioning regimen. Typical dose of ATG at our center is 4 mg/kg total.
Prednisone >0.5mg/kg per day prior to day +30.
Based on CD8+ T-cell responses to CMV-pp65 measured on day +30. High levels of PS were defined as >1.5% for CMV reactivation (any DNAemia), and as >0.45% for clinical significant CMV reactivation.
Based on CD8+ T-cell responses to CMV-pp65 measured on day +30. High levels of NPS were defined as >5.7% for CMV reactivation (any DNAemia), and as >16% for clinical significant CMV reactivation.
Composite biomarker consisted of low levels of the PS (IL-2+IFN-γ+TNF-α+MIP-1β+) and NPS high levels of the (IL-2−IFN-γ+TNF-α−MIP-1β+) NPS using cutoff outlined above.
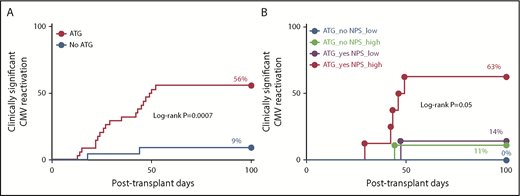
Functional signatures predict risk of clinically significant CMV independent of T-cell depletion. (A) The 100-day cumulative incidence of clinically significant CMV by administration of ATG during conditioning regimen. (B) The 100-day cumulative incidence of clinically significant CMV by administration of ATG and levels of the NPS (IL-2−IFN-γ+TNF-α−MIP-1β+ cells within the CMV(pp65)-specific CD8+ T-cell compartment) using 16% as cutoff is shown. Immunophenotyping was performed on day +30. Note that even though administration of ATG confers significant risk of CMV (A), the risk among ATG-treated patients is higher or lower depending on the levels of the NPS (B). P values correspond to the log-rank test. P value in panel B is for the comparison between ATG_yes NPS_low and ATG_yes NPS_high groups; the P value for overall differences between the 4 groups was P = .001. The number of patients at risk was as follows: 34 and 22 for ATG vs no ATG, respectively, in panel A; and 12, 9, 7, and 8 for ATG_no NPS_low, ATG_no NPS_high, ATG_yes NPS_low, and ATG_yes NPS_high, respectively.
Functional signatures predict risk of clinically significant CMV independent of T-cell depletion. (A) The 100-day cumulative incidence of clinically significant CMV by administration of ATG during conditioning regimen. (B) The 100-day cumulative incidence of clinically significant CMV by administration of ATG and levels of the NPS (IL-2−IFN-γ+TNF-α−MIP-1β+ cells within the CMV(pp65)-specific CD8+ T-cell compartment) using 16% as cutoff is shown. Immunophenotyping was performed on day +30. Note that even though administration of ATG confers significant risk of CMV (A), the risk among ATG-treated patients is higher or lower depending on the levels of the NPS (B). P values correspond to the log-rank test. P value in panel B is for the comparison between ATG_yes NPS_low and ATG_yes NPS_high groups; the P value for overall differences between the 4 groups was P = .001. The number of patients at risk was as follows: 34 and 22 for ATG vs no ATG, respectively, in panel A; and 12, 9, 7, and 8 for ATG_no NPS_low, ATG_no NPS_high, ATG_yes NPS_low, and ATG_yes NPS_high, respectively.
Using logistic regression modeling, we assessed whether the associations between the NPS and PS with CMV outcomes were independent of other known risk factors for CMV infection after HCT. Although unrelated donor, underlying lymphoid malignancy, steroid use, and negative CMV donor serostatus were not associated with the risk of CMV reactivation in this small cohort in univariate analyses (Table 2; supplemental Figure 2), high levels of the NPS within the CMV(pp65)-specific CD8+ T-cell compartment were significantly associated with increased risk of CMV reactivation (HR, 8.2; 95% confidence interval [CI], 1-63; P = .04). After adjusting for unrelated donor or steroid use, high levels of NPS remained associated with increased risk of CMV reactivation (aHR 10.9; 95% CI, 1-88; P = .02; and aHR 8.9; 95% CI, 1.2-69.7; P = .04, respectively; Table 2).
As expected, ATG administration in those receiving an allograft from an unrelated donor was associated with an increased risk of clinically significant CMV infection (Table 2; Figure 7). A composite biomarker consisting of high levels of the NPS and low levels of PS within the CMV-specific CD8+ T-cell compartment was also associated with a sevenfold increase in the probability of clinically significant CMV infection (HR 6.9; 95% CI 1.4-31; P = .01; Table 2). In a multivariate model, after adjusting for unrelated donor graft type or steroid use, this composite biomarker remained associated with a fivefold increased risk of clinically significant CMV infection (aHR, 5.1; 95% CI, 1-23; P = .04; and aHR 5.4; 95% CI, 1.2-24.8; P = .03, respectively; Table 2).
Discussion
Despite recent advancements in monitoring and treatment, CMV disease remains a serious complication of HCT associated with poor outcomes.2,3,14,16 Well-established risk factors for CMV infection in HCT recipients include steroid use, lymphopenia, use of grafts from CMV-seronegative donors, and use of unrelated, haploidentical, cord blood, or T-cell–depleted products.1,3,12,21 However, even in the absence of antiviral prophylaxis, only a subset of individuals with such risk factors develops CMV reactivation,17 suggesting that host factors unique to each individual influence the risk of uncontrolled CMV DNAemia. In an effort to identify immune biomarkers that reliably predict the risk of CMV reactivation following HCT, we used 13-color flow cytometry to study deep phenotypic T-cell responses to CMV-pp65 in 3 clinically distinct subgroups of HCT patients stratified by CMV outcomes. Our data indicate that the risk of CMV reactivation in HCT recipients is poorly captured by assessment of quantitative defects in the T-cell pool or production of IFN-γ alone. Instead, predisposition to clinically significant CMV infection following HCT was strongly associated with skewing of fine functional profiles within the CMV-specific CD8+ T-cell compartment. Using deep immunophenotyping of CMV-specific CD8+ T cells, we identified 2 functional signatures that were highly predictive of CMV outcomes. The first of them, the NPS (IL-2−IFN-γ+TNF-α−MIP-1β+), was found at increased levels among patients who experienced early CMV reactivation. Increased skewing toward this signature was highly predictive for CMV reactivation (any DNAemia) and the risk of progression to clinical significant CMV infection. The second signature associated with risk of CMV was the PS (IL-2+IFN-γ+TNF-α+MIP-1β+); patients whose CD8+ T-cell responses contained a lower relative proportion of cells with this signature had an increased risk of clinically significant CMV. The highest predictive value was observed when these 2 CD8+ T-cell signatures were combined into a composite biomarker. The association between high levels of NPS (alone or in combination with low levels of PS) and risk of CMV was independent of known clinical factors that influence risk of CMV, such as steroid use or T-cell depletion for recipients of unrelated donors. Our findings confirm the hypothesis and our prior experimental findings that inability to control CMV reactivation following allogeneic HCT is linked to the altered function of antigen-specific CD8+ T cells rather than an inability to recover sufficient numbers of CMV-specific T cells.22 Although early donor chimerism was not available for all the patients, the fact that the median donor chimerism posttransplant was >99% suggests that the bulk of the T cells analyzed on day +30 was from donor origin.
It is well known that CMV-specific CD4+ lymphocyte responses play a critical role in susceptibility to CMV infection.18-20 Similar to what we observed with CD8+ T cells, patients with CMV reactivation exhibited an immune profile characterized by high levels of the NPS in the CMV-specific CD4+ T-cell compartment (supplemental Figure 5). Other CD4+ T-cell signatures, such as IL-2−IFN-γ+TNF-α+MIP-1β− were also found at increased levels among patients with CMV reactivation (supplemental Figure 5). However, contrasting with our observations in the CMV-specific CD8+ T-cell compartment, we did not find any CD4+ T-cell functional signature with predictive value for risk of clinically significant CMV infection, although it is possible that larger studies might reveal such associations. Natural killer (NK) cells also play an important role in human immunity against CMV.26,27 In a subset of patients with NK data available, we did not observe differences in NK or NK T-cell compartments between groups, but there the frequency of invariant NK T cells was reduced among patients who experienced CMV reactivation (supplemental Figure 6).
Although IFN-γ was the dominant cytokine produced by T cells in response to CMV, we failed to establish an association between IFN-γ production and the risk of CMV infection. CD8+ T cells that produce only IFN-γ are the most abundant functional subtype in patients with CMV reactivation after HCT.28 However, it is the polyfunctionality of virus-specific T cells that has been shown to be the most important correlate of protective immunity against chronic viral infections such as CMV and HIV.28-32 The presence of polyfunctional CD8+ T cells is associated with lower levels of CMV replication, and higher frequency of self-resolved episodes in HCT recipients.28,32 Robust CMV-specific CD8+ T-cell responses are also characteristic of patients who eradicate DNAemia in <14 days in response to ganciclovir treatment.33 The observed lack of discrimination between clinical groups when using IFN-γ production alone as a predictive biomarker suggests that assessment of T-cell responses at the single cytokine level is simplistic and likely does not adequately the capture risk of CMV infection in immunocompromised subjects.
Consistent with our previous observations that differentiation from naive to the M1/M2 stages of CD8+ T cells is associated with increasing polyfunctionality,24 most cells expressing the PS or NPS corresponded to M1 (CD45RA−CD27+) or M2 (CD45RA−CD27−) memory subsets, with similar memory phenotype distribution across groups (supplemental Figure 7). Although CMV D−R+ serostatus has been associated with reduced number of polyfunctional CMV-specific CD8+ T cells,34 we observed no differences in PS/NPS responses by D/R serostatus (data not shown), a discrepancy that can be attributed to small sample size.
Our findings have important clinical implications with regards to risk assessment, prophylaxis, and preemptive therapy of CMV in HCT: (i) Current laboratory assays available for monitoring of CMV immunity such as the CMV-specific enzyme-linked immunospot assay,35 the T-cell immunity panel (Viracor Eurofins), or the QuantiFERON-CMV assay36,37 are primarily based on production of IFN-γ alone, which, based on our data, does not fully capture risk of CMV in HCT recipients. (ii) Letermovir is a new agent for CMV prophylaxis in seropositive HCT recipients; however, a large proportion of patients at risk will not develop clinically significant CMV17 and may not derive clinical benefit of prophylaxis, or achieve accepted pharmacoeconomic benefit thresholds. We propose that the NPS revealed by deep phenotyping of CMV-specific CD8+ T cells (alone or in combination with the PS) might identify HCT recipients at risk for CMV reactivation who could benefit the most from etermovir prophylaxis, a hypothesis that requires further study. In addition, as with solid organ transplant patients,38 duration of antiviral prophylaxis could be tailored based on CMV-specific deep phenotypic signatures following HCT.39 (iii) Although preemptive therapy is very effective at preventing progression to CMV disease, the optimal CMV level threshold to initiate therapy remains to be defined.3 Low CMV DNA levels frequently pose a therapeutic dilemma, because the salutatory effects of early antiviral therapy must be balanced against the risk of drug-related toxicity due to overtreatment, in addition to the financial burden. The novel CMV-specific CD8+ T-cells functional signatures reported here could facilitate clinical decision making by discriminating between patients who are prone to progress to clinically significant CMV infection vs those likely to spontaneously resolve viremia.
Our study has a number of limitations. We excluded 14 patients with insufficient number of events in the functional signature analyses; however, the rate of CMV reactivation was not different between the assessable (n = 42) and the nonassessable (n = 14) cohorts (69% vs 57%, respectively; P = .52). Small sample size precluded more comprehensive multivariate analyses due to overfitting of the model. We did, however, account for the effects of unrelated donors receiving T-cell depletion as part of conditioning, because this was the only clinical factor significantly associated with risk of CMV in this small cohort. Immunophenotyping was performed on day +30 posttransplant, so it is possible that the NPS phenotype was a result of early exposure to CMV; our group is currently in the process of evaluating earlier time points on samples collected soon after engraftment (ie, day +15 to +20) to assess whether earlier sample collection would yield sufficient events for flow cytometry analysis. Of note, patients in whom CMV reactivation occurred prior to the date of immunophenotyping were excluded from time-to-event and regression analyses to allow proper assessment of the predictive value of T-cell signatures. Because of the cross-sectional nature of the study, we cannot establish causality between immune phenotypes and clinical outcomes. However, our main goal was to identify biomarkers with clinical utility in risk stratification and management of CMV after HCT.
In conclusion, we have identified 2 novel CMV-specific CD8+ T-cell functional signatures with robust predictive value for risk of clinically significant CMV infection following HCT. Although our findings need to be validated in larger prospective studies, we propose that implementation of biomarker-driven strategies based on assessment of CMV-specific CD8+ T-cell functional signatures could guide risk stratification and management of CMV after HCT.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are indebted to all the patients who participated in the study, and thank Gabriela Bravo for assistance with statistical analysis, and Jian Zhang for instrumental assistance in the laboratory analysis.
This work was supported, in part, by a generous grant from the Applebaum Foundation.
Authorship
Contribution: J.F.C., E.D.W., and K.V.K. conceived and designed the study; J.F.C., E.D.W., E.K., C.L.B., and D.S.K. acquired the data; J.F.C., E.D.W., E.K., D.K., X.S.C., and K.V.K. analyzed the data; J.F.C. and K.V.K. prepared the first draft of the manuscript; and all authors were involved in the revision of the draft manuscript and have agreed to the final content.
Conflict-of-interest disclosure: K.V.K. has served as an ad hoc scientific advisor to Kite/Gilead, Novartis, Juno, and Atara. The remaining authors declare no competing financial interests.
Correspondence: Krishna V. Komanduri, Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, 1501 North West 10th Ave, Suite 916, Miami, FL 33136; e-mail: kkomanduri@miami.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal