

Key Points
GWAS in Hispanics identified ERG as a novel ALL risk locus, with effect sizes correlated with Native American ancestry.
ERG risk genotype was underrepresented in ALL with the ETV6-RUNX1 fusion or somatic ERG deletion, but enriched in the TCF3-PBX1 subtype.

Abstract
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children. Characterized by high levels of Native American ancestry, Hispanics are disproportionally affected by this cancer with high incidence and inferior survival. However, the genetic basis for this disparity remains poorly understood because of a paucity of genome-wide investigation of ALL in Hispanics. Performing a genome-wide association study (GWAS) in 940 Hispanic children with ALL and 681 ancestry-matched non-ALL controls, we identified a novel susceptibility locus in the ERG gene (rs2836365; P = 3.76 × 10−8; odds ratio [OR] = 1.56), with independent validation (P = .01; OR = 1.43). Imputation analyses pointed to a single causal variant driving the association signal at this locus overlapping with putative regulatory DNA elements. The effect size of the ERG risk variant rose with increasing Native American genetic ancestry. The ERG risk genotype was underrepresented in ALL with the ETV6-RUNX1 fusion (P < .0005) but enriched in the TCF3-PBX1 subtype (P < .05). Interestingly, ALL cases with germline ERG risk alleles were significantly less likely to have somatic ERG deletion (P < .05). Our results provide novel insights into genetic predisposition to ALL and its contribution to racial disparity in this cancer.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in children, with substantial racial disparities in both disease susceptibility and treatment outcomes.1,2 In particular, Hispanics have a disproportionally higher incidence of ALL with a significantly lower survival than other racial/ethnic groups in the United States (supplemental Figure 1, available on the Blood Web site),3,4 which may be partially attributed to Native American ancestry-related genomic variations.5-7
Through genome-wide association studies (GWASs), a number of risk loci have been identified for childhood ALL.8-10 The majority of these risk genes are transcription factors involved in hematopoietic development, with variable effects by race/ethnicity. For instance, single-nucleotide polymorphisms (SNPs) in ARID5B, GATA3, and PIP4K2A have higher-risk allele frequencies in Hispanics,5,11-13 whereas CEBPE SNP does not contribute to ALL susceptibility in African Americans (AAs).11 However, due to the limited sample size and complex admixture, there is a paucity of genome-wide investigation of ALL risk variants in Hispanics.
In this study, we performed a GWAS in genetically defined Hispanic children with ALL and ancestry-matched controls to systematically identify novel leukemia risk loci in this population and evaluate their associations with ALL clinical features.
Study design
In the discovery GWAS, Hispanic childhood B-cell ALL (B-ALL) cases were from the Children’s Oncology Group (COG) AALL023214 and P9904/P990515 clinical trials (supplemental Figure 2; supplemental Table 1). Non-ALL controls were unrelated subjects from the Multi-Ethnic Study of Atherosclerosis (MESA).12 The replication cohort included 144 Hispanic B-ALL cases from the COG P990615 and St. Jude Total Therapy XIIIB/XV cohorts,16,17 with 441 Hispanic controls from the Genetics of Asthma in Latino Americans (GALA) study.18 For rs2836365, we also examined its allele frequency across populations in Europe and Latino groups in the Americas in the 1000 Genomes Project (supplemental Figure 3), and compared them against allele frequency observed in MESA (supplemental Figure 4), to rule out selection bias in our control subjects. This study was approved by the respective institutional review boards with proper informed consent. Detailed methods are described in supplemental Methods.
Results and discussion
The discovery GWAS was conducted by comparing genotype frequencies of 572 556 SNPs between 940 Hispanic B-ALL cases and 681 controls, with SNP genotype-based principal components representing genetic ancestry included as covariables to control for population structure. Four loci reached genome-wide significance (P < 5 × 10−8, Figure 1A; supplemental Table 2), of which ARID5B, IKZF1, and GATA3 have been reported previously.11,12,19,20 A novel locus was identified in the intronic region of the ERG gene at 21q22.2 (Figure 1A), with the strongest association signal at rs2836365 (P = 3.8 × 10−8; odds ratio [OR] = 1.56, 1.33-1.83; supplemental Table 3). In the replication cohort of 144 Hispanic cases and 441 controls, the association signal was confirmed for rs2836365 (P = .01; OR = 1.43 [1.07-1.89]; supplemental Table 3). To further explore ALL risk variants in ERG, we imputed genotypes at additional SNPs within a 1-Mb region flanking rs2836365 and found 12 variants achieving genome-wide significance (supplemental Table 4). An imputed SNP rs2836371 showed more significant association than the original GWAS top hit (P = 1.42 × 10−9; OR = 1.64 [1.40-1.93]; supplemental Table 4), and it remained significant even after adjusting for rs2836365 (P = .006; OR = 2.03 [1.22-3.37]; supplemental Figure 5). However, no SNP in this region was significant after adjusting for rs2836371, pointing to single plausible causal variant.
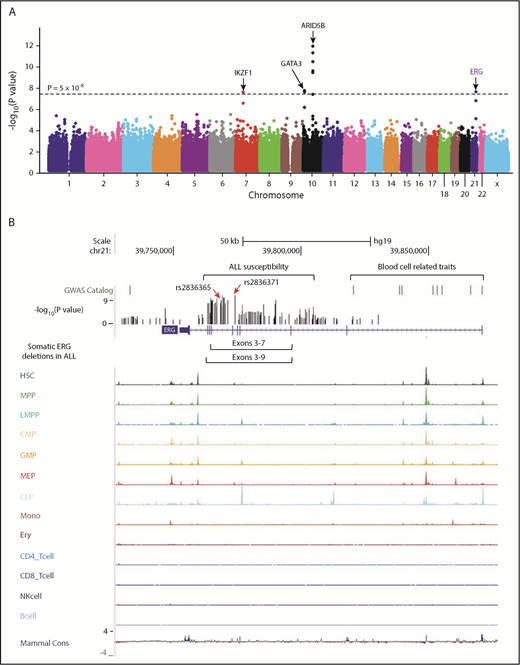
GWAS of ALL susceptibility in Hispanics and functional annotation of genomic variants at the ERG locus. (A) The association between genotype and ALL was evaluated by using a logistic regression model for 572 556 SNPs in 940 Hispanic ALL cases and 681 ancestry-matched non-ALL controls. Hispanics were defined on the basis of Native American genetic ancestry. P values (−log10P, y-axis), estimated from the additive logistic regression test in PLINK, were plotted against respective chromosomal position (x-axis). Gene symbols were indicated for 4 loci achieving genome-wide significance threshold (P < 5 × 10−8, dashed black horizontal line): ARID5B (10q21.2), IKZF1 (7p12.2), GATA3 (10p14), and ERG (21q22.3). The novel risk locus ERG identified in this study is underlined and highlighted in blue. (B) Functional annotation of genomic variants at the ERG locus. The default tracks including genomic positions and scale for the human genome assembly February 2009 (GRCh37/hg19) are shown on the top. The SNPs significantly associated with blood cell–related traits at this locus were marked in the GWAS catalog track. The log-transformed P values for SNPs tested for association with ALL in Hispanics are shown in the bed graph. Somatic ERG deletions in ALL (commonly involving exons 3-7 or 3-9) are indicated below the gene structure. The gene structure, Assay for Transposase-Accessible Chromatin using sequencing signals in different types of hematopoietic cells,21 and placental mammal basewise conservation scores by phyloP are also included. CD4_Tcell, CD4+ T-cell; CD8_Tcell, CD8+ T-cell; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; Ery, erythroid; GMP, granulocyte-macrophage progenitor; HSC, hematopoietic stem cell; LMPP, lymphoid-primed multipotent progenitor; MEP, megakaryocyte-erythroid progenitor; Mono, monocyte; MPP, multipotent progenitor; NK, natural killer cell.
GWAS of ALL susceptibility in Hispanics and functional annotation of genomic variants at the ERG locus. (A) The association between genotype and ALL was evaluated by using a logistic regression model for 572 556 SNPs in 940 Hispanic ALL cases and 681 ancestry-matched non-ALL controls. Hispanics were defined on the basis of Native American genetic ancestry. P values (−log10P, y-axis), estimated from the additive logistic regression test in PLINK, were plotted against respective chromosomal position (x-axis). Gene symbols were indicated for 4 loci achieving genome-wide significance threshold (P < 5 × 10−8, dashed black horizontal line): ARID5B (10q21.2), IKZF1 (7p12.2), GATA3 (10p14), and ERG (21q22.3). The novel risk locus ERG identified in this study is underlined and highlighted in blue. (B) Functional annotation of genomic variants at the ERG locus. The default tracks including genomic positions and scale for the human genome assembly February 2009 (GRCh37/hg19) are shown on the top. The SNPs significantly associated with blood cell–related traits at this locus were marked in the GWAS catalog track. The log-transformed P values for SNPs tested for association with ALL in Hispanics are shown in the bed graph. Somatic ERG deletions in ALL (commonly involving exons 3-7 or 3-9) are indicated below the gene structure. The gene structure, Assay for Transposase-Accessible Chromatin using sequencing signals in different types of hematopoietic cells,21 and placental mammal basewise conservation scores by phyloP are also included. CD4_Tcell, CD4+ T-cell; CD8_Tcell, CD8+ T-cell; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; Ery, erythroid; GMP, granulocyte-macrophage progenitor; HSC, hematopoietic stem cell; LMPP, lymphoid-primed multipotent progenitor; MEP, megakaryocyte-erythroid progenitor; Mono, monocyte; MPP, multipotent progenitor; NK, natural killer cell.
To explore the potential functional effects of ALL risk alleles in the ERG locus, we examined lineage-specific chromatin accessibility data of the human hematopoietic cells,21 and found that rs2836371 resided in a region of open chromatin with a moderate ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) signal in both hematopoietic stem cells and megakaryocyte-erythroid progenitor cells (Figure 1B). More interestingly, the ALL association peak at this locus was located within a ∼150-kb region encompassing genome-wide significant loci for plateletcrit, mean corpuscular volume/hemoglobin, and white blood cell types.
The ERG risk allele at rs2836365 was only modestly associated with ALL susceptibility in European Americans (EAs) (P = .02; OR = 1.12 [1.02-1.22]; N = 2317 cases and 2050 controls) and was not significant in AAs (P > .05, OR = 0.96 [0.74-1.24], N = 227 cases and 1380 controls; supplemental Table 5). In both GWAS discovery and replication series, the ERG risk allele was significantly more common in Hispanics than EAs and AAs, and the allele frequency was positively related to the proportion of Native American ancestry (Figure 2A). The effect size of this variant also increased with Native American ancestry (OR = 1.13, 1.55, and 2.35, respectively; Figure 2B). These results pointed to ERG as a plausibly ancestry-related risk locus for childhood ALL.
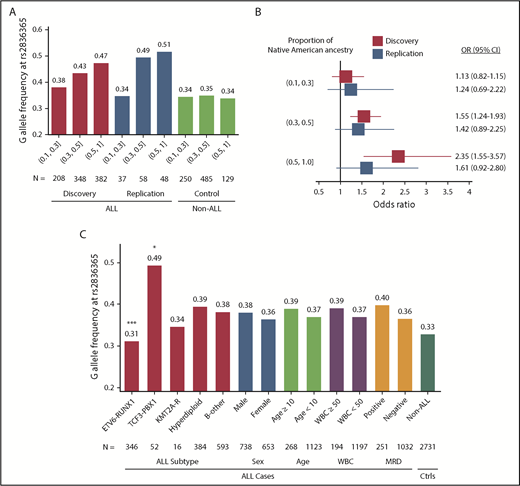
The frequency and effect sizes of the ERG risk allele and ALL features. Risk allele frequency (A) and OR (B) of rs2836365 were estimated for Hispanics with increasing levels of Native American genetic ancestry (10%-30%, 30%-50%, and 50%-100%). In the Forest plot (B), bars indicate 95% confidence intervals (CIs) and the gray vertical line indicates OR of 1. OR was estimated by logistical regression test. (C) Risk allele frequency of ERG SNP rs2836365 and ALL features. The analysis was restricted to the Hispanic Americans and EAs in the COG P9904/9905/9906 cohort because it represents a largely unselected and nationwide patient population. Variant frequency was indicated for ALL molecular subtype, sex, age at diagnosis, presenting white blood cell (WBC; 109 cell/L) count, and MRD at the end of remission induction. Logistic regression test with rs2836365 genotype adjusting for genetic ancestry (eg, ALL with vs without ETV6-RUNX1); *P < .05; ***P < .0005. Ctrls, controls; KMT2A-R, KMT2A rearrangement; MRD, minimal residual disease (at the end of induction therapy on day 29).
The frequency and effect sizes of the ERG risk allele and ALL features. Risk allele frequency (A) and OR (B) of rs2836365 were estimated for Hispanics with increasing levels of Native American genetic ancestry (10%-30%, 30%-50%, and 50%-100%). In the Forest plot (B), bars indicate 95% confidence intervals (CIs) and the gray vertical line indicates OR of 1. OR was estimated by logistical regression test. (C) Risk allele frequency of ERG SNP rs2836365 and ALL features. The analysis was restricted to the Hispanic Americans and EAs in the COG P9904/9905/9906 cohort because it represents a largely unselected and nationwide patient population. Variant frequency was indicated for ALL molecular subtype, sex, age at diagnosis, presenting white blood cell (WBC; 109 cell/L) count, and MRD at the end of remission induction. Logistic regression test with rs2836365 genotype adjusting for genetic ancestry (eg, ALL with vs without ETV6-RUNX1); *P < .05; ***P < .0005. Ctrls, controls; KMT2A-R, KMT2A rearrangement; MRD, minimal residual disease (at the end of induction therapy on day 29).
We next examined whether ERG SNP genotype preferentially predisposes to any ALL subtype, focusing on the COG P9904/9905/9906 series because it represented a large national cohort of ALL patients consecutively enrolled with minimal selection bias, including major subtypes: ETV6-RUNX1, TCF3-PBX1, KMT2A rearrangement, hyperdiploidy, and B-other. Because the ERG risk allele was significant in both Hispanics and EAs, we performed our analyses combining patients from these 2 racial/ethnic groups and adjusted for genetic ancestry (N = 1391). The ERG risk genotype was significantly underrepresented in ETV6-RUNX1 ALL (P = .0003), but enriched in the TCF3-PBX1 subtype (P = .03; Figure 2C). ERG expression also varied significantly across ALL subtypes, with the highest level observed in ETV6-RUNX1 ALL (supplemental Figure 6). Because somatic alterations at the ERG locus have been recently described and define a novel ALL subtype (concomitant with IGH-DUX4 rearrangements),22 we also evaluated its association with ERG risk variants in a subset of 905 ALL cases with both somatic and germline genomic data available. The frequency of ERG risk allele at rs2836365 was significantly lower in cases with somatic ERG deletion than those without (supplemental Figure 7; P = .04 and .02, for with or without adjusting for genetic ancestry, respectively).
The biological basis of racial disparities in cancer is poorly understood, in part because non-European populations are disproportionally underrepresented in cancer genomic studies. Taking a race/ethnicity-specific approach, we identified a novel ALL risk locus in Hispanics, in the ERG intronic region. The ERG risk variant is related to Native American ancestry in that its variant frequency and effect size both increase with the level of Native American ancestry, pointing to a likely ancestry-related effect on ALL susceptibility. The correlation of the ERG risk allele frequency with Native American ancestry was also true in a cohort of Guatemalan children with ALL (supplemental Figure 8). The underlying mechanism for such race/ethnicity-dependent effects of a genetic risk factor is unclear, although it has been reported for other cancers23 (eg, a stronger effect of the ESR1 locus for breast cancer susceptibility in Chinese women compared with Europeans and not significant in Africans24 ). It can be posited that the ERG variant interacts with another yet-to-be-discovered ALL risk allele that is exclusively present in Hispanics and the combination of both is important for ALL susceptibility. Alternatively, the ERG risk variant identified herein tags a causal allele that is absent in non-Hispanics, although this is less likely given the results from the imputation analyses. Future studies are thus warranted to unravel the mechanistic details linking ERG to ALL pathogenesis. We also examined all previously reported ALL susceptibility loci in our Hispanic GWAS (supplemental Table 2).
ERG encodes an ETS domain-containing transcription factor important for normal hematopoietic development.25 Recently, we and others identified a novel ALL subtype characterized by IGH-DUX4 rearrangement in which the overexpression of DUX4 leads to ERG deregulation (primarily the expression of an alternative ERG transcript [ERGalt] with secondary deletion of the wild-type ERG allele in some cases).22 Interestingly, our novel ALL risk variant resides within close proximity to the hotspot of leukemic ERG deletions (Figure 1B), and there was a significant negative correlation between germline and somatic variation at the ERG locus, arguing for similar effects of these variants on ERG function (supplemental Figure 7A).
Our results suggested that there could be a substantial number of genetic variants/loci contributing to racial/ethnic disparities in ALL, and collaborative efforts with larger sample sizes are needed to systematically uncover these molecular determinants in the future.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and parents who participated in the clinical protocols included in this study, and the clinicians and research staff at participating institutions.
This work was partly supported by a St. Baldrick’s Foundation International Scholar award (H.Z.); a St. Baldrick’s Foundation Scholar award and a Robert J. Arceci award (C.G.M.); National Institutes of Health grants P50 GM115279 (National Institute of General Medical Sciences), CA156449, CA21765, CA36401, CA98543, CA114766, CA98413, CA140729, and CA176063 (all from the National Cancer Institute), GM92666 (National Institute of General Medical Sciences), and HHSN261200800001E (National Cancer Institute); the National Key Research and Development Program of China (2016YFC0905000 [2016YFC0905001, 2016YFC0905002]); the National Natural Science Foundation of China (81522028, 81728003, and 81673452); and the American Lebanese Syrian Associated Charities.
Authorship
Contribution: J.J.Y. is the principal investigator of this study, has full access to all of the data in the study, and takes responsibility for the integrity of the data and the accuracy of the data analysis; M.Q., H.X., W.Y., and S.Z. performed data analysis; M.Q., H.X., and J.J.Y. wrote the manuscript; V.P.-A., K.G.R., X.Z., C.S., M.D., J.M.G.-F., E.R., E.L., N.W., F.A.-K., W.P.B., P.L.M., M.B., B.W., E.G.B., C.-H.P., C.G.M., W.E.E., S.P.H., M.V.R., and M.L.L. contributed reagents, materials, and/or data; M.Q., H.X., H.Z., W.Y., and J.J.Y. interpreted the data and the research findings; and all of the coauthors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jun J. Yang, Hematologic Malignancies Program, Comprehensive Cancer Center, Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, MS313, Memphis, TN 38105; e-mail: jun.yang@stjude.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal