

Key Points
Anti-CD117 antibody inhibits and depletes normal human HSCs.
Anti-CD117 antibody depletes human MDS cells and facilitates transplantation of normal human HSCs in the MDS xenograft model.

Abstract
The myelodysplastic syndromes (MDS) represent a group of clonal disorders that result in ineffective hematopoiesis and are associated with an increased risk of transformation into acute leukemia. MDS arises from hematopoietic stem cells (HSCs); therefore, successful elimination of MDS HSCs is an important part of any curative therapy. However, current treatment options, including allogeneic hematopoietic cell transplantation (HCT), often fail to ablate disease-initiating MDS HSCs, and thus have low curative potential and high relapse rates. Here, we demonstrate that human HSCs can be targeted and eliminated by monoclonal antibodies (mAbs) that bind cell-surface CD117 (c-Kit). We show that an anti-human CD117 mAb, SR-1, inhibits normal cord blood and bone marrow HSCs in vitro. Furthermore, SR-1 and clinical-grade humanized anti-human CD117 mAb, AMG 191, deplete normal and MDS HSCs in vivo in xenograft mouse models. Anti-CD117 mAbs also facilitate the engraftment of normal donor human HSCs in MDS xenograft mouse models, restoring normal human hematopoiesis and eradicating aggressive pathologic MDS cells. This study is the first to demonstrate that anti-human CD117 mAbs have potential as novel therapeutics to eradicate MDS HSCs and augment the curative effect of allogeneic HCT for this disease. Moreover, we establish the foundation for use of these antibody agents not only in the treatment of MDS but also for the multitude of other HSC-driven blood and immune disorders for which transplant can be disease-altering.
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of related, clonal disorders that affect hundreds of thousands of people and are characterized by ineffective mature blood cell production and increased risk of progression to acute myeloid leukemia (AML). According to the Revised International Prognostic Scoring System (IPSS-R), the standard scoring system used to predict MDS patient survival, median life expectancy, ranges from only 9.6 months in patients with very-high-risk MDS to 8.8 years in patients with very-low-risk disease.1 Currently available therapies rarely confer long-term benefit. Allogeneic hematopoietic cell transplantation (HCT) is the only treatment that can cure MDS and AML arising from MDS, but outcomes from HCT are limited by high rates of relapse, morbidity, and mortality associated with the transplant procedure itself, including toxicities associated with the conditioning regimen as well as graft-versus-host disease.2-6 In the current clinical practice of HCT, elimination of host hematopoietic stem cells (HSCs) is accomplished by chemotherapy and/or radiation-based regimens, all of which have substantial off-target toxicities. Furthermore, because MDS patients are predominantly elderly, HCT-associated toxicities preclude many of these patients from undergoing this potentially life-saving therapy. Hence, there is a pressing need to develop safer and more effective HCT methods for patients with MDS as well as other blood and immune diseases for whom HCT could be beneficial.
We previously established a robust MDS xenograft model, in which human HSCs taken from primary bone marrow (BM) samples from MDS patients are purified by fluorescence-activated cell-sorting (FACS) and transplanted into immunodeficient NOD/SCID/IL2-Rγ null (NSG) newborn mice, recapitulating several aspects of MDS disease phenotype.7 We showed that MDS HSCs produce myeloid progeny susceptible to programmed cell removal via recognition of cell-surface calreticulin by macrophages,7 likely a significant pathological mechanism explaining the cytopenias observed in MDS. We and others also previously showed that HSCs are the disease-initiating cells in MDS, and that these pathogenic clonal MDS HSCs outcompete normal HSCs present in the BM of affected individuals, leaving minimal (<5%) normal HSCs.7-13 Therefore, elimination of MDS HSCs and replacement with unaffected healthy HSCs during allogeneic HCT can result in cure of MDS.
Here, we sought to determine whether human CD117 is a viable target that will safely permit depletion of pathogenic MDS HSCs and allow their replacement by normal HSCs. CD117 (c-Kit) is a cell-surface receptor on HSCs14,15 that, upon interaction with its ligand stem cell factor (SCF), provides important cellular signals for survival, proliferation, and differentiation of HSCs and hematopoietic progenitor cells.16 We previously showed in mice that a monoclonal antibody (mAb) that targets CD117 can be used instead of chemoradiation to prepare hosts for transplant and achieve engraftment of donor HSCs.17,18 A 1-time dose of an anti-mouse CD117 mAb, ACK2, which reportedly blocks SCF binding to CD117, resulted in endogenous HSC depletion and depletion of the downstream CD117-expressing progeny, permitting engraftment of congenic HSCs in immune-deficient Rag2−/− or Rag2−/−IL2Rγc−/− mice without notable toxicity.17 In addition, we and others have shown that, when combined with low-dose irradiation or CD47 blockade, ACK2 can be used to condition immunocompetent wild-type mice and permit engraftment of congenic and allogeneic donor HSCs.18-20 Given these findings, we have developed anti-human CD117 mAbs for human transplantation to treat diverse blood and immune diseases, and show the applicability of these agents for the treatment of MDS.
Methods
Human samples
Normal human BM samples were purchased from AllCells (Alameda, CA). Human umbilical cord blood (UCB) samples and human BM samples from MDS patients were obtained according to institutional review board–approved protocols, with informed consent, where applicable. MDS samples represented the full spectrum of IPSS-R risk groups. Samples were either processed and cryopreserved as mononuclear cell fractions for future analysis or evaluated immediately. Mononuclear cells were isolated using Ficoll-Paque Plus (GE Healthcare, Chicago, IL). Thawed or fresh mononuclear cells were CD34+ enriched using CD34+ microbeads (Miltenyi Biotec, Auburn, CA) on LS magnetic columns (Miltenyi Biotec).
Flow cytometry and FACS
Antibodies to stain human UCB and BM mononuclear cells for analysis and sorting of hematopoietic stem and progenitor subpopulations were as previously described.7,21 Other antibodies included: PERCP/Cy5.5- or BV421- or BV605-conjugated anti-human CD117 104D2 (BioLegend, San Diego, CA). Cells were analyzed and sorted using a FACSAriaII cytometer (BD Biosciences, San Jose, CA). FlowJo software (TreeStar, Ashland, OR) was used to analyze flow cytometry data.
SR-1 production
Assessment of SCF blockade
hSCF-A488 was generated through conjugation of human SCF (R&D Systems, Minneapolis, MN) to Alexa fluorophore 488 using the Alexa Fluor 488 Antibody Labeling kit (Molecular Probes, Eugene, OR) according to the manufacturer’s instructions. SR-1–A647 was generated through similar conjugation of SR-1 to Alexa fluorophore 647 using the Alexa Fluor 647 Antibody Labeling kit (Molecular Probes) according to the manufacturer’s instructions. Flow cytometry was used to assess hSCF-A488 binding to human CD117-expressing fibroblasts or HMC-1 cells. Inhibition of hSCF-A488 binding to human CD117 by SR-1 and other anti-human CD117 mAbs was assessed by first incubating human CD117+ fibroblasts with antibodies, and subsequently staining with hSCF-A488 and observing decreased A488 fluorescence. To confirm binding of anti-human CD117 mAbs, cells were then stained with goat-anti-mouse A647 secondary antibodies (Molecular Probes).
In vitro liquid culture of human HSCs and hematopoietic progenitor cells
In vitro methylcellulose culture of human HSCs and hematopoietic progenitor cells was performed as previously described.24 Briefly, either 10 or 25 immunophenotypic HSCs (lineage-negative [Lin−] CD34+CD38−CD90+CD45RA−)25-28 or 10 immunophenotypic hematopoietic progenitors (Lin−CD34+) from normal human UCB or BM samples were clone sorted by FACS into individual wells of a 96-well round-bottom plates each containing 100 μL of serum-free StemSpan media (StemCell Technologies, Vancouver, BC, Canada), supplemented with 40 μg/mL human low-density lipoprotein (Sigma-Aldrich, Saint Louis, MO) and cytokines (R&D Systems): 100 ng/mL Flt-3 ligand, 100 ng/mL SCF, 50 ng/mL thrombopoietin, 20 ng/mL interleukin 3 (IL-3), and 20 ng/mL IL-6. HSCs treated with mAb were cultured in the presence of various concentrations of mAb as indicated at 37°C, and viable cells were counted under light microscopy on days 1 to 7.
In vitro methylcellulose assay of human HSCs
In vitro methylcellulose culture of human HSCs and hematopoietic progenitor cells was performed as previously described.24 Briefly, FACS-purified human UCB-derived HSCs were plated via plate-sorting with 100 cells per well in methylcellulose supplemented with granulocyte colony-stimulating factor (G-CSF) and incubated with various concentrations of SR-1 as indicated at 37°C. Colony-forming unit–granulocyte macrophage (CFU-GM) colony counts were performed 2 weeks after culture initiation.
Generation of human HSC-xenografted mice
Transplantation of FACS-sorted normal human UCB-derived, normal human BM-derived, and MDS HSCs into NSG pups was performed as previously described.7,21 Briefly, NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) prenatal day 0 (P0) to P3 newborn pups were preconditioned with 100 cGy γ irradiation 4 to 24 hours before transplantation. Cells were suspended in phosphate-buffered saline containing 2% (vol/vol) fetal calf serum and were transplanted IV via the anterior facial vein by using a 30- or 31-gauge needle.
Anti-human CD117 mAb treatment of xenografted mice
Five hundred micrograms of SR-1 or control isotype immunoglobulin G (IgG) antibody were administered IV via retro-orbital injection to xenografted mice on days 1, 3, 5, and 7. Seventy-five micrograms of AMG 191 were administered IV via retro-orbital injection to xenografted mice on day 1. In mice that received a second normal human UCB HSC graft, 500 μg of ACK2 was also administered IV on day 1 to limit mouse hematopoiesis.
Second transplantation of allogeneic human UCB HSCs into antibody-treated xenografted mice
MDS-xenografted mice were treated with SR-1 mAb or control isotype IgG antibody as detailed in “Anti-human CD117 mAb treatment of xenografted mice.” Subsequently, these same animals were transplanted IV via retro-orbital injection 15 days later with 1000 to 5000 FACS-purified UCB-derived HSCs.
Assessment of human chimerism in xenografted mice
Antibodies used for analysis and sorting of BM obtained from xenografted mice were as previously described.7,21 BM from transplanted mice was obtained through femoral aspiration on indicated days or from femurs, tibias, and/or pelvic bones of euthanized mice. Red blood cells were lysed by using hypotonic solution. For analyses and sorting, cells were stained with the appropriate antibody combinations for 30 to 60 minutes on ice, and dead cells were excluded by propidium iodide staining.
Human chimerism from femoral aspirates was measured on day 0 of the second graft administration, which was ∼12 weeks after initial xenografting. Total human chimerism was assessed as human CD45+ cells among total human CD45+ and mouse CD45+ cells. Human B-cell (human CD45+CD19+) and human T-cell (human CD45+CD3+) chimerism were assessed among total human CD45+ and mouse CD45+ cells. Human myeloid (human CD45+CD13/33+) chimerism was assessed as human CD45+CD13/33+ cells among total human CD45+ and mouse CD45+ cells. This measurement was used as the baseline human myeloid chimerism prior to antibody treatment. Human myeloid chimerism from BM aspirates from the contralateral femur was again measured either on day 8 (for SR-1–treated xenografted animals) or day 21 (for AMG 191 treated xenografted animals). This measurement represented the human myeloid chimerism shortly following completion of antibody treatment. Human myeloid chimerism from BM from the femurs, tibias, and pelvis of euthanized mice was measured either 8 weeks, 12 weeks, 20 weeks, 24 weeks, 28 weeks, or 8 months after completion of antibody treatment. In mice that received a second normal human UCB HSC graft, total human chimerism (human CD45+), as well as human myeloid human B-cell (human CD45+CD19+) and human T-cell (human CD45+CD3+) chimerism was also measured at the final time point. Immature cells, which include blasts, were approximated by forward scatter and side scatter and percentage of human CD45+CD34+ among human CD45+ cells.
Fluorescence in situ hybridization
Human CD45+ cells were sorted by FACS directly onto a slide for cytogenetic analysis by interphase fluorescence in situ hybridization (FISH) using Vysis LSI EGFR SpectrumOrange/CEP 7 SpectrumGreen Probe, Vysis CEP 8 SpectrumGreen Probe, Vysis LSI CSF1R SpectrumOrange/D5S23, D52721 SpectrumGreen Probe, Vysis CEP X SpectrumOrange/Y SpectrumGreen Probe, and TelVysion 20q SpectrumOrange/20p SpectrumGreen Probe (Abbott Molecular, Abbott Park, IL), according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis using the Student t test was done using Microsoft Excel and/or GraphPad Prism software.
Results
Normal and MDS human HSCs express CD117 (c-Kit), which is recognized by anti-human CD117 mAb SR-1
To evaluate CD117 as a potential target on HSCs, we examined, by flow cytometry, HSCs (Lin−CD34+CD38−CD90+CD45RA−)25-28 and Lin−CD34+ progenitor cells obtained from BM of healthy individuals and MDS patients. All samples tested from healthy donor and MDS BM, irrespective of IPSS-R score1 or cytogenetics (supplemental Table 1, available on the Blood Web site), stained brightly for CD117 (Figure 1A). Although other groups have reported changes in CD117 antigen clustering or expression, we did not observe significant differences in CD117 expression between healthy and MDS HSCs or CD34+ hematopoietic progenitors. To target CD117 on normal and MDS HSCs, we produced and surveyed several mouse anti-human CD117 mAbs, and determined that clone SR-122,23,29 bound to both normal and MDS human HSCs, and uniquely inhibited binding of SCF to CD117 (Figure 1B; supplemental Figure 1). Therefore, given the ability of SR-1 to block SCF binding to CD117, we elected to use SR-1 in our subsequent studies.
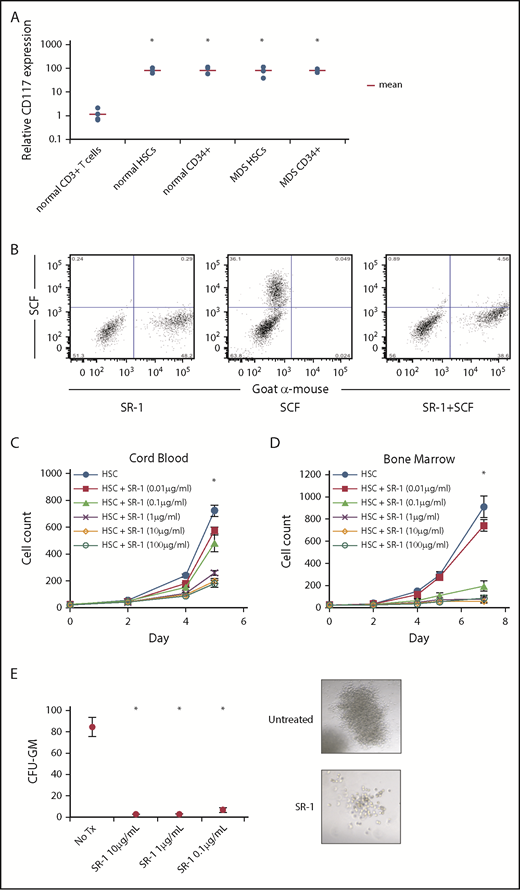
Anti-human CD117 mAb SR-1 blocks SCF binding to CD117 on human HSCs and inhibits human HSC proliferation in vitro. (A) Relative CD117 expression of HSCs (Lin−CD34+CD38−CD90+CD45RA−) and progenitors (Lin−CD34+) from normal BM samples and MDS BM samples from all IPSS-R risk groups. CD117 expression was assessed by mean fluorescence intensity (MFI) by flow cytometry using a commercially available anti-human CD117 mAb, clone 104D2. MFI was normalized to BM CD3+ T cells from healthy subjects, which served as staining control. *P < .001 compared with control CD3+ T cells (Student t test). (B) Pretreatment with SR-1 mAb inhibits binding of fluorescently labeled SCF (SCF-A488) to human CD117. A mixture of CD117-expressing and untransfected fibroblasts were stained with SR-1 mAb (left), SCF-A488 alone (center), or first stained with SR-1 and then subsequently stained with SCF-A488 (right). SR-1 mAb was detected using a fluorescently labeled goat-anti-mouse secondary mAb. (C) SR-1 mAb inhibits proliferation of FACS-purified human UCB-derived HSCs (Lin−CD34+CD38−CD90+CD45RA−) in liquid culture. UCB HSCs were plated via plate-sorting at a density of 25 cells per well in StemSpan media supplemented with SCF, TPO, FLT3L, IL-3, and IL-6, and treated with varying concentrations of SR-1. Cell counts were performed on days 2, 4, and 5 postplating. *P < .001 comparing 100 μg/mL, 10 μg/mL, and 1 μg/mL conditions compared with untreated on day 7 (Student t test). Error bars indicate 1 standard error of the mean. (D) SR-1 mAb inhibits proliferation of FACS-purified human BM-derived HSCs (Lin−CD34+CD38−CD90+CD45RA−) in liquid culture. BM HSCs were plated via plate sorting at a density of 25 cells per well in StemSpan media supplemented with SCF, TPO, FLT3L, IL-3, and IL-6, and treated with varying concentrations of SR-1. Cell counts were performed on days 2, 4, 5, and 7 postplating. *P < .001 comparing 100 μg/mL, 10 μg/mL, 1 μg mL, and 0.1 μg/mL conditions compared with untreated on day 7 (Student t test). Error bars indicate 1 standard error of the mean. (E) SR-1 mAb inhibits colony formation of FACS-purified human UCB-derived HSCs (Lin−CD34+CD38−CD90+CD45RA−) in methylcellulose supplemented with G-CSF and treated with indicated concentrations of SR-1. CFU-GM colony counts were performed 2 weeks after culture initiation. Representative images of the colonies present at 2 weeks are shown (for untreated and 1 μg/mL SR-1). *P < .001 compared with day 0 (Student t test). Error bars indicate 1 standard error of the mean.
Anti-human CD117 mAb SR-1 blocks SCF binding to CD117 on human HSCs and inhibits human HSC proliferation in vitro. (A) Relative CD117 expression of HSCs (Lin−CD34+CD38−CD90+CD45RA−) and progenitors (Lin−CD34+) from normal BM samples and MDS BM samples from all IPSS-R risk groups. CD117 expression was assessed by mean fluorescence intensity (MFI) by flow cytometry using a commercially available anti-human CD117 mAb, clone 104D2. MFI was normalized to BM CD3+ T cells from healthy subjects, which served as staining control. *P < .001 compared with control CD3+ T cells (Student t test). (B) Pretreatment with SR-1 mAb inhibits binding of fluorescently labeled SCF (SCF-A488) to human CD117. A mixture of CD117-expressing and untransfected fibroblasts were stained with SR-1 mAb (left), SCF-A488 alone (center), or first stained with SR-1 and then subsequently stained with SCF-A488 (right). SR-1 mAb was detected using a fluorescently labeled goat-anti-mouse secondary mAb. (C) SR-1 mAb inhibits proliferation of FACS-purified human UCB-derived HSCs (Lin−CD34+CD38−CD90+CD45RA−) in liquid culture. UCB HSCs were plated via plate-sorting at a density of 25 cells per well in StemSpan media supplemented with SCF, TPO, FLT3L, IL-3, and IL-6, and treated with varying concentrations of SR-1. Cell counts were performed on days 2, 4, and 5 postplating. *P < .001 comparing 100 μg/mL, 10 μg/mL, and 1 μg/mL conditions compared with untreated on day 7 (Student t test). Error bars indicate 1 standard error of the mean. (D) SR-1 mAb inhibits proliferation of FACS-purified human BM-derived HSCs (Lin−CD34+CD38−CD90+CD45RA−) in liquid culture. BM HSCs were plated via plate sorting at a density of 25 cells per well in StemSpan media supplemented with SCF, TPO, FLT3L, IL-3, and IL-6, and treated with varying concentrations of SR-1. Cell counts were performed on days 2, 4, 5, and 7 postplating. *P < .001 comparing 100 μg/mL, 10 μg/mL, 1 μg mL, and 0.1 μg/mL conditions compared with untreated on day 7 (Student t test). Error bars indicate 1 standard error of the mean. (E) SR-1 mAb inhibits colony formation of FACS-purified human UCB-derived HSCs (Lin−CD34+CD38−CD90+CD45RA−) in methylcellulose supplemented with G-CSF and treated with indicated concentrations of SR-1. CFU-GM colony counts were performed 2 weeks after culture initiation. Representative images of the colonies present at 2 weeks are shown (for untreated and 1 μg/mL SR-1). *P < .001 compared with day 0 (Student t test). Error bars indicate 1 standard error of the mean.
Anti-CD117 mAb SR-1 inhibits normal human HSC proliferation in vitro
The effect of SR-1 on hematopoiesis in vitro was examined by co-incubating FACS-purified human UCB or BM HSCs with SR-1 in liquid culture.24 SR-1, but not another anti-CD117 mAb (4F7), inhibited proliferation of HSCs and progenitors in a dose-dependent manner (Figure 1C-D; supplemental Figure 2), suggesting that in vitro inhibition of HSC proliferation by SR-1 is determined by its ability to block SCF binding to CD117. SR-1 also reduced the number and size of colonies formed by UCB HSCs in methylcellulose (Figure 1E). SR-1 did not induce significant apoptosis of UCB HSCs after 3 and 7 days of culture (supplemental Figure 3A). Addition of caspase inhibitor Z-VAD-FMK also did not abrogate inhibition of proliferation by SR-1 (supplemental Figure 3B), suggesting that a functional caspase pathway is not necessary for the inhibitory activity of SR-1.
Anti-CD117 mAb SR-1 depletes normal human HSCs in vivo
To examine whether SR-1 can deplete human HSCs in vivo, stably engrafted human-mouse hematopoietic chimeras were generated by transplanting >1000 purified human UCB HSCs into sublethally irradiated newborn NSG mice. BM aspirates were obtained from these animals and assayed for human chimerism by flow cytometry to establish baseline engraftment. Mice with >15% human CD45+ chimerism in the BM at 12 weeks posttransplant were selected for further experimentation. The average pretreatment baseline level of total human BM engraftment (percentage of human CD45+) was 58.6%, with myeloid chimerism (percentage of human CD13/33+) of 25.4% (supplemental Figure 4A). These mice also exhibited robust engraftment by human B cells and T cells as well (supplemental Figure 4A), as has been previously reported with this model.24 These humanized mice were treated IV with 500 μg of SR-1 every other day for 1 week. SR-1 treatment resulted in significant depletion of human CD45+CD13/33+ cells (Figure 2A; supplemental Figure 4B). Consistent with depletion of human HSCs, human myeloid engraftment 8 weeks posttreatment remained suppressed at a decrease of 96.9% from pretreatment levels. The persisting human cells were primarily mature B cells and T cells (supplemental Figure 4C), populations that do no express CD117 and are not constantly maintained by HSCs, and thus were unaffected by this therapy. Collectively, these data show that SR-1 can effectively deplete human HSCs, and suggest that anti-human CD117 mAbs may be useful, targeted conditioning agents for HCT.
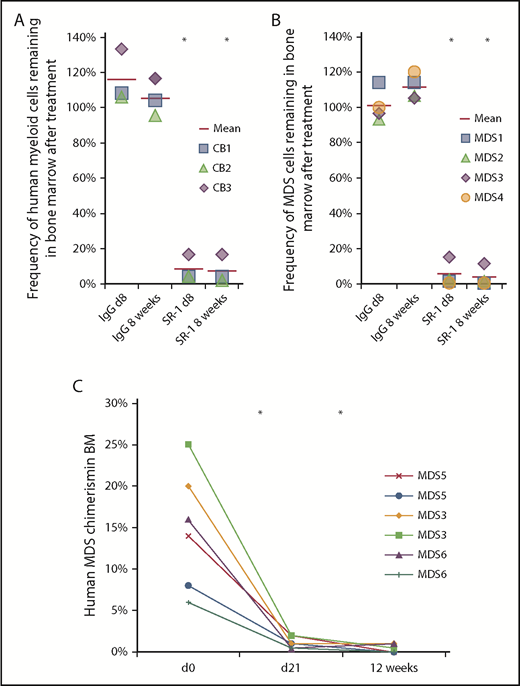
Anti-human CD117 antibodies deplete normal and MDS HSCs in vivo. (A) SR-1 depletes normal human UCB-derived HSCs in vivo, as shown by percent depletion from baseline of human myeloid (human CD45+CD13/33+) chimerism on day 8 and 8 weeks after completion of SR-1 treatment; baseline human myeloid chimerism was determined on day 0, prior to treatment. FACS-purified human UCB HSCs (Lin−CD34+CD38−CD90+CD45RA−) were transplanted into sublethally irradiated NSG pups and baseline myeloid chimerism was obtained on day 0 of the second graft administration, which was ∼12 weeks after initial establishment of xenografts. Xenografted mice were treated with 500 μg of SR-1 or control isotype IgG antibody administered IV on day 1, day 3, day 5, and day 7. *P < .001 compared with IgG control at same time point (Student t test) (n = 3, IgG treated; n = 3, SR-1 treated). (B) SR-1 depletes low-risk MDS HSCs in vivo, as shown by percent depletion from baseline of human myeloid (human CD45+CD13/33+) chimerism on day 8 and 8 weeks after completion of SR-1 treatment; baseline human myeloid chimerism was determined on day 0, prior to treatment. FACS-purified low-risk MDS HSCs (Lin−CD34+CD38−CD90+CD45RA−) were transplanted into sublethally irradiated NSG pups and baseline myeloid chimerism was obtained on day 0 of the second graft administration, which was ∼12 weeks after initial establishment of xenografts. Xenografted mice were treated with 500 μg of SR-1 or control isotype IgG antibody administered IV on day 1, day 3, day 5, and day 7. *P < .001 compared with IgG control at same time point (Student t test) (n = 4, IgG treated; n = 4, SR-1 treated). (C) AMG 191 depletes low-risk MDS HSCs in vivo, as shown by human myeloid (human CD45+CD13/33+) chimerism on prior to treatment on day 0 and then at day 21 and 12 weeks after completion of treatment with AMG 191. FACS-purified low-risk MDS HSCs (Lin−CD34+CD38−CD90+CD45RA−) were transplanted into sublethally irradiated NSG pups and baseline myeloid chimerism was obtained on “day 0,” which was ∼12 weeks after initial establishment of xenografts. Xenografted mice were treated with 75 μg of AMG 191 administered IV on day 1. *P < .001 compared with pretreatment day 0 (Student t test; n = 6, AMG 191 treated).
Anti-human CD117 antibodies deplete normal and MDS HSCs in vivo. (A) SR-1 depletes normal human UCB-derived HSCs in vivo, as shown by percent depletion from baseline of human myeloid (human CD45+CD13/33+) chimerism on day 8 and 8 weeks after completion of SR-1 treatment; baseline human myeloid chimerism was determined on day 0, prior to treatment. FACS-purified human UCB HSCs (Lin−CD34+CD38−CD90+CD45RA−) were transplanted into sublethally irradiated NSG pups and baseline myeloid chimerism was obtained on day 0 of the second graft administration, which was ∼12 weeks after initial establishment of xenografts. Xenografted mice were treated with 500 μg of SR-1 or control isotype IgG antibody administered IV on day 1, day 3, day 5, and day 7. *P < .001 compared with IgG control at same time point (Student t test) (n = 3, IgG treated; n = 3, SR-1 treated). (B) SR-1 depletes low-risk MDS HSCs in vivo, as shown by percent depletion from baseline of human myeloid (human CD45+CD13/33+) chimerism on day 8 and 8 weeks after completion of SR-1 treatment; baseline human myeloid chimerism was determined on day 0, prior to treatment. FACS-purified low-risk MDS HSCs (Lin−CD34+CD38−CD90+CD45RA−) were transplanted into sublethally irradiated NSG pups and baseline myeloid chimerism was obtained on day 0 of the second graft administration, which was ∼12 weeks after initial establishment of xenografts. Xenografted mice were treated with 500 μg of SR-1 or control isotype IgG antibody administered IV on day 1, day 3, day 5, and day 7. *P < .001 compared with IgG control at same time point (Student t test) (n = 4, IgG treated; n = 4, SR-1 treated). (C) AMG 191 depletes low-risk MDS HSCs in vivo, as shown by human myeloid (human CD45+CD13/33+) chimerism on prior to treatment on day 0 and then at day 21 and 12 weeks after completion of treatment with AMG 191. FACS-purified low-risk MDS HSCs (Lin−CD34+CD38−CD90+CD45RA−) were transplanted into sublethally irradiated NSG pups and baseline myeloid chimerism was obtained on “day 0,” which was ∼12 weeks after initial establishment of xenografts. Xenografted mice were treated with 75 μg of AMG 191 administered IV on day 1. *P < .001 compared with pretreatment day 0 (Student t test; n = 6, AMG 191 treated).
Anti-CD117 mAbs SR-1 and AMG 191 deplete MDS cells in vivo
To examine the capability of SR-1 to deplete MDS HSC in vivo, we generated mice xenografted with primary BM samples obtained from patients with very-low-risk or low-risk MDS (designated as “low-risk MDS”) as well as patients with high-risk and very-high-risk disease (designated as “high-risk MDS”), as classified by the IPSS-R1 (supplemental Table 1). Newborn NSG mice were sublethally irradiated and transplanted with >1000 FACS-purified MDS HSCs. These HSCs from MDS patients engraft in NSG mice and recapitulate salient features of the disease, including the inability to generate lymphoid progeny, and produce only myeloid progeny predominantly from the abnormal MDS clone (supplemental Figure 5). Consistent with our previous findings, >90% of xenografted MDS human cells were derived from the mutant clone7 and this predominance was independent of cytogenetic abnormality (supplemental Table 1). BM aspirates obtained at 12 weeks posttransplant were assessed for the presence of human cells by flow cytometry. The human cells were further purified by FACS and analyzed by FISH to determine the frequency of the clonal MDS-associated cytogenetic (−Y, del(20q), or +8) abnormalities (supplemental Figure 5B). The blood of MDS-xenografted mice had only human myeloid cells and no lymphoid cells (supplemental Figure 5C). Immature cells, including blasts, were not increased in these low-risk MDS HSC-engrafted mice (supplemental Figure 5D).
Low-risk MDS-xenografted mice with >4% human CD45+ BM chimerism at 12 weeks were selected for antibody treatment. Mice received either 500 μg of SR-1 or control isotype IgG antibody IV every other day for 1 week. Dosage was selected to maximize the effect, as a full-dose titration was not possible due to scarcity of patient samples. At this dosing, SR-1 robustly depleted cells from low-risk MDS subtypes, as demonstrated by marked reduction of human CD45+ chimerism (Figure 2B; supplemental Figure 6A). In contrast, control isotype IgG antibody treatment had no significant effect (Figure 2B; supplemental Figure 6A). The depletion of human cells from the low-risk MDS group was persistent, as sustained depletion was noted at 8 weeks and 8 months post–SR-1 treatment (supplemental Figure 6B).
In a separate set of studies, we evaluated the ability of AMG 191, a humanized mAb of SR1, to deplete low-risk MDS HSCs in vivo. AMG 191 similarly binds to and inhibits human HSC proliferation in vitro, and depletes normal human UCB HSCs in xenografted mice.30 Here, mice engrafted with HSCs derived from low-risk MDS patients were treated with AMG 191 at a single IV dose of 75 μg. This dosage was selected based on pharmacokinetic studies performed in NSG mice.30 Similar to SR-1, AMG 191 depleted low-risk MDS cells in vivo, as evidenced by significant reduction of human CD45+ chimerism in the BM 3 weeks after treatment (Figure 2C). Depletion of low-risk MDS cells was sustained as determined by analyses performed at 12 weeks after AMG 191 treatment (Figure 2C). Taken together, these data show that targeting human HSCs and hematopoietic progenitors with either anti-human CD117 mAb SR-1 or AMG 191 significantly depletes low-risk MDS HSCs in the xenografted mice and that this depletion is maintained long-term. Nevertheless, depletion is incomplete, as minimal residual MDS cells persist despite anti-CD117 mAb treatment, and it is also possible that there are residual HSCs that are not part of the dominant MDS clone detectable by FISH but carry other MDS-associated mutations.
Anti-CD117 mAb SR-1 permits engraftment of normal HSCs after depleting HSCs from low-risk MDS subtypes in vivo
We next tested whether SR-1 can be used as to prepare mice for transplant and permit engraftment of normal HSCs in MDS-xenografted mice, analogous to an allogeneic HCT in humans. MDS-xenografted mice with >10% total chimerism (all xenografted human cells were of myeloid CD13/33+ cells) at 8 weeks posttransplantation were used. SR-1 or control isotype IgG antibody (500 μg given every other day for 1 week) was injected into mice bearing HSC grafts from low-risk MDS patients. One week after completion of antibody treatment, mice were transplanted with 1000 FACS-purified normal human UCB HSCs. To limit endogenous murine HSCs from filling the empty HSC niches, mice were concurrently treated with anti-mouse CD117 mAb ACK2. Twelve weeks after transplantation of human UCB HSCs, mice treated with SR-1, but not mice treated with control isotype IgG antibody, generated human myeloid and lymphoid cells in the BM (Figure 3A), indicative of engraftment of healthy human HSCs. To confirm the non-MDS origin of these cells, human CD45+ cells from these mice were FACS-isolated and analyzed for clonal cytogenetic (−Y, del(20q), or +8) abnormalities by FISH. Predominantly cytogenetically normal human cells were found (>97% of human cells) in the low-risk MDS-xenografted mice treated with SR-1 and subsequently transplanted with human UCB HSCs (Figure 3B; supplemental Table 2). In contrast, in MDS-xenografted mice treated with control isotype IgG antibody and transplanted with normal human UCB HSCs, abnormal MDS cells comprised >91% of human cells (Figure 3B; supplemental Table 2). As a control, low-risk MDS-xenografted mice treated with SR-1 and ACK2, but not transplanted with normal human UCB HSCs, demonstrated sustained depletion of the MDS cells up to 8 months after completion of antibody treatment (supplemental Figure 6C). These data indicate that anti-CD117 mAb SR-1 not only depletes low-risk MDS HSCs but also sufficiently clears HSC niches, allowing subsequent engraftment of healthy human HSCs and restoration of normal human hematopoiesis.
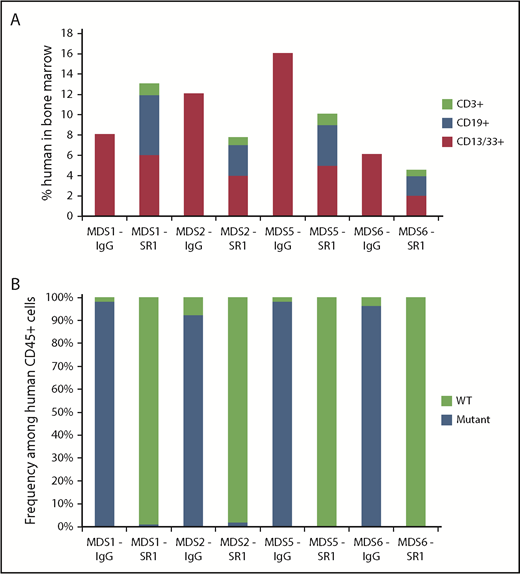
SR-1 permits engraftment of second human UCB HSC graft in low-risk MDS-xenografted mice. (A) Total human chimerism (human CD45+) as well as distribution of myeloid (human CD45+CD13/33+), B-cell (human CD45+CD19+), and T-cell (human CD45+CD3+) chimerism in BM of low-risk MDS-xenografted mice treated with control isotype IgG antibody or SR-1 mAb on day 1, 3, 5, and 7, and then transplanted on day 15 with a second normal human UCB HSC graft. Chimerism was measured 12 weeks after second normal human UCB HSC transplant (n = 4, IgG treated; n = 4, SR-1 treated). (B) Frequency of cytogenetically abnormal clones (−Y, del(20q), or +8), as detected by FISH within human cells FACS-isolated from BM of low-risk MDS-xenografted mice treated with IgG control antibody or SR-1 mAb on days 1, 3, 5, and 7 and then transplanted on day 15 with a second normal human UCB HSC graft. Cells were analyzed 12 weeks after second normal human UCB HSC transplant (n = 4, IgG treated; n = 4, SR-1 treated).
SR-1 permits engraftment of second human UCB HSC graft in low-risk MDS-xenografted mice. (A) Total human chimerism (human CD45+) as well as distribution of myeloid (human CD45+CD13/33+), B-cell (human CD45+CD19+), and T-cell (human CD45+CD3+) chimerism in BM of low-risk MDS-xenografted mice treated with control isotype IgG antibody or SR-1 mAb on day 1, 3, 5, and 7, and then transplanted on day 15 with a second normal human UCB HSC graft. Chimerism was measured 12 weeks after second normal human UCB HSC transplant (n = 4, IgG treated; n = 4, SR-1 treated). (B) Frequency of cytogenetically abnormal clones (−Y, del(20q), or +8), as detected by FISH within human cells FACS-isolated from BM of low-risk MDS-xenografted mice treated with IgG control antibody or SR-1 mAb on days 1, 3, 5, and 7 and then transplanted on day 15 with a second normal human UCB HSC graft. Cells were analyzed 12 weeks after second normal human UCB HSC transplant (n = 4, IgG treated; n = 4, SR-1 treated).
Anti-CD117 mAb SR-1 transiently depletes cells from high-risk MDS subtypes in vivo
We next evaluated whether SR-1 could effectively deplete transplanted MDS HSCs from high-risk MDS patients. Unlike the sustained response seen with SR-1 treatment of low-risk MDS, mice xenografted with HSCs from high-risk MDS patients (>5% blasts) achieved only transient depletion of human cells with SR-1 treatment (Figure 4A), and no depletion with IgG control antibody treatment (supplemental Figure 7A). Recovery of human myeloid cells in the BM was uniformly evident by 8 weeks following exposure to SR-1 (Figure 4A), and the recovering human cells were derived entirely from the abnormal MDS clone (supplemental Figure 7B). Blasts, as assessed by percentage of CD34+ cells among human CD45+ cells, also rebounded by 8 weeks following SR-1 treatment (supplemental Figure 7C), suggesting that anti-CD117 mAb treatment alone will likely be ineffective as stand-alone therapy for high-risk MDS patients. We have previously shown that CD47 expression is elevated in high-risk MDS cells compared with low-risk MDS cells,7 and this upregulation of CD47 could be a potential evasion mechanism enabling high-risk MDS HSCs to recover after antibody treatment.

SR-1 transiently depletes high-risk MDS cells and permits engraftment of second human UCB HSC graft in high-risk MDS-xenografted mice. (A) SR-1 transiently depletes human blood cells derived from high-risk MDS HSCs in vivo, as shown by chimerism of human myeloid (hCD13/CD33+) cells on pretreatment day 0 and 8 days and 8 weeks after completion of treatment with 500 μg of SR-1 administered IV on days 1, 3, 5, and 7. *P < .001 compared with day 0 (Student t test) (n = 4, SR-1 treated). (B) Total human chimerism (human CD45+) as well as distribution of myeloid (human CD45+CD13/33+), B-cell (human CD45+CD19+), and T-cell (human CD45+CD3+) chimerism in BM of high-risk MDS-xenografted mice treated with SR-1 mAb on days 1, 3, 5, and 7, and then transplanted on day 15 with a second normal human UCB HSC graft. Chimerism was measured 12 weeks after second normal human UCB HSC transplant (n = 4, SR-1 treated). (C) Frequency of cytogenetically abnormal clone (−7, del(5q), or +8), as detected by FISH within human cells FACS-isolated from BM of high-risk MDS-xenografted mice treated with SR-1 mAb on days 1, 3, 5, and 7 and then transplanted on day 15 with a second normal human UCB HSC graft. Cells were analyzed 12 weeks after second normal human UCB HSC transplant (n = 4, SR-1 treated).
SR-1 transiently depletes high-risk MDS cells and permits engraftment of second human UCB HSC graft in high-risk MDS-xenografted mice. (A) SR-1 transiently depletes human blood cells derived from high-risk MDS HSCs in vivo, as shown by chimerism of human myeloid (hCD13/CD33+) cells on pretreatment day 0 and 8 days and 8 weeks after completion of treatment with 500 μg of SR-1 administered IV on days 1, 3, 5, and 7. *P < .001 compared with day 0 (Student t test) (n = 4, SR-1 treated). (B) Total human chimerism (human CD45+) as well as distribution of myeloid (human CD45+CD13/33+), B-cell (human CD45+CD19+), and T-cell (human CD45+CD3+) chimerism in BM of high-risk MDS-xenografted mice treated with SR-1 mAb on days 1, 3, 5, and 7, and then transplanted on day 15 with a second normal human UCB HSC graft. Chimerism was measured 12 weeks after second normal human UCB HSC transplant (n = 4, SR-1 treated). (C) Frequency of cytogenetically abnormal clone (−7, del(5q), or +8), as detected by FISH within human cells FACS-isolated from BM of high-risk MDS-xenografted mice treated with SR-1 mAb on days 1, 3, 5, and 7 and then transplanted on day 15 with a second normal human UCB HSC graft. Cells were analyzed 12 weeks after second normal human UCB HSC transplant (n = 4, SR-1 treated).
Anti-CD117 mAb SR-1 permits stable engraftment of normal HSCs after depleting high-risk MDS HSCs
Because SR-1 alone was insufficient to yield long-lasting depletion of high-risk MDS clones, we tested whether anti-CD117 treatment in conjunction with transplantation of normal allogeneic HSCs could prevent the return of MDS cells. Mice bearing high-risk MDS xenografts were treated with SR-1 concurrently with anti-mouse CD117 mAb ACK2. One week after completion of SR-1 treatment, xenografted mice were transplanted with 3000 to 5000 allogeneic UCB HSCs. At 12 weeks posttransplantation, BM aspirates showed normal multilineage hematopoiesis (Figure 4B). Human CD45+ cells from the BM aspirates were purified by FACS and analyzed by FISH to determine the frequency of the MDS clones. The human cells present in SR-1–treated xenografts were predominantly (>95%) cytogenetically normal, lacking the cytogenetic markers of the primary MDS (Figure 4C). However, low numbers of abnormal MDS cells remained at detectable levels (Figure 4C), suggesting an incomplete clearance of MDS cells. Nevertheless, the MDS clones remained stably reduced even after 18 weeks posttransplantation (supplemental Figure 8). High-risk MDS-xenografted mice treated with control IgG antibody and transplanted with allogeneic UCB HSCs had persistent MDS cells and did not demonstrate any engraftment of normal HSCs (supplemental Figure 9). Thus, the combination of antibody-mediated depletion followed by transplantation of allogeneic UCB HSCs resulted in long-term suppression and/or eradication of MDS HSCs from both low-risk and high-risk groups, and sustained production of normal human hematopoiesis.
Discussion
In this study, we show that anti-CD117 mAbs inhibit human HSC proliferation in vitro and deplete normal human HSCs in humanized mice, positioning such antibodies as next-generation targeted conditioning agents. Importantly, we also show that MDS disease-initiating HSCs express CD117, and that anti-CD117 mAbs can target and eradicate these pathogenic cells. In vitro assessment of candidate therapeutic anti-CD117 mAbs performed here by us and previously by others29 single out clone SR-1 as potently inhibitory of hematopoietic cell proliferation. Through binding to CD117, SR-1 has been shown to block SCF’s interaction with this receptor, inhibiting SCF-mediated receptor internalization and decreasing autophosphorylation, thereby inhibiting SCF-dependent proliferation.29 Humanized SR-1, AMG 191, which has also been demonstrated to have similar in vitro effects, is aglycosylated,31 and therefore unable to be recognized by Fc receptors and promote downstream effector functions including antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Yet, AMG 191 is still capable of depleting MDS HSCs (and normal human HSCs30 ) in our in vivo xenograft model. Given the previously published findings on SR-1,29 as well as our current findings, we hypothesize that 1 likely mechanism of action by which these antibodies deplete normal and MDS HSCs is by impeding binding of SCF to CD117, leading to decreased CD117 internalization and decreased CD117 autophosphorylation.
The unmet medical need for better treatments for MDS and secondary AML is immense. The cure rate for these disorders remains dismal because current therapies, including HCT-conditioning regimens, often do not eradicate MDS HSCs,32 which harbor relative resistance to chemotherapy,10,12 and these treatments have off-target activities preventing their use in older and/or fragile individuals who comprise the majority of patients affected by MDS. Development of anti-CD117 targeted agents, which can be safely used for the eradication of MDS clones would represent a major step forward for this disease. Our studies demonstrate that the efficacy of anti-CD117 mAbs for the treatment of MDS will be optimized by using these agents in conjunction with HSC replacement as afforded by an allogeneic HCT. We and others previously showed that the HSC population in MDS patients is composed almost entirely of abnormal MDS HSCs and very few, if any, detectable healthy HSCs remain.7-12,33,34 Although the exact contributions to the maintenance of hematopoiesis by individual cells within the HSC pool remains controversial, in a diseased setting such as MDS where the vast majority of HSCs and progenitors are abnormal, healthy HSCs are likely critical to reestablishing functional hematopoiesis after any form of treatment, including HCT.
Sustained suppression and, in some cases, apparent eradication of MDS clones was achieved by transplantation of allogeneic human UCB HSCs following the anti-CD117 mAb treatment. That said, in mice bearing xenografts from high-risk patients, MDS cells remained detectable, albeit at low levels, after mAb treatment and transplantation of a second normal HSC graft, suggesting that not all MDS HSCs are eliminated by this therapeutic strategy. Possible mechanisms to explain why MDS cells are unable to recover after SR-1 treatment in the second normal human UCB HSC-transplanted animals include: (1) UCB HSCs occupy the human HSC niches thereby excluding and outcompeting MDS HSCs and/or (2) functional immune cells derived from the allogeneic UCB HSC graft suppress MDS HSC-driven hematopoiesis. It is possible that MDS cells could eventually overtake the second human HSC allografts, if given sufficient time. Alternative strategies we are pursuing to enhance elimination of MDS HSCs include generating directly cytotoxic antibody-drug conjugates or combining anti-CD117 mAbs with other agents, such as anti-CD47 mAbs, which we have shown to enhance anti-CD117–mediated clearance of HSCs.18
Notably, in our xenograft studies, following anti-CD117 mAb treatment, successful suppression of MDS was accomplished by transplantation of grafts composed of purified allogeneic human HSCs, which are depleted of human T cells that cause graft-versus-host disease.35-37 The transplantation of purified allogeneic HSCs appeared to suppress the MDS HSCs, without reliance on the direct presence of alloreactive T cells from the donor graft. Our results suggest that it may be possible to use anti-CD117 mAb conditioning in HCT to permit engraftment of purified donor HSCs that can outcompete the MDS HSC clones, thereby precluding the development of graft-versus-host disease in HCT recipients. On the other hand, addition of alloreactive T cells to the normal HSC graft, in particular T cells that can specifically target MDS cells, could provide additional graft-versus-tumor effects, potentially enhancing MDS HSC eradication and/or continued MDS HSC suppression.
In parallel ongoing studies, we have found that clinical-grade AMG 191 mAb is similarly effective at depleting normal human and nonhuman primate HSCs, and facilitates engraftment of allogeneic HSCs.30 Therefore, we anticipate our findings using SR-1 against MDS will be directly translatable to study treatment of MDS patients using AMG 191 or similar agents. Taken together, our studies show that CD117 is a novel therapeutic target that can be harnessed to deplete normal and abnormal disease-causing HSCs and allow the engraftment of healthy donor HSCs. The addition of anti-CD117 mAbs as agents to target HSCs has the potential not only to enhance safely the curative potential HCT for MDS and secondary AML, but also to improve HCT outcomes and broaden the use of HCT to the many other blood and immune disorders of HSC origin, where current conditioning regimens significantly limit the use of this therapeutic strategy.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank members of the Shizuru and Weissman laboratories for helpful advice, critical discussion, and technical assistance. The authors especially thank Hye-sook Kwon, Charles Chan, and Ravindra Majeti for helpful advice and discussions; Theresa Storm and Libuse Jerabek for excellent laboratory management; Steven Jungers, Aaron McCarty, Adriane Mosley, and Allison Rainey for animal husbandry and assistance with animal immunizations; Susan Prohaska for facilitating testing with AMG 191; and Chrissy Muscat, Teja Naik, Adriel Cha, and Matt Inlay for antibody production and generation assistance. The authors thank the AMG 191 team at Amgen Inc for their important insights on study design, review of the manuscript, supply of AMG 191, and continued support for these studies. The authors also thank the Stanford Hematology Tissue Bank and Parveen Abidi.
This work was supported by: the Walter V. and Idun Berry Postdoctoral Fellowship (W.W.P.), Howard Hughes Medical Institute Medical Research Training Fellowship (A.C.), Stanford University Medical Scholars Fellowship (A.C.), The Paul and Daisy Soros Fellowship for New Americans (A.C.), the Virginia and D. K. Ludwig Fund for Cancer Research (J.A.S. and I.L.W.), the California Institute for Regenerative Medicine (grants RT3-07683 [I.L.W. and J.A.S.] and DR2A-05365 [J.A.S.]), the National Institutes of Health (National Cancer Institute grant R01 CA86065 and National Heart, Lung, and Blood Institute grant R01HL058770) (I.L.W.), the Gunn/Olivier Research Fund (I.L.W. and J.A.S.), the H. L. Snyder Medical Foundation (J.A.S.), and the Stinehart-Reed Foundation (I.L.W. and J.A.S.).
Authorship
Contribution: W.W.P., A.C., and C.Y.P. conceived the project, designed the studies, performed the experiments, and analyzed the data involving normal human HSCs; A.C.L. designed the studies, performed experiments, and analyzed the data involving normal human HSCs; W.W.P. conceived the project, designed the studies, performed the experiments, and analyzed the data involving MDS HSCs; J.P. and R.B. performed experiments and provided technical assistance; C.Y.P. provided some of the MDS samples; W.W.P. wrote the manuscript; A.C., A.C.L., and C.Y.P. edited the manuscript; and I.L.W. and J.A.S. supervised the project and edited the manuscript.
Conflict-of-interest disclosure: I.L.W. and A.C. are inventors on patents that include the use of anti-CD117 antibodies in HCT conditioning. I.L.W. and J.A.S. are inventors on patents that pair anti-CD47 agents and anti-CD117 antibodies for HCT conditioning. I.L.W. is a cofounder, stockholder, and Director of Forty Seven, Inc, which has licensed these patents from Stanford University. J.A.S. has equity ownership in Forty Seven, Inc. A.C. has equity ownership in Forty Seven, Inc; Magenta Therapeutics; Beam Therapeutics; Editas Medicines; and Global Blood Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Judith A. Shizuru, Stanford University, 269 Campus Dr West #2205a, Stanford, CA 94305; e-mail: jshizuru@stanford.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal