

Key Points
FXII with Lys/Arg substitutions for Thr309 can be cleaved by thrombin and FXIa to generate the truncated species δFXII.
δFXII activation by kallikrein is markedly enhanced when compared with FXII, increasing kininogen cleavage in plasma and in vivo.

Abstract
The plasma proteins factor XII (FXII) and prekallikrein (PK) undergo reciprocal activation to the proteases FXIIa and kallikrein by a process that is enhanced by surfaces (contact activation) and regulated by the serpin C1 inhibitor. Kallikrein cleaves high-molecular-weight kininogen (HK), releasing the vasoactive peptide bradykinin. Patients with hereditary angioedema (HAE) experience episodes of soft tissue swelling as a consequence of unregulated kallikrein activity or increased prekallikrein activation. Although most HAE cases are caused by reduced plasma C1-inhibitor activity, HAE has been linked to lysine/arginine substitutions for Thr309 in FXII (FXII-Lys/Arg309). Here, we show that FXII-Lys/Arg309 is susceptible to cleavage after residue 309 by coagulation proteases (thrombin and FXIa), resulting in generation of a truncated form of FXII (δFXII). The catalytic efficiency of δFXII activation by kallikrein is 15-fold greater than for full-length FXII. The enhanced rate of reciprocal activation of PK and δFXII in human plasma and in mice appears to overwhelm the normal inhibitory function of C1 inhibitor, leading to increased HK cleavage. In mice given human FXII-Lys/Arg309, induction of thrombin generation by infusion of tissue factor results in enhanced HK cleavage as a consequence of δFXII formation. The effects of δFXII in vitro and in vivo are reproduced when wild-type FXII is bound by an antibody to the FXII heavy chain (HC; 15H8). The results contribute to our understanding of the predisposition of patients carrying FXII-Lys/Arg309 to angioedema after trauma, and reveal a regulatory function for the FXII HC that normally limits PK activation in plasma.
Introduction
The plasma kallikrein-kinin system (KKS) is composed of the protease precursors prekallikrein (PK) and factor XII (FXII) and the cofactor/substrate high-molecular-weight kininogen (HK).1-4 PK and FXII undergo reciprocal conversion to the proteases kallikrein and αFXIIa by a process that is accelerated by a variety of natural and artificial “surfaces.” The surface-mediated reactions are referred to as contact activation.1,2,5,6 Kallikrein cleaves HK to release the nanopeptide bradykinin, which contributes to inflammation, vasodilatation, and vascular permeability through interactions with specific cellular receptors.3,4,6,7 Excessive bradykinin formation due to accelerated activation or dysregulation of the KKS contributes to a range of pathologies including angioedema (rapid soft tissue swelling).4,7,8 In hereditary angioedema (HAE), which affects ∼1 in 50 000 individuals,9,10 patients experience episodic swelling of the face, airway, limbs, or gastrointestinal tract. Consistent with a role for bradykinin, kallikrein inhibitors reduce the frequency and severity of angioedema in HAE patients.11-13
HAE is usually caused by reduced plasma activity of the serpin C1 inhibitor (C1-INH), the main regulator of kallikrein and αFXIIa.8-10 However, HAE does occur in patients with normal C1-INH activity.14,15 In some, Thr309 in FXII is replaced with lysine or arginine.16-19 de Maat et al showed that these substitutions create a novel cleavage site for the protease plasmin, and that inducing plasmin generation in patient plasma results in cleavage of FXII-Lys/Arg309 that enhances kallikrein and bradykinin generation.20 Angioedema in patients with FXII-Lys/Arg309 often follows trauma,18,19 suggesting a relationship with thrombin generation. Here, we show that FXII-Lys/Arg309 is cleaved after residue 309 by proteases generated during blood coagulation, removing the protein’s noncatalytic heavy chain (HC) region. The resulting truncated FXII is activated by kallikrein more rapidly than is FXII, accelerating kallikrein and bradykinin generation in a surface-independent manner. The findings explain the predisposition of patients with FXII-Lys/Arg309 to develop angioedema after trauma, and support an important role for elements of the FXII HC in regulating bradykinin production.
Methods
Materials
The following materials were used (obtained from the companies shown in parentheses): normal and FXII-deficient plasma (George King Bio-Medical); FXII, αFXIIa, βFXIIa, PK, HK, thrombin, and corn trypsin inhibitor (CTI; Enzyme Research Laboratory); FXI and FXIa (Haematologic Technologies); plasmin (Athens Research & Technology); C1-INH (Sigma-Aldrich); RecombiPlasTin human tissue factor (TF; Instrumentation Laboratory); recombinant hirudin (Bayer); human rhinovirus 3C protease (3CP; ACRO Biosystems); PTT-A reagent (Diagnostica Stago); argatroban (GlaxoSmith Kline); and S-2302 (H-d-prolyl-l-phenylalanyl-l-arginine-p-nitroanilide dihydrochloride) and S-2366 (l-pyro-Glu-L-Pro-L-Arg-p-nitroanilide) (DiaPharma). Polyphosphate (200-1300 U; ILC Performance Products) was provided by James Morrissey (University of Michigan, Ann Arbor, MI).
Antibodies
The following antibodies were used (obtained from the companies shown in parentheses): goat horseradish peroxidase (HRP)–anti-human FXII, sheep HRP–anti-human PK, and goat HRP–anti-human C1-INH immunoglobulin Gs (IgGs) (Enzyme Research Laboratories); goat anti-human HK IgG (Nordic Immunology); and rabbit anti-plasminogen IgG 1677-1-AP (Proteintech). Rabbit anti-mouse (anti-mHK) HK IgG was raised against a peptide representing residues 607-638 of mHK. Monoclonal IgGs 15H8 (FXII HC),21 1B2 (FXII catalytic domain),22 and H03 (kallikrein active site)23 have been described.
Recombinant proteins
Diagrams of human FXII and its activation products are shown in Figure 1A. FXII (FXII–wild type [WT]) and FXII with Lys, Arg, or Ala replacing Thr309 (FXII-Lys309, FXII-Arg309, or FXI-Ala309; Figure 1B) were expressed in HEK293 cells as described (Figure 1C).24 FXII-Arg309 in which Arg334 is replaced with alanine (FXII-Arg309, Ala334; supplemental Figure 1A [available on the Blood Web site]), FXII containing Leu-Glu-Val-Leu-Phe-Gln-Gly inserted between FXII Gln307 and Pro308 to introduce a 3CP cleavage site (FXII-3C; Figure 1C), and FXII-3C in which Arg334, Arg343, and Arg353 are replaced with alanine (FXII-T)24 were also prepared. FXII was purified by anion exchange chromatography from conditioned media,24 and stored in 4 mM sodium acetate, 150 mM NaCl, pH 5.2 at −80°C.
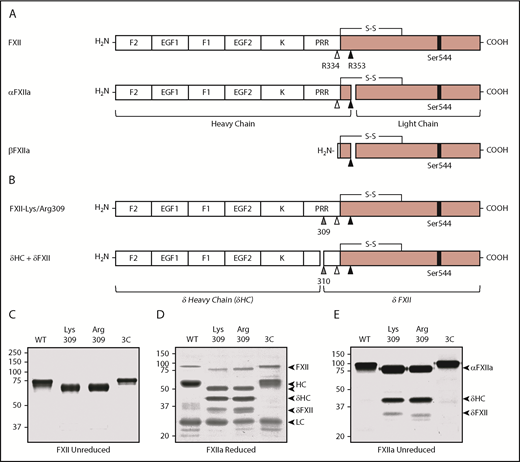
Recombinant FXII. (A) Schematic diagrams of FXII, αFXIIa, and βFXIIa showing noncatalytic (white boxes) and protease (pink boxes) domains. Positions of the active site serine (Ser544) are indicated by black bars. Sites of proteolysis during activation are indicated by arrows. Cleavage after Arg353 (black arrow) converts FXII to αFXIIa. Cleavage of αFXIIa after Arg334 (white arrow) separates the noncatalytic and protease domains, forming βFXIIa. FXII noncatalytic domains are the fibronectin type 2 (F2), epidermal growth factor (EGF), fibronectin type 1 (F1), and kringle (K) domains, and a proline-rich region (PRR). (B) Schematic diagrams of FXII with lysine or arginine replacement of Thr309 (gray arrow). Cleavage after Lys/Arg309 creates 2 proteins, δHC and δFXII. (C) Nonreducing SDS-PAGE of purified FXII (∼3 μg per lane). Shown are FXII-WT, FXII-Lys309, FXII-Arg309, and FXII-3C. Reducing (D) and nonreducing (E) SDS-PAGE of the FXIIa (activated) forms of FXII shown in panel C. Proteins were activated by incubation with dextran sulfate. Positions of molecular mass standards in kilodaltons are shown to the left of the images in panels C-E. To the right of these images are markers for FXII, activated FXII (αFXIIa), HC of αFXIIa (HC), LC of αFXIIa (LC), HC of FXII-Lys309 or FXII-Arg309 cleaved after residue 309 (δHC), and FXII residues Thr310 to Ser596 (δFXII).
Recombinant FXII. (A) Schematic diagrams of FXII, αFXIIa, and βFXIIa showing noncatalytic (white boxes) and protease (pink boxes) domains. Positions of the active site serine (Ser544) are indicated by black bars. Sites of proteolysis during activation are indicated by arrows. Cleavage after Arg353 (black arrow) converts FXII to αFXIIa. Cleavage of αFXIIa after Arg334 (white arrow) separates the noncatalytic and protease domains, forming βFXIIa. FXII noncatalytic domains are the fibronectin type 2 (F2), epidermal growth factor (EGF), fibronectin type 1 (F1), and kringle (K) domains, and a proline-rich region (PRR). (B) Schematic diagrams of FXII with lysine or arginine replacement of Thr309 (gray arrow). Cleavage after Lys/Arg309 creates 2 proteins, δHC and δFXII. (C) Nonreducing SDS-PAGE of purified FXII (∼3 μg per lane). Shown are FXII-WT, FXII-Lys309, FXII-Arg309, and FXII-3C. Reducing (D) and nonreducing (E) SDS-PAGE of the FXIIa (activated) forms of FXII shown in panel C. Proteins were activated by incubation with dextran sulfate. Positions of molecular mass standards in kilodaltons are shown to the left of the images in panels C-E. To the right of these images are markers for FXII, activated FXII (αFXIIa), HC of αFXIIa (HC), LC of αFXIIa (LC), HC of FXII-Lys309 or FXII-Arg309 cleaved after residue 309 (δHC), and FXII residues Thr310 to Ser596 (δFXII).
δFXII-3C and δFXIIa-3C
FXII-3C (8 μM) was incubated with 3CP (25 U/mL) in 50 mM Tris-HCl, 150 mM NaCl overnight at 4°C. 3CP was removed with a glutathione column (Thermo Scientific). Cleaved FXII-3C was dialyzed into sodium acetate buffer and stored at −80°C. Activated δFXII (truncated form of FXIIa-3C [δFXIIa-3C]) was prepared by converting FXII-3C (10 μM) to αFXIIa-3C by incubation with 175 μg/mL dextran sulfate (500 kDa). αFXIIa-3C was dissociated from dextran sulfate with polybrene (500 μM), and isolated on a 15H8 IgG column.21 After elution with 2M NaSCN, αFXIIa-3C was cleaved with 3CP to generate δFXIIa-3C. Proteins were dialyzed into sodium acetate buffer and stored at −80°C.
Chromogenic assays
Assays were performed in PEG-20000–coated microtiter plates.
Continuous assays
Continuous assays for FXII and/or PK activation were in 100-μL standard buffer (20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES], pH 7.4, 100 mM NaCl, 0.1% PEG-8000, 10 μM ZnCl2) with 200 μM S-2302. Some reactions contained polyphosphate (70 μM) or 15H8 (400 nM). Changes in optical density (OD) at 405 nm at 37°C were monitored.
Discontinuous assays
Discontinuous assays for FXII, PK, and FXI activation were run in 100-μL standard buffer at 37°C. FXII cleavage by PK, kallikrein (10-20 nM), FXIa (10 nM), or plasmin (20 or 500 nM) with or without polyphosphate (70 μM) or 15H8 was assessed. For measuring FXIIa, reactions were stopped with polybrene (0.1 mg/mL) and H03 (100 nM for reactions with kallikrein) or aprotinin (10-40 μM for reactions with FXIa or plasmin) and S-2302 (500 μM) was added. For measuring kallikrein, reactions were stopped with CTI (500 nM) and polybrene (0.1 mg/mL) and S-2302 (200 μM) was added. For partial cleavage of FXII-Lys309 or FXII-Arg309 (100 nM) by thrombin (25 nM), reactions were stopped with argatroban (125 μM) and 10 μL of reactions were incubated with 60 nM PK. For measuring FXIa, reactions were stopped with CTI (500 nM) and polybrene (0.1 mg/mL), and S-2366 (250 μM) was added. For all reactions, ΔOD 405 nm was followed, and amounts of FXIIa, kallikrein, or FXIa extrapolated from standard curves prepared with pure proteases.
FXII autoactivation
FXII (1.25 μM) was incubated with dextran sulfate (20 μg/mL) in standard buffer. At various times, aliquots were removed into reducing or nonreducing sample buffer, size-fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and stained with Coomassie blue.
FXII cleavage in plasma
FXII-deficient plasma was supplemented with 400 nM FXII and 10 mM H-Gly-Pro-Arg-Pro-OH to prevent fibrin polymerization. Coagulation was initiated with 2.5 pM human TF and 6.25 mM CaCl2 (final concentrations). At various times, aliquots were removed into nonreducing sample buffer, size-fractionated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with HRP-conjugated goat anti-human FXII IgG, using chemiluminescence.
FXII cleavage by coagulation proteases
FXII (200 nM) was incubated with 10 nM thrombin, FXIa, plasmin, FVIIa/TF, FIXa, or FXa, or with 50 nM kallikrein, at 37°C in standard buffer with 5 mM CaCl2. At various times, aliquots were mixed with nonreducing sample buffer, size-fractionated by SDS-PAGE, transferred to nitrocellulose, and probed with 15H8 and 1B2. HRP-conjugated goat anti-mouse IgG and chemiluminescence were used for detection.
Reciprocal FXII and PK activation
FXII (200 nM) and PK (200 nM) were incubated in standard buffer at 37°C. At various times, aliquots were mixed with reducing sample buffer, size-fractionated by SDS-PAGE, transferred to nitrocellulose, and probed with HRP-conjugated goat anti-human FXII or sheep anti-human PK IgG and chemiluminescence.
HK cleavage in human plasma
FXII-deficient plasma was supplemented with FXII (400 nM), with or without PTT-A reagent (10% final volume), and incubated at 37°C. At various times, aliquots were mixed with nonreducing sample buffer, size-fractionated by SDS-PAGE, transferred to nitrocellulose, and probed with goat anti-human HK IgG or anti-human C1-INH and chemiluminescence.
HK cleavage in mice
Procedures with mice were approved by the Vanderbilt University Animal Care and Use Committee. FXII (0.6 mg/kg) in phosphate-buffered saline (PBS) was infused into WT, FXI-deficient (F11−/−),25 or PK-deficient (Klkb1−/−)26 C57Bl/6 mice through a tail vein. In some experiments, mice also received infusions of human TF (70 ng/kg), IgG 1B2 (1 mg/kg), IgG 15H8 (1 mg/kg), or hirudin (10 mg/kg). Blood samples were collected from tail veins into capillary tubes, and plasma was prepared by centrifugation. Samples underwent SDS-PAGE, then transfer to nitrocellulose. Blots were probed with anti-mHK IgG using HRP-conjugated goat anti-rabbit IgG, and chemiluminescence. In some experiments, animals were pretreated with antisense oligonucleotide (ASO) to mouse plasminogen messenger RNA (mRNA) (AGTGATGGTCTATTGTCACA, provided by Alexey Revenko and Gourab Bhattacharejee, Ionis Pharmaceuticals, Carlsbad, CA), 330 mg/kg/week for 2 weeks. Plasminogen in plasma was assessed by western blot.
Results
Recombinant FXII
FXII is an 80-kDa protein containing several noncatalytic domains and a C-terminal protease domain (Figure 1A).27,28 Kallikrein cleaves FXII after Arg353 to form αFXIIa, and subsequent cleavage after Arg334 forms βFXIIa (Figure 1A; supplemental Figure 1B). FXII with Lys or Arg replacing Thr309 (Figure 1B) migrate slightly faster than FXII-WT on nonreducing SDS-PAGE (Figure 1C). Lys/Arg309 substitutions disrupt 1, and perhaps 2, glycosylation site(s) at Thr309 and Thr310,27,29 which may account for the apparent mass loss. Cleavage of FXII after Arg353 during autoactivation or activation by kallikrein generates the 50- and 30-kDa HC and light chain (LC) of αFXIIa seen on reducing SDS-PAGE (Figure 1D).24,27,28 On nonreducing gels (Figure 1E), αFXIIa runs as a single 80-kDa band because the HC and LC are connected by a disulfide bond (Figure 1A). With FXIIa-Lys309 and FXIIa-Arg309, additional bands appear on reducing and nonreducing gels (Figure 1D-E). Cleavage after Lys/Arg309 would generate fragments unconnected by a disulfide bond (Figure 1A), explaining this pattern. Hereafter, the term “HC” will be used to indicate the N-terminal noncatalytic portion of FXII as well as the larger polypeptide generated when FXII is cleaved after Arg353 or Arg309.
FXII-Lys/Arg309 cleavage during coagulation
FXII-WT, FXII-Lys309, FXII-Arg309 (Figure 2A), and FXII-Ala309 (supplemental Figure 1C) shorten the activated partial thromboplastin time (aPTT) of FXII-deficient plasma comparably, and autoactivate in the presence of polyphosphate similarly (Figure 2B), indicating that Thr309 substitutions do not compromise basic function. When thrombin generation was induced with TF in FXII-deficient plasma reconstituted with FXII-Lys309 or FXII-Arg309 (Figure 2C), bands appear on FXII western blots that are not present in plasma containing FXII-WT (Figure 2C). The novel bands are similar to those generated when FXII-Lys/Arg309 is incubated with plasmin (Figure 2D), as reported by de Maat et al,20 and to those generated during autoactivation (Figure 1D-E). They do not migrate in the same position as βFXIIa (supplemental Figure 1B), indicating that they are not due to cleavage after Arg334. Consistent with this, FXII-Arg309 in which Arg334 was replaced with alanine (FXII-Arg309, Ala334) is cleaved similarly to FXII-Arg309 (supplemental Figure 1A,D). This indicates that 1 or more proteases generated during coagulation cleave FXII after Lys/Arg309. In reactions with pure proteases, thrombin and FXIa (FXIa) cleave FXII-Lys/Arg309 to generate patterns identical to those in plasma (Figure 2E), but do not cleave FXII-WT (Figure 2E) or FXII-Ala309 (supplemental Figure 1A). Thrombin appears to be more potent than plasmin as a cleaver of FXII-Lys/Arg309 (Figure 2E). Mass spectrometry verified that thrombin cleaves FXII-Lys309 after residue 309 (supplemental Figure 2). FXII-Lys/Arg309 was not cleaved by FVIIa/TF, IXa, or Xa (supplemental Figure 3). Kallikrein cleaves FXII-Lys/Arg309 after residue 309, while also cleaving after Arg353 to form FXIIa (Figure 2F).
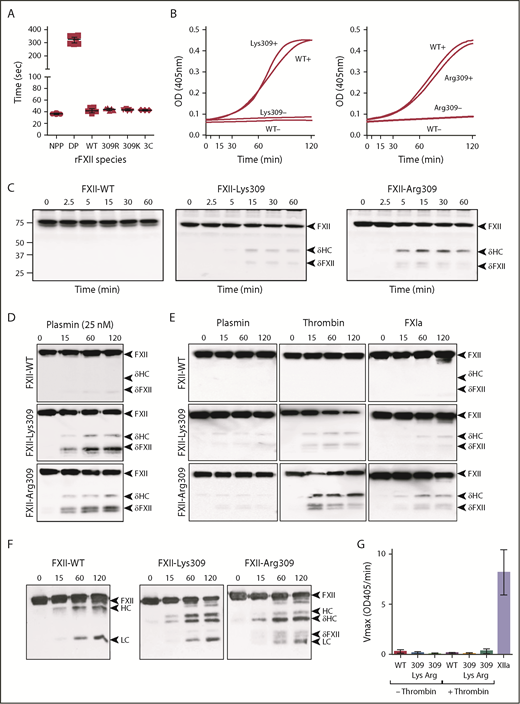
FXII cleavage by coagulation proteases. (A) Clotting times in an aPTT assay for normal plasma (NPP), FXII-deficient plasma (DP), or FXII-deficient plasma supplemented with recombinant FXII-WT (WT), FXII-Lys309 (309K), FXII-Arg309 (309R), or FXII-3C (3C). Each symbol indicates 1 clotting time. (B) Recombinant FXII (200 nM) incubated in standard buffer with 200 μM S-2302 at 37°C in the presence (+) or absence (−) of 70 μM polyphosphate. Left panel, FXII-Lys309; right panel, FXII-Arg309. Changes in OD 405 nm were continuously monitored. Curves are means of 3 independent runs. (C) Western blot of FXII-deficient plasma supplemented with 400 nM FXII-WT, FXII-Lys309, or FXII-Arg309 induced to clot with TF (2.5 pM) and CaCl2 (6.25 mM). Fibrin formation was prevented with H-Gly-Pro-Arg-Pro-OH. Western blots of time courses were probed with polyclonal IgG to human FXII. Positions of molecular mass standards in kilodaltons are shown to the left of the images. (D-F) FXII (200 nM) in standard buffer incubated with 25 nM plasmin (D), 10 nM plasmin, thrombin, or FXIa (E), or α-kallikrein (50 nM) (F). Western blots of time courses were probed with a mixture of monoclonal IgGs to the FXII HC (15H8) and LC (1B2). To the right of images in panels C-F are markers for FXII (FXII), the HC and LC of αFXIIa, HC of FXII-Lys309 or FXII-Arg309 cleaved after residue 309 (δHC) and FXII residues Thr310 to Ser596 (δFXII). (G) FXII (100 nM) incubated in standard buffer with (+) or without (−) 25 nM thrombin for 120 minutes at 37°C. Thrombin was inhibited with argatroban (125 μM) and FXIIa cleavage of S-2302 (500 μM) was determined. The proteins are FXII-WT (WT), FXII-Lys309 (309 Lys), and FXII-Arg309 (309 Arg). Control (XIIa) is 12 nM αFXIIa. Error bars ± 1 standard deviation (SD); n = 6.
FXII cleavage by coagulation proteases. (A) Clotting times in an aPTT assay for normal plasma (NPP), FXII-deficient plasma (DP), or FXII-deficient plasma supplemented with recombinant FXII-WT (WT), FXII-Lys309 (309K), FXII-Arg309 (309R), or FXII-3C (3C). Each symbol indicates 1 clotting time. (B) Recombinant FXII (200 nM) incubated in standard buffer with 200 μM S-2302 at 37°C in the presence (+) or absence (−) of 70 μM polyphosphate. Left panel, FXII-Lys309; right panel, FXII-Arg309. Changes in OD 405 nm were continuously monitored. Curves are means of 3 independent runs. (C) Western blot of FXII-deficient plasma supplemented with 400 nM FXII-WT, FXII-Lys309, or FXII-Arg309 induced to clot with TF (2.5 pM) and CaCl2 (6.25 mM). Fibrin formation was prevented with H-Gly-Pro-Arg-Pro-OH. Western blots of time courses were probed with polyclonal IgG to human FXII. Positions of molecular mass standards in kilodaltons are shown to the left of the images. (D-F) FXII (200 nM) in standard buffer incubated with 25 nM plasmin (D), 10 nM plasmin, thrombin, or FXIa (E), or α-kallikrein (50 nM) (F). Western blots of time courses were probed with a mixture of monoclonal IgGs to the FXII HC (15H8) and LC (1B2). To the right of images in panels C-F are markers for FXII (FXII), the HC and LC of αFXIIa, HC of FXII-Lys309 or FXII-Arg309 cleaved after residue 309 (δHC) and FXII residues Thr310 to Ser596 (δFXII). (G) FXII (100 nM) incubated in standard buffer with (+) or without (−) 25 nM thrombin for 120 minutes at 37°C. Thrombin was inhibited with argatroban (125 μM) and FXIIa cleavage of S-2302 (500 μM) was determined. The proteins are FXII-WT (WT), FXII-Lys309 (309 Lys), and FXII-Arg309 (309 Arg). Control (XIIa) is 12 nM αFXIIa. Error bars ± 1 standard deviation (SD); n = 6.
FXII-Lys/Arg309 cleaved by thrombin does not hydrolyze the tripeptide S-2302 (Figure 2G), indicating that it is a form of FXII and not FXIIa (ie, it is not cleaved after Arg353). Cleavage after Lys/Arg309, therefore, produces a truncated FXII (Thr310 to Ser596), which we designated δFXII, and a δHC (Ile1 to Lys/Arg309) (Figure 1B). Western blot analyses indicate that the ∼45-kDa band is δHC and the ∼35-kDa doublet is δFXII (supplemental Figure 4A). δFXII derived from FXII-Lys/Arg309 may run as a doublet due to variable glycosylation around residues 309 and 310 (supplemental Figure 4B).29 These results show that FXII-Lys/Arg309 may be cleaved after residue 309 by plasma proteases generated during TF-induced coagulation (Figure 2C) or during contact activation (Figures 1D and 2E).
PK activation is accelerated by cleaving FXII after Lys/Arg309
FXII and PK undergo reciprocal conversion to FXIIa and kallikrein in the absence of a surface (Figure 3A).24,30-32 The process is supported similarly by FXII-WT, FXII-Lys309, FXII-Arg309 (Figure 3B white symbols), and FXII-Ala309 (supplemental Figure 5). Preincubating FXII with thrombin enhances kallikrein generation in reactions with FXII-Lys309 or FXII-Arg309, but not FXII-WT (Figure 3B black symbols) or FXII-Ala309 (supplemental Figure 5). As thrombin does not activate PK,33 this shows PK activation is enhanced in reactions in which thrombin cleaves FXII-Lys309/Arg309 to form δFXII.

PK activation by FXII and δFXII. (A) Western blot of FXII/PK reciprocal activation. Plasma-derived FXII (200 nM) and PK (200 nM) were incubated in standard buffer at 37°C. At indicated times, samples were removed into reducing sample buffer. Western blots were probed with goat anti-human FXII IgG or sheep anti-human PK IgG. Positions of markers for FXII, PK, and the HC and LC of αFXIIa and kallikrein are indicated to the right of each image. (B) Kallikrein generation in reciprocal reactions with FXII. FXII-WT (, ), FXII-Lys309 (, ), or FXII-Arg309 (, ), 100 nM in standard buffer was incubated with (, , ) or without (, , ) thrombin (25 nM) for 2 hours at 37°C. Reactions were stopped with argatroban (125 μM). PK (60 nM) was mixed with 12.5 nM of the preincubated FXII. At various times, aliquots were removed, FXIIa was inhibited with CTI, and kallikrein concentration was determined by chromogenic assay. (C) FXII amino acid sequence within the proline-rich region showing the amino acids inserted to create a cleavage site for 3CP in the protein FXII-3C. The image to the right of the arrow shows the N and C termini after cleavage of FXII-3C with 3CP. (D) Schematic diagrams of FXII with the sequence Leu-Glu-Val-Leu-Phe-Gln-Gly inserted between FXII residues Gln307 and Pro308 to create a cleavage site for 3C protease in FXII-3C. Cleavage of FXII-3C with 3C creates a mixture of δHC (with a slightly different C terminus than in panel B), and δFXII-3C. The latter differs from δFXII in having an additional Gly-Pro at the N terminus. Cleavage of δFXII after Arg353 creates the fully active species δFXIIa-3C. (E) Nonreducing 20% Tris–2-(N-morpholino)ethanesulfonic acid (MES) SDS-PAGE of FXII-3C cleaved by 3C protease and the purified FXII-3C δHC and δFXII-3C. Positions of molecular mass standards in kilodaltons are shown to the left of the image. (F) Kallikrein generation in reciprocal reactions with FXII-3C and δFXII-3C. PK (60 nM) was incubated in standard buffer at 37°C with 12.5 nM FXII-WT (), FXII-3C (□), FXII-3C cleaved with 3C protease () or purified δFXII-3C (), or in the absence of FXII (). Reactions were run in the absence (left panel) or presence (right panel) of 70 μM Poly-P. At the indicated times, aliquots were removed and tested for kallikrein generation by chromogenic assay. For panels B and F, error bars ± 1 SD; n = 5 or 6.
PK activation by FXII and δFXII. (A) Western blot of FXII/PK reciprocal activation. Plasma-derived FXII (200 nM) and PK (200 nM) were incubated in standard buffer at 37°C. At indicated times, samples were removed into reducing sample buffer. Western blots were probed with goat anti-human FXII IgG or sheep anti-human PK IgG. Positions of markers for FXII, PK, and the HC and LC of αFXIIa and kallikrein are indicated to the right of each image. (B) Kallikrein generation in reciprocal reactions with FXII. FXII-WT (, ), FXII-Lys309 (, ), or FXII-Arg309 (, ), 100 nM in standard buffer was incubated with (, , ) or without (, , ) thrombin (25 nM) for 2 hours at 37°C. Reactions were stopped with argatroban (125 μM). PK (60 nM) was mixed with 12.5 nM of the preincubated FXII. At various times, aliquots were removed, FXIIa was inhibited with CTI, and kallikrein concentration was determined by chromogenic assay. (C) FXII amino acid sequence within the proline-rich region showing the amino acids inserted to create a cleavage site for 3CP in the protein FXII-3C. The image to the right of the arrow shows the N and C termini after cleavage of FXII-3C with 3CP. (D) Schematic diagrams of FXII with the sequence Leu-Glu-Val-Leu-Phe-Gln-Gly inserted between FXII residues Gln307 and Pro308 to create a cleavage site for 3C protease in FXII-3C. Cleavage of FXII-3C with 3C creates a mixture of δHC (with a slightly different C terminus than in panel B), and δFXII-3C. The latter differs from δFXII in having an additional Gly-Pro at the N terminus. Cleavage of δFXII after Arg353 creates the fully active species δFXIIa-3C. (E) Nonreducing 20% Tris–2-(N-morpholino)ethanesulfonic acid (MES) SDS-PAGE of FXII-3C cleaved by 3C protease and the purified FXII-3C δHC and δFXII-3C. Positions of molecular mass standards in kilodaltons are shown to the left of the image. (F) Kallikrein generation in reciprocal reactions with FXII-3C and δFXII-3C. PK (60 nM) was incubated in standard buffer at 37°C with 12.5 nM FXII-WT (), FXII-3C (□), FXII-3C cleaved with 3C protease () or purified δFXII-3C (), or in the absence of FXII (). Reactions were run in the absence (left panel) or presence (right panel) of 70 μM Poly-P. At the indicated times, aliquots were removed and tested for kallikrein generation by chromogenic assay. For panels B and F, error bars ± 1 SD; n = 5 or 6.
Preparing δFXII
Isolating δFXII for detailed analyses by cleaving FXII-Lys309/Arg309 with thrombin or plasmin proved difficult because complete cleavage after Lys/Arg309 does not occur before cleavage at other sites causes degradation and/or activation. To address this, we prepared FXII with a unique cleavage site for human rhinovirus 3CP near residue 309 (Figure 3C-D). FXII-3C has comparable activity to FXII-WT in aPTT (Figure 2A) and autoactivation (supplemental Figure 6) assays. Cleaving FXII-3C with 3CP produces a form of δFXII (δFXII-3C) comprising FXII residues Pro308 to Ser596 with an additional glycine at the N terminus (Figure 3C). δFXII-3C and the accompanying δHC (Figure 3D) can be separated chromatographically (Figure 3E), but as PK activation by δFXII-3C is the same with or without δHC (Figure 3F), subsequent experiments were performed without separating the polypeptides. As expected, polyphosphate did not induce δFXII-3C autoactivation (supplemental Figure 6), and δFXII-3C did not cleave S-2302 because it is not a form of FXIIa (data not shown). When mixed with PK, δFXII-3C induced kallikrein generation more rapidly than FXII-WT or full-length FXII-3C (Figure 3F left). Polyphosphate enhanced kallikrein generation in reactions with FXII-WT or FXII-3C,31,32 but not δFXII-3C (Figure 3F right), consistent with the importance of the FXII HC in surface-mediated reactions.28,34,35
δFXII-3C activation and activity
Kallikrein converted δFXII-3C to δFXIIa-3C with an initial rate ∼40-fold faster than it activated FXII-WT or FXII-3C (Figure 4A), and at least 100-fold faster than plasmin activated δFXII-3C (Figure 4B).36 Fitting curves for FXII activation by kallikrein to the Michaelis-Menten equation revealed a catalytic efficiency (kcat/Km) ∼15-fold higher for δFXII-3C than for FXII-WT or FXII-3C (65 800 M−1s−1 vs 4400 and 3400 M−1s−1, respectively; supplemental Figure 7A). kcat/Km values correspond to the initial slopes of the v0 dependences on the substrate concentration. Kms for the reactions appear ≥3.5 μM, but could not be accurately determined because reactions did not reach saturation. FXIa, a homolog of kallikrein, also converted δFXII-3C to δFXIIa-3C ∼50-fold faster than full-length FXII (Figure 4C). Previously, we showed that FXII locked into its precursor form by replacing Arg353 with alanine has intrinsic proteolytic activity that initiates reciprocal PK/FXII activation.24,32,37 We created similar versions of FXII-3C and δFXII-3C (FXII-T and δFXII-T) that cannot be converted to αFXIIa and δFXIIa, respectively.24 δFXII-T catalyzed PK conversion to kallikrein more rapidly than FXII-T (Figure 4D). kcat/Km for activation by δFXII-T was ∼2.5-fold higher than by FXII-T (67 M−1s−1 and 26 M−1s−1, respectively; supplemental Figure 7B). Thus, δFXII-3C has 2 properties that enhance KKS activation relative to FXII-WT. δFXII-3C initially converts PK to kallikrein more rapidly than does FXII, and then kallikrein converts δFXII-3C to δFXIIa more rapidly than FXII to αFXIIa.

Properties of δFXII-3C and its active form δFXIIa-3C, and their effects on HK cleavage. (A-C) Activation of FXII. FXII-WT (), FXII-3C (□), or δFXII-3C (), 200 nM, were incubated in standard buffer at 37°C with kallikrein (10 nM) (A), plasmin (500 nM) (B), or FXIa (10 nM, subunit concentration) (C). Kallikrein was inhibited with HO3, and FXIa and plasmin were inhibited with aprotinin, and FXIIa generation was determined by chromogenic substrate cleavage. (D) PK activation by FXII-T or δFXII-T. PK (200 nM) was incubated in standard buffer at 37°C with vehicle (), 200 nM FXII-T (□), or δFXII-T (), and cleavage of chromogenic substrate was continuously monitored. (E) PK activation by FXIIa. PK (60 nM) was incubated in standard buffer at 37°C with 50 pM αFXIIa (), βFXIIa (□), or δFXIIa-3C (), or in the absence of FXIIa (). At various times, reactions were stopped with CTI, and kallikrein generation was determined by chromogenic assay. (F) FXI (30 nM) was incubated in standard buffer at 37°C with 10 nM αFXIIa (), βFXII (), or δFXIIa-3C (), or in the absence of FXIIa (). At various times, reactions were stopped with CTI, and FXIa generation was determined by chromogenic assay. For panels A and B, n = 4; C, n = 6; D, n = 3; E, n = 9; and F, n = 6. Error bars ± 1 SD for all panels. (G) HK cleavage in human plasma. Shown are western blots of human FXII-deficient plasma supplemented with FXII-WT, FXII-3C, or δFXII-3C (400 nM) in the absence (−) or presence (+) of PTT-A silica-based reagent. At indicated times, samples were removed into nonreducing sample buffer. Western blots were probed with goat anti-human HK IgG (HK) or goat anti-human C1-INH IgG (C1-INH). Positions of standards for HK, the 2 bands of cleaved HK (HKa), and kallikrein or αFXIIa in complex with C1-INH are shown on the right. Positions of molecular mass standards in kilodaltons are shown to the left of the images.
Properties of δFXII-3C and its active form δFXIIa-3C, and their effects on HK cleavage. (A-C) Activation of FXII. FXII-WT (), FXII-3C (□), or δFXII-3C (), 200 nM, were incubated in standard buffer at 37°C with kallikrein (10 nM) (A), plasmin (500 nM) (B), or FXIa (10 nM, subunit concentration) (C). Kallikrein was inhibited with HO3, and FXIa and plasmin were inhibited with aprotinin, and FXIIa generation was determined by chromogenic substrate cleavage. (D) PK activation by FXII-T or δFXII-T. PK (200 nM) was incubated in standard buffer at 37°C with vehicle (), 200 nM FXII-T (□), or δFXII-T (), and cleavage of chromogenic substrate was continuously monitored. (E) PK activation by FXIIa. PK (60 nM) was incubated in standard buffer at 37°C with 50 pM αFXIIa (), βFXIIa (□), or δFXIIa-3C (), or in the absence of FXIIa (). At various times, reactions were stopped with CTI, and kallikrein generation was determined by chromogenic assay. (F) FXI (30 nM) was incubated in standard buffer at 37°C with 10 nM αFXIIa (), βFXII (), or δFXIIa-3C (), or in the absence of FXIIa (). At various times, reactions were stopped with CTI, and FXIa generation was determined by chromogenic assay. For panels A and B, n = 4; C, n = 6; D, n = 3; E, n = 9; and F, n = 6. Error bars ± 1 SD for all panels. (G) HK cleavage in human plasma. Shown are western blots of human FXII-deficient plasma supplemented with FXII-WT, FXII-3C, or δFXII-3C (400 nM) in the absence (−) or presence (+) of PTT-A silica-based reagent. At indicated times, samples were removed into nonreducing sample buffer. Western blots were probed with goat anti-human HK IgG (HK) or goat anti-human C1-INH IgG (C1-INH). Positions of standards for HK, the 2 bands of cleaved HK (HKa), and kallikrein or αFXIIa in complex with C1-INH are shown on the right. Positions of molecular mass standards in kilodaltons are shown to the left of the images.
δFXIIa-3C activity
δFXIIa-3C, the activated (cleaved after Arg353) form of δFXII-3C, and the truncated species βFXIIa activate PK similarly, and at slightly reduced rates compared with αFXIIa (Figure 4E). Enhanced KKS activity with δFXII, therefore, is not due to δFXIIa being a better PK activator than αFXIIa. During contact activation in plasma, αFXIIa converts FXI to FXIa.5,6,38 δFXIIa-3C, like βFXIIa, activated FXI poorly compared with αFXIIa (Figure 4F), and rates were not enhanced by polyphosphate (data not shown), consistent with loss of the FXII HC.
δFXII and HK cleavage in plasma
In FXII-deficient plasma supplemented with FXII-WT or FXII-3C, rapid HK conversion to cleaved HK (HKa, an indicator of kallikrein-mediated proteolysis and a surrogate for bradykinin release)39,40 was evident when contact activation was induced with a silica-based reagent, but not in its absence (Figure 4G). δFXII-3C, in contrast, induced HK cleavage without silica, and silica did not enhance cleavage. This indicates that δFXII-3C supports PK activation in plasma by a surface-independent process that overcomes regulation by C1-INH. C1-INH inhibition of δFXIIa-3C is only slightly slower than inhibition of αFXIIa (supplemental Figure 8), indicating that δFXIIa-3C is not resistant to C1-INH. Indeed, complexes between C1-INH and kallikrein or FXIIa appear in plasma with δFXII-3C in the absence of a surface as they do with full-length FXII with a surface (Figure 4G). Most likely, the enhanced reciprocal PK/δFXII activation simply exceeds the capacity of C1-INH to control the rate of the process.
δFXII and HK cleavage in vivo
Plasma HK in mice is primarily the product of the mKng1 gene.41 An antibody specific for the high-molecular-weight species encoded by mKng1 recognizes 2 prominent bands on plasma western blots that are not present in mKng1-null mice (supplemental Figure 9A). Inducing contact activation in plasma results in reductions in the apparent sizes of these bands (supplemental Figure 9B), consistent with HK conversion to HKa and bradykinin release.39,40
Infusing FXII-WT or FXII-3C into WT mice so that the FXII in plasma is ∼25% (140 nM) recombinant protein does not alter the baseline HK pattern (Figure 5A). In contrast, 140 nM δFXII-3C induced rapid complete HK cleavage. Significant cleavage occurs when δFXII-3C is only ∼10% (40 nM) of plasma FXII. δFXII-3C–induced HK cleavage was reduced by coinfusing an antibody to human FXII (1B2) (Figure 5B).22 HK cleavage did not occur when δFXII-3C was infused into mice lacking PK (Klkb1−/−) but did occur in mice lacking FXI (F11−/−) (Figure 5B), consistent with a major role for kallikrein in converting δFXII-3C to δFXIIa-3C, and in subsequent HK cleavage. These findings support a role for δFXII in HAE, and suggest that the FXII HC serves a role in regulating bradykinin generation.
![Figure 5. Effect of δFXII on HK cleavage in mice. (A) FXII-WT, FXII-3C, or δFXII-3C were administered IV to WT C57Bl/6 mice to an estimated final plasma concentration of 140 nM or 40 nM. Shown are nonreducing western blots of plasma collected 0, 15, 30 minutes, or ∼18 hours (overnight [ON]) after FXII infusion. Blots were developed with anti-murine HK IgG (anti-mHKFXI). (B) As in panel A. δFXII-3C was infused into WT, PK-deficient (Klkb1−/−) mice), or FXI-deficient (F11−/−) mice (140 nM final plasma concentration). FXII-WT mice also received an IV infusion of vehicle (Control) or monoclonal anti-human FXII IgG 1B2. Samples were analyzed as in panel A. (C-D) WT mice received IV infusions of FXII-WT (C) or FXII-Arg309 (D) (140 nM final plasma concentration). Mice were then infused with vehicle (−TF) or 70 ng/kg human TF (+TF). Samples of plasma collected 0, 15, or 30 minutes postvehicle/TF infusion were analyzed as in panel A. (E) WT mice were treated with vehicle or 70 ng/kg human TF after infusion of FXII-Arg309, as in panel D. Some mice were pretreated with the thrombin inhibitor hirudin (10 mg/kg). FXI-deficient (F11−/−) mice were treated with 70 ng/kg human TF after FXII-Arg309 infusion. The 2 left panels in E are the same as in panel D. (F) Western blot for plasminogen in plasma from WT C57Bl/6 mice before (C) or after (+ASO) subcutaneous injections of an ASO directed at the mRNA of plasminogen. (G) Western blots of plasma HK in WT mice treated with anti-plasminogen ASO. Mice received FXII-WT (left) or FXII-Arg309 (right) prior to infusion of vehicle (−TF) or 70 ng/kg human TF (+TF). For blots in panels A-E and G, arrows on the right indicate the positions of cleaved HK (HKa), and positions of molecular mass standards in kilodaltons are shown to the left of the images. Plg, the position of the band representing plasminogen.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/10/10.1182_blood-2018-06-860270/3/m_blood860270f5.png?Expires=1769119419&Signature=JtLpzNfBVGPJLWIjVC7-ERAWgAY87TXBGK9Iw9WvKRjfKxh23Bs91QCHeX02GmdDC97LlB12b8-RXPC7CZ3OmxyzsXdO00CR8sO4W1ZUCLN4roJcb~pm6q2yoCQLPNrxAV1O7wHsZm6~EsgOS7FNvkEhEkf3lJGkyXSwmVEvfhhgjfcs0tf6UfR~ljBpSW6r3-ISqtIu0143Is14zSRGtUaEpnbovuW-kvlVVE8itUIrTX5noPkuMFydZ6NLUiiS27oqB1JNqfo0Zq8276~m2yjCzu0f5mIohgZdk3-vti63QtM38UZJ-PCkA4HbKqVEMr0P7SCaUZCRm1YXt6EqSQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of δFXII on HK cleavage in mice. (A) FXII-WT, FXII-3C, or δFXII-3C were administered IV to WT C57Bl/6 mice to an estimated final plasma concentration of 140 nM or 40 nM. Shown are nonreducing western blots of plasma collected 0, 15, 30 minutes, or ∼18 hours (overnight [ON]) after FXII infusion. Blots were developed with anti-murine HK IgG (anti-mHKFXI). (B) As in panel A. δFXII-3C was infused into WT, PK-deficient (Klkb1−/−) mice), or FXI-deficient (F11−/−) mice (140 nM final plasma concentration). FXII-WT mice also received an IV infusion of vehicle (Control) or monoclonal anti-human FXII IgG 1B2. Samples were analyzed as in panel A. (C-D) WT mice received IV infusions of FXII-WT (C) or FXII-Arg309 (D) (140 nM final plasma concentration). Mice were then infused with vehicle (−TF) or 70 ng/kg human TF (+TF). Samples of plasma collected 0, 15, or 30 minutes postvehicle/TF infusion were analyzed as in panel A. (E) WT mice were treated with vehicle or 70 ng/kg human TF after infusion of FXII-Arg309, as in panel D. Some mice were pretreated with the thrombin inhibitor hirudin (10 mg/kg). FXI-deficient (F11−/−) mice were treated with 70 ng/kg human TF after FXII-Arg309 infusion. The 2 left panels in E are the same as in panel D. (F) Western blot for plasminogen in plasma from WT C57Bl/6 mice before (C) or after (+ASO) subcutaneous injections of an ASO directed at the mRNA of plasminogen. (G) Western blots of plasma HK in WT mice treated with anti-plasminogen ASO. Mice received FXII-WT (left) or FXII-Arg309 (right) prior to infusion of vehicle (−TF) or 70 ng/kg human TF (+TF). For blots in panels A-E and G, arrows on the right indicate the positions of cleaved HK (HKa), and positions of molecular mass standards in kilodaltons are shown to the left of the images. Plg, the position of the band representing plasminogen.
Effect of δFXII on HK cleavage in mice. (A) FXII-WT, FXII-3C, or δFXII-3C were administered IV to WT C57Bl/6 mice to an estimated final plasma concentration of 140 nM or 40 nM. Shown are nonreducing western blots of plasma collected 0, 15, 30 minutes, or ∼18 hours (overnight [ON]) after FXII infusion. Blots were developed with anti-murine HK IgG (anti-mHKFXI). (B) As in panel A. δFXII-3C was infused into WT, PK-deficient (Klkb1−/−) mice), or FXI-deficient (F11−/−) mice (140 nM final plasma concentration). FXII-WT mice also received an IV infusion of vehicle (Control) or monoclonal anti-human FXII IgG 1B2. Samples were analyzed as in panel A. (C-D) WT mice received IV infusions of FXII-WT (C) or FXII-Arg309 (D) (140 nM final plasma concentration). Mice were then infused with vehicle (−TF) or 70 ng/kg human TF (+TF). Samples of plasma collected 0, 15, or 30 minutes postvehicle/TF infusion were analyzed as in panel A. (E) WT mice were treated with vehicle or 70 ng/kg human TF after infusion of FXII-Arg309, as in panel D. Some mice were pretreated with the thrombin inhibitor hirudin (10 mg/kg). FXI-deficient (F11−/−) mice were treated with 70 ng/kg human TF after FXII-Arg309 infusion. The 2 left panels in E are the same as in panel D. (F) Western blot for plasminogen in plasma from WT C57Bl/6 mice before (C) or after (+ASO) subcutaneous injections of an ASO directed at the mRNA of plasminogen. (G) Western blots of plasma HK in WT mice treated with anti-plasminogen ASO. Mice received FXII-WT (left) or FXII-Arg309 (right) prior to infusion of vehicle (−TF) or 70 ng/kg human TF (+TF). For blots in panels A-E and G, arrows on the right indicate the positions of cleaved HK (HKa), and positions of molecular mass standards in kilodaltons are shown to the left of the images. Plg, the position of the band representing plasminogen.
FXII-Arg309 and HK cleavage in mice
Thrombin generation was induced in mice by TF infusion. This did not result in HK cleavage in mice given FXII-WT (Figure 5C), but did in mice receiving FXII-Arg309 (Figure 5D). TF-induced HK cleavage was blocked by the thrombin inhibitor hirudin and did not occur when experiments were run in F11−/− mice (Figure 5E), supporting the conclusion that FXII-Arg309 is cleaved after residue 309 during coagulation to form δFXII. In this model, intravascular thrombin generation could have stimulated fibrinolysis, with plasmin subsequently cleaving FXII-Arg309.20 We analyzed HK cleavage in mice in which plasminogen was reduced to undetectable levels with an ASO (Figure 5F). TF induced HK cleavage in plasminogen-depleted mice receiving FXII-Arg309, but not FXII-WT (Figure 5G), indicating that plasmin is not required for HK cleavage in this model.
Anti-FXII IgG 15H8 and HK cleavage
IgG 15H8 binds the EGF2 domain of human FXII (Figure 1A), blocking contact activation and inhibiting collagen-induced thrombosis in baboons.21 In the absence of surface, however, 15H8 enhanced reciprocal PK/FXII activation, and PK activation by FXII-T (Figure 6A), suggesting that it blocks an inhibitory activity intrinsic to the FXII HC. Consistent with this, 15H8 induced HK cleavage in mice supplemented with FXII-WT (Figure 6B). In vitro, 15H8 did not induce FXII autoactivation (Figure 6C), but did enhance FXII activation by kallikrein (Figure 6D) and plasmin (Figure 6E) ∼14-fold. Therefore, 15H8 appears to neutralize an activity that normally restricts PK/FXII activation in the absence of a surface, producing an effect similar to loss of the HC in δFXII.

Effect of anti-FXII IgG 15H8 on protease activity and HK cleavage. (A) Effect of IgG 15H8 on activity of FXII-WT and FXII-T. PK (50 nM) was incubated in standard buffer at 37°C with 200 μM S-2302 and 50 nM FXII-WT (left) or FXII-T (right) in the absence () or presence () of 15H8 IgG (50 nM). For FXII-WT, a control reaction without FXII was run (). Continuous cleavage of S-2302 at 405 nM was followed. Data are means of 3 runs. (B) Western blot showing the effect of 15H8 antibody infusion on HK in WT mice supplemented with FXII-WT. The blot was processed as in Figure 5. Arrows on the right indicate the positions of cleaved HK (HKa), and positions of molecular mass standards in kilodaltons are shown to the left of the image. (C-E) Effect of IgG 15H8 on FXIIa generation. Plasma FXII (200 nM) was incubated in standard buffer at 37°C with vehicle (control) (C), 20 nM kallikrein (D), or 20 nM plasmin (E) in the absence () or presence () of 15H8 (400 nM). At various time points, aliquots were mixed with stop solution (containing HO3 for kallikrein, aprotinin for plasmin) and FXIIa concentration was determined by chromogenic assay. Data show means ± 1 SD.
Effect of anti-FXII IgG 15H8 on protease activity and HK cleavage. (A) Effect of IgG 15H8 on activity of FXII-WT and FXII-T. PK (50 nM) was incubated in standard buffer at 37°C with 200 μM S-2302 and 50 nM FXII-WT (left) or FXII-T (right) in the absence () or presence () of 15H8 IgG (50 nM). For FXII-WT, a control reaction without FXII was run (). Continuous cleavage of S-2302 at 405 nM was followed. Data are means of 3 runs. (B) Western blot showing the effect of 15H8 antibody infusion on HK in WT mice supplemented with FXII-WT. The blot was processed as in Figure 5. Arrows on the right indicate the positions of cleaved HK (HKa), and positions of molecular mass standards in kilodaltons are shown to the left of the image. (C-E) Effect of IgG 15H8 on FXIIa generation. Plasma FXII (200 nM) was incubated in standard buffer at 37°C with vehicle (control) (C), 20 nM kallikrein (D), or 20 nM plasmin (E) in the absence () or presence () of 15H8 (400 nM). At various time points, aliquots were mixed with stop solution (containing HO3 for kallikrein, aprotinin for plasmin) and FXIIa concentration was determined by chromogenic assay. Data show means ± 1 SD.
Discussion
Angioedema refers to rapid submucosal or subcutaneous soft tissue swelling that can be life-threatening if the airway or gastrointestinal tract is involved.42-44 It is common in allergic reactions, where it is triggered by histamine release from IgE-activated mast cells. Angioedema initiated by bradykinin is less common, and is usually associated with acquired or hereditary C1-INH deficiency.4,8-10 More rarely, HAE occurs in individuals with normal C1-INH activity, and has been linked to mutations in other proteins, including FXII, plasminogen, and angiopoietin 1.9,10,14,15 Lysine or arginine replacements for Thr309 in the FXII proline-rich region have been identified in several families with HAE and normal C1-INH.16-20 Trypsin-like enzymes including coagulation and fibrinolytic proteases cleave substrates after lysine or arginine.45-47 Indeed, the X-Pro-Arg sequence created by Arg309 in FXII is present in many thrombin substrates.33 The fibrinolytic protease plasmin activates FXII by cleavage after Arg353.3,10,48 de Maat et al showed that plasmin also cleaves FXII-Lys/Arg309 after residue 309, and that inducing plasminogen activation in plasma containing FXII-Lys/Arg309 enhances HK cleavage.20 Here, we show that FXII is cleaved after Lys/Arg309 by proteases generated during blood coagulation, including thrombin. This may explain trauma-induced bouts of angioedema in patients carrying these mutations.18,19 In contrast to plasmin, which cleaves FXII after Arg353 and Lys/Arg309, thrombin does not activate FXII by cleavage after Arg353,33 indicating that FXII-Lys/Arg309 activation requires a second protease.
Our data support a mechanism for enhanced bradykinin production in patients carrying FXII-Lys/Arg309 that involves a specific sequence of proteolytic reactions, as proposed recently by Jukema et al.49 The initial step is generation of a truncated form of FXII (δFXII) and not FXII activation. This could be catalyzed by any protease that attacks the Lys/Arg309-Thr310 peptide bond, including plasmin, thrombin, or FXIa. Two properties of δFXII then accelerate KKS activation. First, the intrinsic capacity of δFXII to convert PK to kallikrein is greater than that of FXII,24,33,37 leading to more kallikrein generated early during reciprocal activation. Second, δFXII is a better kallikrein substrate than is FXII. Although αFXIIa and δFXIIa are inhibited by C1-INH at roughly similar rates, the accelerated PK/FXII activation with δFXII appears to overwhelm the regulatory function of C1-INH at normal plasma levels of the serpin. Consistent with this, C1-INH infusion to achieve supraphysiologic plasma concentrations can be effective in treating angioedema in patients with FXII-Lys/Arg309.50 Our findings do not support a process in which FXII is converted first to αFXIIa, followed by truncation to δFXIIa. δFXIIa, like βFXIIa, converts PK to kallikrein, but its ability to do so is, at best, comparable to that of αFXIIa, and would not account for the increased kallikrein generation associated with FXII-Lys/Arg309. It is the superiority of δFXII as a kallikrein substrate, and not a property of δFXIIa as an enzyme, that appears to accelerate KKS activation with FXII-Lys/Arg309.
Removal of the FXII HC is central to the proposed mechanism. During contact activation, FXII, PK, and FXI bind to an activating surface.1-3,6 HK functions as a cofactor that facilitates PK and FXI surface binding.5 For FXII, the comparable cofactor activity resides in the protein’s HC region,28,34,35 and its loss greatly reduces activity in surface-dependent processes. This is illustrated by βFXIIa, a protease formed by cleaving αFXIIa after Arg334.4,7,51 βFXIIa converts PK to kallikrein in solution, but does not support surface-dependent FXII, PK, or FXI activation. δFXIIa, the activated form of δFXII, has similarly restricted activity. For enhancing reciprocal FXII/PK activation, then, there is no advantage to removing the HC after αFXIIa is generated, and such a process would not explain enhanced KKS activation associated with FXII-Lys/Arg309.
Although removing the FXII HC compromises performance in surface-dependent reactions, it enhances surface-independent FXII activation. This points to a role for this part of the protein in restricting FXII activation and, consequently, PK activation in solution. Removing the HC may expose the Arg353-Val354 peptide bond, facilitating activation. The antibody 15H8, which binds to the FXII HC, may produce a similar effect. 15H8 inhibits contact activation-induced thrombosis in mice and primates,21 but in the absence of a surface it enhances PK/FXII reciprocal activation in vitro and in vivo, mimicking loss of the HC. The results are similar to those reported by Ravon et al in 1995 for an antibody to the FXII kringle domain.36 Previously, Jukema et al proposed that removing the HC with neutrophil elastase produces a FXII fragment with increased susceptibility to activation by kallikrein or plasmin.49 It seems likely that removal of the HC can be catalyzed by several proteases that use different cleavage sites, all producing an effect similar to the one observed with δFXII. Cleavage after FXII-Lys/Arg309 in HAE patients, then, may be an example of a general process relevant to regulating FXII and perhaps other proteases. It is interesting to note that kallikrein removes the HC from hepatocyte growth factor activator, a FXII homolog, creating a “short form” of the protein that may be the major functional species.32,52 Similarly, removing the plasminogen HC with elastase results in “mini-plasminogen,” which is activated by urokinase faster than full-length plasminogen.53,54 As an aside, if truncated FXIIs with similar properties are created by cleavage at multiple sites, the term δFXII may not be ideal for the group as a whole and a revised nomenclature would be required going forward.
δFXII does not participate in contact activation because it lacks an HC. A reasonable conclusion stemming from this is that angioedema in patients with FXII-Lys/Arg309 results from solution-phase PK activation. The broader implications, however, remain uncertain. Bradykinin is almost certainly generated at a basal rate in normal health,4,7,30,55 and symptoms in patients with FXII-Lys/Arg309 may reflect loss of the regulatory activity of the FXII HC on this process. Alternatively, normal basal bradykinin generation may occur primarily on surfaces, and loss of the HC would transfer the process to the fluid phase where it may not be regulated properly, and could more easily spread systemically.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Jon Schoenecker (Vanderbilt University Medical Center, Nashville, TN) and Alexey Revenko and Gourab Bhattacharjee (Ionis Pharmaceuticals, Carlsbad, CA) for discussions on plasminogen, and supplying plasminogen ASO.
This work was supported by awards HL81326, HL58837, and HL140025 (D.G.) and HL130081 (I.M.V.) from the National Institutes of Health, National Heart, Lung, and Blood Institute.
Authorship
Contribution: I.I. designed and conducted experiments with recombinant FXII, and contributed to data interpretation and to the writing of the manuscript; A.M. contributed to experimental design and data interpretation; M.-f.S. and S.K.D. developed systems for expressing recombinant proteins and carried out initial characterizations of recombinant FXII; B.M.M. and Q.C. contributed to design and conducted animal studies; S.K. developed the antibody to murine HK and characterized its performance; I.M.V. conducted kinetic analyses; A.G. developed the anti-FXII antibody 15H8 and contributed to experimental design, data interpretation, and the writing of the manuscript; K.M. contributed to experimental design, data interpretation, and the writing of the manuscript; and D.G. oversaw experimental design and data interpretation and the writing of the manuscript.
Conflict-of-interest disclosure: D.G. is a consultant for and receives consultant’s fees from several pharmaceutical companies (Aronora, Bayer, Dyax, Ionis, Merck, Novartis, Ono) with an interest in inhibition of contact activation proteases for therapeutic purposes. A.G. and Oregon Health and Sciences University may have financial interest in the results of this study. The remaining authors declare no competing financial interests.
Correspondence: David Gailani, Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, 4918 TVC, 1301 Medical Center Dr, Nashville, TN 37232-5310; e-mail: dave.gailani@vanderbilt.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal