

Abstract
Neutrophils are an absolutely essential part of the innate immune system, playing an essential role in the control of infectious diseases but more recently are also being viewed as important players in tissue repair. Neutrophils are able to counteract an infection through phagocytosis and/or the release of neutrophil extracellular traps (NETs). By contrast, neutrophils help repair damaged tissues, limiting NET production but still phagocytosing debris. However, when inflammation is recurrent, or the inciting agent persists, neutrophils through a frustrated inability to resolve the problem can release NETs to exacerbate tissue damage during inappropriate inflammation. In this review, we discuss the mechanisms of NET formation, as well as the apparent paradoxical role of neutrophils and NETs in host defense, chronic inflammation, and tissue disrepair.
Introduction
Neutrophils are the predominant leukocyte population in human blood and among the first cells recruited to an inflammatory site. During an infection or tissue damage, pathogen-associated molecular patterns and/or damage-associated molecular patterns (DAMPs) are sensed by pattern recognition receptors as well as DAMP receptors, which can activate resident cells to produce inflammatory mediators, such as chemokines CXCL1 and CXCL2, which bind to and activate G-protein–coupled receptors on neutrophils.1 In addition, DAMPs and pathogen-associated molecular patterns may also directly activate receptors on neutrophils to induce recruitment.2 Neutrophils display a wide range of effector mechanisms to counteract pathogens that include phagocytosis and the production of reactive oxygen species (ROS), proteases, and neutrophil extracellular traps (NETs).3 Thus, impairment of neutrophil recruitment to an infectious nidus facilitates pathogen dissemination into blood and vital organs, development of systemic infection, and eventual death.4,5 This is best exemplified by the fact that neutropenia is the biggest risk factor for infection.6
During sterile tissue injuries, neutrophils are also essential to participate in the clearance of cellular debris, returning tissue to homeostasis.7 From an evolutionary standpoint, it would seem reasonable to conclude that the system has been optimized to allow for bacterial clearance while minimizing surrounding collateral damage. However, pathogens have also evolved mechanisms to subvert the immune system, causing neutrophils to respond in a more aggressive manner, leading to untoward tissue injury.8 Moreover, lack of resolution or persistence of sterile injury may also lead to inappropriate inflammation.9 This type of persistent injury is very different from normal repair and involves either the presence of foreign substances for which our immune system has not evolved an appropriate response (eg, recurrent smoke inhalation, prolonged alcohol and drug overdose, inappropriate high-fat or high-cholesterol diet) and surgical procedures, including transplantation, ischemia reperfusion, or foreign body implantation. We would also include among these nonevolutionary situations, experimental models created by investigators to study inappropriate inflammation, including exposure of animals to carbon tetrachloride, bleomycin, acetaminophen (Tylenol), and many other experimental substances. It is under these different conditions that the neutrophils, our greatest allies, in an attempt to eradicate what they perceive as danger, respond overexuberantly or inappropriately, causing bystander tissue damage and contributing to immunopathology. In this review, we describe the mechanisms underlying NET formation in response to natural stimuli like bacteria and also highlight the paradoxical role of neutrophils and NETs in unnatural situations.
Lytic vs nonlytic NET release
Until the year 2004, phagocytosis and subsequent oxidant- and protease-dependent killing of pathogens inside of phagolyososomes were thought to be the major neutrophil effector mechanisms. This concept was changed by Brinkmann and colleagues,10 who showed that neutrophil stimulation with phorbol myristate acetate (PMA) released weblike structures of DNA coated with histones, elastase, myeloperoxidase (MPO), and cathepsin G. These structures were given the name neutrophil extracellular traps (NETs), which were initially proposed to immobilize or trap and kill bacteria extracellularly.10 Although in that first publication there was no mention of neutrophil death during NET release, the preface to that paper suggested the name NETosis, implying neutrophil death.11 The same investigators in subsequent work avidly dissected the role of NETosis using PMA and showed NET release was associated with the rupture of the neutrophil.12 As such, NETosis was further characterized as a cell death program distinct from apoptosis and necrosis and dependent on the generation of ROS by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.11,12 PMA-induced NETosis was dependent on plasma membrane lysis, which followed chromatin decondensation and chromatin mixing with cytoplasmic granule proteins.12 As such, NETosis was retained and was identified by extracellular DNA decorated with various neutrophil-derived granular proteins, including MPO and elastase. Although many groups have reproduced NETosis with PMA in vitro, to our knowledge, no one has been able to visualize exploding or NETosing neutrophils in vivo (despite sincere attempts).
By contrast, in parallel studies in vitro, it was demonstrated that NET formation induced by natural stimuli can occur entirely independent of cell lysis and subsequent cell death.13,14 Although the composition of NETs is assumed to be similar regardless of stimulus, proteomics have only been done on PMA-induced NETs. However, 1 group showed that nonlytic NET release was mitochondrial15 not nuclear, and by definition, these NETs would lack histones, a major antimicrobial and host cell toxin. A second group using primarily bacteria or bacterial products with or without platelets reported that nonlytic NETs were made of nuclear not mitochondrial DNA, which had reduced proteolytic activity.13,14 It is our personal view that PMA likely activates many pathways in neutrophils and en masse causes NET formation, oxidant production as well as a necrotic cell death that may complicate the study of NET release. In fact, NETs can be detected at 30 minutes with PMA, whereas cell death occurs at 3 hours, at which point an unregulated release of all intracellular contents is seen and perhaps makes NETs much more toxic. Alternatively, PMA-induced NET release and PMA-induced lysis are causally unrelated events. As a final point, mainly for clarity, rather than using the term lytic and nonlytic NETosis (the latter being a contradiction in terms), we will use the term lytic and nonlytic NET formation.
Lytic NET formation
NET formation followed by cell death or lytic NET release was the first to be described (Figure 1A). Stimulation of neutrophils with PMA resulted in the activation of NADPH oxidase, via PKC and Raf-MEK-ERK signaling pathway and consequent ROS generation.16 This activated protein-arginine deiminase 4 (PAD4), which hypercitrullinated histones, causing chromatin decondensation,17,18 while simultaneously MPO and neutrophil elastase (NE) were released from cytoplasmic azurophilic granules.19 Overall, 24 different proteins have been described in the NETome.20 MPO was reported to bind to chromatin and activate NE in small azurosome structures that can be seen in vitro as well as in vivo.21 NE degraded actin filaments in the cytoplasm, translocated to the nucleus, and cleaved histones.21 Subsequently, the nuclear envelope broke down via cell-cycle proteins,22 releasing the chromatin in the cytosol, which mixed with cytosolic proteins.12 The mechanism involved in cell lysis and NET release involves gasdermin D (GSDMD),23,24 a recently identified factor that mediates pyroptosis in macrophages.25 During PMA-inducing NET release, NE cleaves GSDMD to its active form (GSDMD-NT),26 which forms pores in the plasma membrane and granule membranes, enhancing NE and other granule content release.23
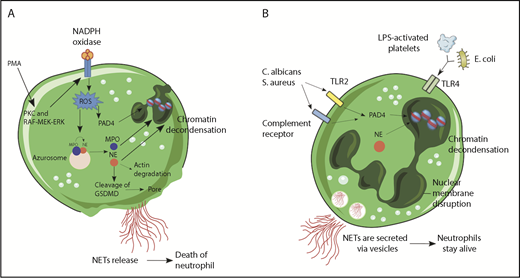
Mechanisms of NET formation. (A) PMA and other stimuli induce lytic-NET formation. Stimulation of neutrophils with PMA resulted in the activation of NADPH oxidase, via PKC and Raf-MEK-ERK signaling pathway and consequent ROS generation. Afterward, PAD4 is activated and citrullinates arginine on histones causing chromatin decondensation. MPO and NE are released from cytoplasmic azurophilic granules and then translocated to the nucleus contributing to unfolding of chromatin. Subsequently, the nuclear envelope broke down, releasing the chromatin in the cytosol, which mixed with cytosolic proteins. NE also cleaves GSDMD in the cytosol to its active form (GSDMD-NT), which, besides forming pores in the plasma membrane, also mediates pore formation in nuclear and granule membranes, enhancing NE and other granular content release. Finally, NETs are released, and the neutrophil dies. (B) Nonlytic NET formation is induced by the recognition of stimuli through Toll-like receptor 2 (TLR2), TLR4, or complement receptors independent of NAPDH oxidase activation. S aureus and C albicans activate TLR2 and complement receptors, respectively, and E coli or LPS-activated platelets activate TLR4. Along with PAD4 activation and NE translocation to the nucleus, chromatin decondensation proceeds and protein-decorated chromatin is expelled via vesicles without plasma membrane disruption. After the release of NETs, neutrophils are still alive for further functions.
Mechanisms of NET formation. (A) PMA and other stimuli induce lytic-NET formation. Stimulation of neutrophils with PMA resulted in the activation of NADPH oxidase, via PKC and Raf-MEK-ERK signaling pathway and consequent ROS generation. Afterward, PAD4 is activated and citrullinates arginine on histones causing chromatin decondensation. MPO and NE are released from cytoplasmic azurophilic granules and then translocated to the nucleus contributing to unfolding of chromatin. Subsequently, the nuclear envelope broke down, releasing the chromatin in the cytosol, which mixed with cytosolic proteins. NE also cleaves GSDMD in the cytosol to its active form (GSDMD-NT), which, besides forming pores in the plasma membrane, also mediates pore formation in nuclear and granule membranes, enhancing NE and other granular content release. Finally, NETs are released, and the neutrophil dies. (B) Nonlytic NET formation is induced by the recognition of stimuli through Toll-like receptor 2 (TLR2), TLR4, or complement receptors independent of NAPDH oxidase activation. S aureus and C albicans activate TLR2 and complement receptors, respectively, and E coli or LPS-activated platelets activate TLR4. Along with PAD4 activation and NE translocation to the nucleus, chromatin decondensation proceeds and protein-decorated chromatin is expelled via vesicles without plasma membrane disruption. After the release of NETs, neutrophils are still alive for further functions.
Studies demonstrated that the mechanism of NET formation could vary depending on the initial stimulus that activates neutrophils. Interestingly, NET formation is inhibited in the absence of NADPH oxidase in both patients with chronic granulomatous diseases27 and in knockout mice28 stimulated with Aspergillus nidulans or PMA, respectively. However, during neutrophil stimulation with Staphylococcus aureus,14 NET formation was independent of oxidant production. This was subsequently shown to also be the case in Candida albicans29 ; ionomycin30,31 and nicotine32 induced NET release where the process occurred independent of NADPH oxidase. However, the mechanism proposed for NET formation by nicotine32 and ionomycin31 was dependent on AKT signaling and calcium-induced mitochondrial ROS-release (ie, oxidant dependent but via a different mechanism). Numerous studies have also demonstrated that the pharmacological or genetic blockade of MPO and PAD4 reduce or impair NET release.19,33,34 In contrast, NET formation in response to Pseudomonas aeruginosa seems to be independent of MPO activity,30 whereas C albicans,35 Klebsiella pneumoniae,36 and cholesterol crystals37 induce NET release independently of PAD4. Recent data indicate that histone citrullination is not enough to promote chromatin decondensation. The inhibition of NE blocked chromatin decondensation without interfering with histone citrullination.38 PMA required PKC to induce NETs, while Helicobacter pylori–induced NETs were independent of PKC.16 Altogether, the mechanisms by which NETs were formed depended on the stimulus. However, such huge discrepancies beg the question whether different processes are being studied.
Nonlytic NET release
The time required for lytic NET formation has been reported to occur primarily at 3 to 4 hours. By contrast, neutrophils can also release NETs in a very rapid (5-60 minutes) and cell death–independent manner (Figure 1B). Indeed, neutrophils bound by lipopolysaccharide (LPS)-activated platelets in vivo or in vitro formed NETs within a few minutes but restricted Sytox green entry, a live cell-impermeant nucleic acid staining dye.13 During human sepsis, nonlytic NET release occurred via TLR4 activation of platelets that then bound to the neutrophils. Similar results were seen when Escherichia coli was administered in vivo, with platelets immediately tethering to neutrophils to induce NET formation while lysis was not observed.39 Other bacteria also induced nonlytic NET release. Mechanistically, it was demonstrated that NADPH oxidase was not required during nonlytic NET formation in response to S aureus,14 and neutrophils were still able to migrate and phagocytose.40,41 In addition, nonlytic NET formation required specific receptors, including activation of TLRs and complement receptors during infection with S aureus40 and C albicans.29 Moreover, human neutrophils primed with granulocyte macrophage colony–stimulating factor and subsequently stimulated with LPS or complement factor 5a for a short period also released NETs, but in a process dependent on mitochondrial, instead of nuclear, DNA.15 Interestingly, optic atrophy 1, a mitochondrial inner membrane protein that is important for mitochondrial biogenesis, was also important for NETosis through the stimulation of microtubule network formation and subsequent DNA release.42 In a recent study, that in our opinion provides important insight regarding NET formation, Branzk and colleagues38 demonstrated that neutrophils were able to sense microbe size and selectively released NETs in response to large pathogens, such as C albicans hyphae, but not in response to small yeast or single bacterium. The mechanism reported in this study was that phagocytosis prevented NET release through downregulation of NE translocation to the nucleus.38 This would likely extend to bacteria like S aureus that in vivo are found as clumps and to biofilms,43 which would both induce NET release rather than phagocytosis. It is also worth mentioning that NET release can be prevented by phagocytosis of platelets,44 suggesting that regardless of the particulate matter that is phagocytosed, NET release will be blocked. It also explains why some but not other neutrophils make NETs in infections and why neutrophils that phagocytose bacteria do not subsequently release bacteria during lytic NET formation.
The work with PMA has informed studies on nonlytic NET formation. For example, NE is also translocated to the nucleus during nonlytic NET formation, and PAD4 is activated, inducing chromatin decondensation.45 However, instead of plasma membrane disruption for NET release, protein-decorated chromatin is secreted via vesicles, allowing neutrophils to stay alive for further functions.40 Because neutrophils have very low to nonexistent transcriptional activity, loss of the nucleus did not impair processes such as phagocytosis, release of cytotoxic molecules, or motility, although the latter was altered because neutrophils use the nucleus as a fulcrum during crawling.40 Nevertheless, much like their close relatives red blood cells and platelets, neutrophils devoid of nuclei, known as cytoplasts, still performed important functions.46 Recently, it was demonstrated that cytoplasts derived from neutrophils that had released NETs were able to activate lung dendritic cells to differentiate naive CD4+ T cells to antigen-specific T-helper 17 effectors in a murine model of severe asthma, identifying a potential pathogenic role for these cellular remnants.47 Using sandwich enzyme-linked immunosorbent assays employing antibodies against MPO and N-terminal histone tails as a measure of nonlytic NETs, it was demonstrated that circulating NETs from septic patients are derived from an NADPH oxidase–independent nonlytic pathway,48 corroborating previous findings from experimental sepsis.13,39 However, nonlytic NET formation has been less studied and (1) how vesicles of DNA get formed and released, (2) what happens to anuclear neutrophils/cytoplasts, and (3) what are the signaling pathways need to be determined.
Influence of neutrophil heterogeneity on NET formation
There has been a large effort to identify neutrophil subsets and delineate which ones release NETs. Indeed, not all neutrophils release NETs; only 20% to 25% of neutrophils release NETs after S aureus stimulation.41 Circulating neutrophils can be easily separated from peripheral blood mononuclear cells by density gradient differences after centrifugation. However, during an inflammatory process, a population of neutrophils with altered density colocalizes with peripheral blood mononuclear cells density fractions, which have been called low-density neutrophils (LDNs).49 LDNs are a heterogeneous population containing both immature and mature neutrophils, and their functions differ depending on the inflammatory stimulus. Interestingly, it has been demonstrated that LDNs have an increased capacity to generate NETs in autoimmune diseases50,51 and in a model of spontaneous small intestinal tumors,52 raising the possibility that these are a distinct proinflammatory neutrophil subset.53 Numerous molecular markers have been proposed to delineate different neutrophil populations with respect to NET production. CD177− neutrophils stimulated with LPS were not able to release NETs.54,55 Olfactomedin 4 (OLFM4), a matrix glycoprotein predominantly found within specific granules, is also differentially expressed on 10% to 30% of neutrophils,56 aligning nicely with the percentage of neutrophils that make NETs. NET formation leads to OLFM4 secretion,57 and a recent study demonstrated that septic patients with a high percentage of OLFM4+ neutrophils were at higher risk of organ failure and death.58 However, direct evidence linking OLFM4-positive neutrophils and NET releasing neutrophils has yet to be confirmed. As such, molecular markers that designate NET-producing neutrophils are not available, begging the question whether NET-producing neutrophils are simply older, more mature, or more primed neutrophils, but all 1 population.59,60
Neutrophil NETs in host defense
During infection, NETs mediate host defense by trapping and killing microorganisms.41 Indeed, the first study that described NETs demonstrated that NETs were important for the sequestration and killing of bacteria by delivering a high local concentration of antimicrobial molecules.10 Subsequent studies confirmed that NETs were also formed in vivo, mediating the trapping of bacteria. Using in vivo intravital microscopy, it was possible to visualize NET trapping of E coli in hepatic sinusoids, and it was demonstrated that the disruption of NETs resulted in the spread of the bacteria systemically.13,39 In addition, the systemic treatment of S aureus–infected mice with DNase also resulted in the spread of bacteria from the infection site to the circulation.40 Likewise, fungi61,62 and virus63 were visualized being trapped by NETs in vivo and in vitro. C albicans61 and Aspergillus fumigatus62 were trapped by human neutrophil-released NETs and by NETs formed in the lung of infected mice, respectively; PMA-activated human neutrophils were also able to capture HIV-1 in vitro, an event inhibited after DNase treatment.63 However, the molecular mechanisms by which NETs bind and trap microorganisms are poorly understood, although charge has been proposed.12 Once in a NET, there is growing evidence that this structure is capable of killing bacteria. Several studies have demonstrated that NETs kill microorganisms in vitro through the action of microbicidal components. Indeed, histones have a potent antimicrobial capacity.64 Moreover, NE actively targets bacterial virulence factors from Shigella flexneri, culminating in bacteria killing in vitro.65 Calprotectin is also present in NETs, and its inhibition reduced the antifungal activity of NETs in vitro.20
Not surprisingly, as is the case with any important mechanism, pathogens have learned to counter and/or subvert NET production to favor their survival. Indeed, some of the earliest studies on NETs revealed that microbial DNase was a virulence factor that could help pathogens escape NETs, making them more apt to disseminate.66 Moreover, pathogens have learned to express virulence factors that suppress or induce NET release. Recently, it was demonstrated that pore-forming leukocidins PVL and HlgAB, toxins released from S aureus biofilms, induce both neutrophil death and NET formation. Whether this was a form of lytic NET formation or just neutrophil lysis due to these pore-forming molecules was difficult to delineate. Nevertheless, the NET release was implicated in the persistence of S aureus biofilms in vitro and in a chronic model of skin infection in vivo.8
Neutrophils and NETs mediate tissue damage during acute and chronic inflammation
Acute inflammation
However, like all processes, too many NETs in infection can be detrimental to the host. One of the earliest studies on NETs showed that the interaction between neutrophils and activated platelets induced NET formation, and this led to endothelial cell damage and organ injury after E coli infection.13 Other studies have also demonstrated NET-induced tissue injury.39,67 Acute respiratory distress syndrome (ARDS) is a life-threatening disorder characterized by widespread inflammatory lung injury often caused by neutrophil-pathogen interactions. The partial reduction of NETs by DNase I treatment or partial PAD4-deficiency (Pad4+/−) reduced acute lung injury induced by bacteria and improved survival, whereas complete NET inhibition by PAD4 deficiency (Pad4−/−) reduced lung injury, but was counterbalanced by increased bacterial load and inflammation.68 There is growing evidence that histones in NETs may be the cytotoxic component that harms the endothelium and epithelium. Interestingly, the cytotoxic activity of NETs on lung epithelial cells was suppressed after histone, but not DNA inhibition.69 NETs contribute to hepatic damage during bloodstream infection with methicillin-resistant S aureus.70 However, DNase treatment only partially reduced the injury, because DNase failed to remove histones that were attached to the vessel wall via von Willebrand factor. Indeed, S aureus–induced tissue damage was almost abolished in PAD-4−/− and Elane−/−(NE−/−) mice, whose neutrophils are unable to release NETs.70 Corroborating these data, it was demonstrated that during NADPH oxidase-dependent NET formation, elastase degraded the N-terminal histone tail within NETs, which only happened during lytic NET release.48 Moreover, nonlytic NET release exhibited increased immunostimulatory effects on endothelial cells compared with lytic NETs, suggesting that the N-terminal histone tail could be responsible for cytotoxicity to host cells.48 Histones are also involved with the formation of microaggregates in the circulation. The activation of TLR by extracellular histone proteins leads to the generation of thrombin and activation of platelets, resulting in microaggregates that contribute to organ damage.71 Importantly, NETs form clots in the circulation of patients and mice with sepsis, an emerging noncanonical mechanism for vascular occlusion and organ damage.72
The role of neutrophils and NETs during sepsis remains enigmatic. It is well known that the failure of neutrophil migration to the infectious nidus is associated with dissemination and sepsis. However, the systemic activation of neutrophils results in their accumulation in secondary organs, such as lung, leading to bystander organ damage by mechanisms that include NETs.5,39 Therefore, the inhibition of NETs may lead to beneficial and detrimental outcomes. The depletion of NETs by recombinant human DNase delayed bacterial clearance and aggravated the pathology during polymicrobial sepsis in mice.73 Pad4−/− mice exhibited some protection from LPS-induced endotoxemia, suggesting that NETs do cause damage in this model74 ; however, this is not a live microbial infection. Moreover, no protection and no effect on bacteremia were noted from PAD4-deficient mice exposed to polymicrobial sepsis.75 The treatment of mice with DNase or histone-neutralizing antibodies, in association with antibiotics, reduced organ damage and improved survival of mice submitted to polymicrobial sepsis.76,77 In addition, NET-containing HMGB-1 induced peritoneal macrophage pyroptosis (a form of cell death) through the activation of caspase-1.78 Although these studies indicate that the combination of microbicidal agents with inhibitors of NET activity could be a possible strategy to minimize the detrimental tissue damage caused by NETs during sepsis, with so many different toxic products it seems inconceivable that DNase alone would be sufficient to reduce tissue damage.
Little is known about NET clearance, and it is proposed that endogenous DNase simply chops up NETs. However, this could potentially release the remaining toxic NET proteins. Perhaps other mechanisms of clearance exist. Monocyte-derived macrophages from healthy but not ARDS patients efficiently phagocytose NETs and apoptotic neutrophils.78 In addition, activation of AMP-activated protein kinase, which is a metabolic sensor that regulates cellular energy production in macrophages, or neutralization of HMGB1 in bronchial-alveolar lavage fluids improved efferocytosis and NET clearance. This represents an important strategy to limit the exacerbated inflammatory response and organ damage induced by NETs during ARDS.
Chronic inflammation
Several chronic inflammatory diseases are also characterized by a sustained influx of neutrophils, and persistent NET release. During respiratory chronic diseases, such as cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD),80 neutrophils and NETs contribute to the reduction of pulmonary function by blocking airways.81 Chronic infection of the lungs is associated with sputum that is rich in neutrophil proteins and DNA, which are thought to arise from NETs.82 Indeed, persistent NET formation has been found in patients with CF and COPD, which is associated with inflammation and disease severity.80,83-85 Although there is a prevalence of recurrent infection in patients with CF and COPD, whether NETs are detrimental or beneficial remains unclear.86 During CF, P aeruginosa is often present in the lung of patients and can induce NET formation. However, a number of clinical isolates of P aeruginosa have been shown to be resistant to NET-mediated killing, and the NETs appear to contribute to the generation of fibrotic areas in the lung, which may become a replicative niche for the bacteria.87 Neutrophils from CF subjects live longer due to decreased apoptosis and form more NETs.88 Apart from causing direct damage, NETs also provide proinflammatory stimuli to macrophages, boosting inflammation in CF subjects.88 Interestingly, inhaled recombinant DNase treatment helped to solubilize sputum from CF patients and improved lung function in CF mice.89 However, DNase treatment carries the risk of liberating highly active enzymes and toxic molecules like histones, which can damage the lung epithelium.90 Therefore, despite its widespread use to help clear airways, other benefits or detrimental side effects need to be further assessed.
Neutrophils and NETs may also play critical roles during autoimmune diseases.45 These inflammatory diseases are defined as pathological conditions in which the immune system is intolerant to autoantigens, leading to effector mechanisms, such as autoantibodies and autoreactive lymphocytes. This deteriorates tissues progressively and culminates in organ failure and death. NETs may play significant roles in the initiation phase of autoimmune disorders by exposing intracellular endogenous components to the immune system, which exacerbates inflammation or even results in the production of autoantibodies.45 Patients with systemic lupus erythematosus (SLE) and rheumatoid arthritis exhibited elevated levels of NETs in the serum and in the synovial fluid, respectively.51,91,92 Interestingly, those patients but not healthy controls have a distinct low-density granulocyte population in the circulation with enhanced capacity to produce NETs spontaneously. Accordingly, SLE patients have high levels of anti-ribonucleoprotein and anti-DNA antibodies in their serum, and rheumatoid arthritis patients present autoantibodies directed against citrullinated proteins, such as histones.93,94
The role of NETs during SLE is complicated. Blocking mitochondrial ROS production blocked NET formation in a mouse model of lupus and reduced disease severity.53 However, mice that do not express NADPH oxidase exhibit more severe disease.95 This apparent contradiction can be explained by a study showing that circulating NETs from patients with lupus erythematosus are derived from nonlytic NET formation, that form independent of NADPH oxidase.48 This may explain why individuals with chronic granulomatous disease, who fail to produce ROS through NADPH oxidase, might still make NETs and have a propensity for developing SLE. In addition, a subset of patients with lupus has an accumulation of NETs due to a DNase impairment and a reduction in DNA clearance.91 Although some of these diseases clearly suggest neutrophil-derived autoantigens as potential mediators, there are studies with opposing results, and it certainly is unclear whether targeting NETs once the disease is established will be a viable medical intervention.
Neutrophils and tissue repair
It is always worth identifying situations where a process does not occur. Indeed, during healthy wound repair, neutrophils are involved but do not make NETs.96 After the release of DAMPs by damaged cells, neutrophils are recruited to the site of injury, where they remove cellular debris.7 They also harbor enzymatic activity (matrix metalloproteinases that activate vascular endothelial growth factor) that is important for the revascularization of damaged tissues and/or for the recruitment or activation of repair promoting cells. After executing their functions, neutrophils must be cleared either by macrophages that leads to the release of anti-inflammatory cytokines7 or by reentering the vasculature, in a process called reverse migration, homing to the bone marrow, where they are thought to die by apoptosis.97 It is likely critical that neutrophils are not left to linger in these inflammatory sites because this has been shown to lead to poor healing.97 During a normal healing process, neutrophils release very few if any NETs.96 In diabetes, NETs may delay wound healing,98 whereas NET aggregates may actually contribute to the resolution of inflammation by degrading cytokines and chemokines in a murine model of gout.99 This mechanism could account for the spontaneous remission of acute inflammatory attacks elicited by monosodium urate crystals in individuals with gout.99
Conclusion
As such, inhibiting NETs may benefit in some diseases, but we have evolved this system to help us survive, suggesting according to Darwin’s theory of evolution that NET production has been selected for, and as such, the good properties of NETs likely outweigh the bad. Of course, many of the proposed detrimental effects of NETs occur in diseases caused by bad behavior practiced by the past 2 to 3 generations (fat rich diets, smoking, alcohol, etc), making these “nonevolutionary diseases.” Even so, we will need to identify intelligent ways of preventing NET release and not just getting rid of DNA and hoping that magically histones and proteases will also disappear. Finally, it is our view that the host has evolved to make NETs to help trap and kill pathogens, and it is sufficiently important that pathogens have evolved the production of DNases in an attempt to thwart this important antimicrobial defense mechanism. Interfering with this pathway could help pathogens evade host immunity leading to dire consequences.
Acknowledgments
The authors thank Luiz Gustavo Gardinassi and Gustavo Quirino for proofreading the manuscript and Bruna Araujo David for providing assistance with the figure.
This work is supported by grants from the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada, and the Canada Research Chairs program.
Authorship
Contribution: F.V.S.C. and P.K. wrote, discussed, and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paul Kubes, Calvin, Phoebe, and Joan Snyder Institute for Chronic Diseases, University of Calgary, HRIC 3280 Hospital Dr NW, Calgary, AB T2N 4N1, Canada; e-mail: pkubes@ucalgary.ca.
