

Abstract
In cancer biology, tumor-promoting inflammation and an inflammatory microenvironment play a vital role in disease pathogenesis. In the past decade, aberrant innate immune activation and proinflammatory signaling within the malignant clone and the bone marrow (BM) microenvironment were identified as key pathogenic drivers of myelodysplastic syndromes (MDS). In particular, S100A9-mediated NOD-like receptor protein 3 (NLRP3) inflammasome activation directs an inflammatory, lytic form of cell death termed pyroptosis that underlies many of the hallmark features of the disease. This circuit and accompanying release of other danger-associated molecular patterns expands BM myeloid-derived suppressor cells, creating a feed-forward process propagating inflammasome activation. Furthermore, somatic gene mutations of varied functional classes license the NLRP3 inflammasome to generate a common phenotype with excess reactive oxygen species generation, Wnt/β-catenin–induced proliferation, cation flux-induced cell swelling, and caspase-1 activation. Recent investigations have shown that activation of the NLRP3 inflammasome complex has more broad-reaching importance, particularly as a possible disease-specific biomarker for MDS, and, mechanistically, as a driver of cardiovascular morbidity/mortality in individuals with age-related, clonal hematopoiesis. Recognition of the mechanistic role of aberrant innate immune activation in MDS provides a new perspective for therapeutic development that could usher in a novel class of disease-modifying agents.
Introduction
Proinflammatory cytokines have long been implicated in the ineffective hematopoiesis that characterizes the myelodysplastic syndromes (MDS). Specifically, early insights into the pathogenesis of MDS highlighted elevations of inflammatory cytokines including tumor necrosis factor-α (TNF-α) and interleukin 1 (IL-1) in MDS patients, which appeared to contribute to bone marrow (BM) progenitor cell death.1 Whether the inflammatory microenvironment in MDS was reactive or part of a central pathogenic process was only recently realized. Comprehensive molecular interrogation of blood or BM by next-generation sequencing (NGS) has identified somatic gene mutations in the majority of patients, which ushered in a paradigm shift in the use of NGS in the diagnosis, prognostic assessment, and selection of treatment of patients with MDS. At the same time, the fundamental role of innate immunity as a key driver of inflammatory signals offered new insight as to how such heterogeneous somatic genetic events in MDS converge upon a common hematological phenotype. Indeed, the extraordinary medullary expansion of innate immune effectors, myeloid-derived suppressor cells (MDSCs), and the disease-specific role of a novel inflammatory form of programmed cell death, pyroptosis, are key features of the disease that when successfully targeted, offer the prospect for development of new, biologically rational therapeutic strategies. Aberrant activation of innate immune networks by reciprocal interactions of cell-intrinsic genetic events and cell-extrinsic microenvironmental pressures is now recognized not only as a fundamental driver of MDS pathogenesis, but also as a critical driver in the cardiovascular (CV) morbidity and mortality that accompanies age-related clonal hematopoiesis. Recognition that these divergent pathogenic processes are integrally linked offers new avenues for therapeutic exploitation.
Innate immune signaling in MDS
The innate immune system is activated through the interaction of pathogen-associated molecular patterns (PAMPs) or host cell–derived danger-associated molecular patterns (DAMPs) with pattern recognition receptors (PRRs), with the Toll-like receptors (TLRs) representing the most extensively studied PRR family. TLR activation initiates a complex signaling cascade that is critical to antimicrobial host defense and adaptive immune response.2,3 TLRs, together with the IL-1 receptors, are members of a superfamily known as the “IL-1 receptor/TLR superfamily,” which characteristically has a so-called Toll–IL-1 receptor (TIR) domain. TLR signaling largely occurs via the cytoplasmic adapter myeloid differentiation primary response (MyD88) and less commonly with TLR3 through TIR domain–containing adapter-inducing interferon-β–dependent pathways, ultimately leading to interleukin receptor–associated kinase-1 (IRAK1) and IRAK4 phosphorylation and the recruitment of TNF receptor–associated factor 6 (TRAF6), followed by NF-κB and MAPK activation, respectively (Figure 1). Unrestrained TLR signaling, however, has been implicated in autoimmune and inflammatory diseases, including MDS, which was recently reviewed.4-6 TLRs are overexpressed in hematopoietic stem and progenitor cells (HSPCs) in MDS compared with age-matched controls. TLR-4 expression and signaling, in particular, play an important role in CD34+ cell death in MDS.7,8 TLR-2 is also deregulated in BM CD34+ cells, particularly in lower-risk disease, which can induce cell death via β-arrestin 1, leading to histone H4 acetylation,9,10 whereas transcriptional silencing of TLR-2 restores effective erythopoesis.10
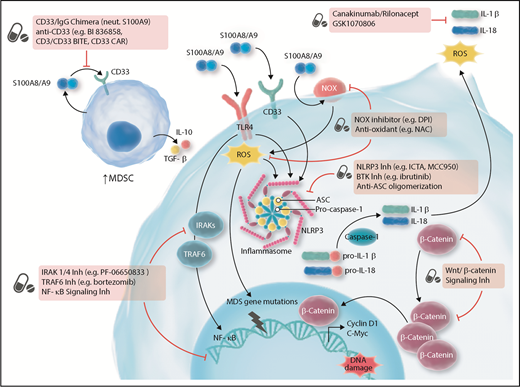
Targeting innate and inflammatory signaling for the treatment of MDS. ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain; BiTE, bispecific T-cell engager; BTK, Bruton tyrosine kinase; CAR, chimeric antigen receptor; DPI, diphenyleneiodonium; IgG, immunoglobulin G; Inh, inhibitor; NAC, N-acetyl-l-cysteine; neut, neutralize; NLRP3, NOD-like receptor protein 3; NOX, nicotinamide-adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species; TGF, transforming growth factor. Adapted from Sallman et al.78 Professional illustration by Somersault18:24.
Targeting innate and inflammatory signaling for the treatment of MDS. ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain; BiTE, bispecific T-cell engager; BTK, Bruton tyrosine kinase; CAR, chimeric antigen receptor; DPI, diphenyleneiodonium; IgG, immunoglobulin G; Inh, inhibitor; NAC, N-acetyl-l-cysteine; neut, neutralize; NLRP3, NOD-like receptor protein 3; NOX, nicotinamide-adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species; TGF, transforming growth factor. Adapted from Sallman et al.78 Professional illustration by Somersault18:24.
IRAK1, a serine/threonine kinase, and TRAF6, an E3 ubiquitin ligase, are key signaling intermediates that play a fundamental role in MDS pathobiology. Elegant investigations into the pathogenesis of deletion 5q (del(5q)) MDS have identified that genetic alterations can directly activate innate immune signaling (Table 1). Specifically, del(5q) MDS results in miR-145 and miR-146 haploinsufficiency leading to TRAF6 overexpression.11 In vivo, knockdown of miR-145/146 or overexpression of TRAF6 recapitulated features of the del(5q) phenotype, including megakaryocytic dysplasia, thrombocytosis, and neutropenia.11 Del(5q) also leads to haploinsufficiency of TRAF-interacting protein with forkhead-associated domain B, which cooperates with miR-146 haploinsufficiency to further increase TRAF6 with consequent activation of TLR signaling and hematopoietic impairment.8 Additionally, in a mDia1/mir-146a dual-deficient mouse model, “inflammaging” was shown to drive ineffective erythropoiesis via DAMP induction of TNF-α and IL-6, and secondary generation of reactive oxygen species (ROS).12 Furthermore, mir-146 is also a negative regulator of IRAK1.13 Subsequently, Rhyasen and colleagues found that IRAK1 overexpression and hyperactivation occurs routinely in MDS.14 More importantly, small molecule inhibition of IRAK1/4 blocked downstream TRAF6/NF-κB activation and was selectively toxic to MDS cells while sparing normal CD34+ cells (Figure 1).14 Notably, IRAK1 inhibition resulted in a compensatory increase in B-cell lymphoma-2 (BCL-2) expression, highlighting a potential resistance mechanism. Notably, coadministration of BCL-2/IRAK inhibitors more effectively suppressed the MDS clone.
Impact of genetic events on innate and inflammasome signaling
| Genetic abnormality | Gene class | Mutant gene/ Chromosome alteration | Innate immune-signaling effect* | Inflammasome-signaling effect† | Ref. |
|---|---|---|---|---|---|
| Somatic mutations | Epigenetic modifiers | TET2 | ↑ IL-6 production via ↓ HDAC2 recruitment; ↑ IL-1β | ↑ Pyroptosis and β-catenin signaling | 28, 30, 40 |
| DNMT3A | ↑ Type 1 IFN production via ↑ HDAC9 expression | 29 | |||
| ASXL1 | ↑ NADPH oxidase ROS; ↑ TLR4, TICAM2 | ↑ Pyroptosis and β-catenin signaling | 31, 40 | ||
| EZH2 | ↑ S100A8/A9 via NF-κB derepression | 25 | |||
| Spliceosomes | SF3B1 | ↑ NF-κB activation via ↓ MAP3K7 | ↑ Pyroptosis and β-catenin signaling | 32, 40 | |
| SRSF2 | ↑ S100A8 and S100A9, DNA-RNA hybrids; ↑ NF-κB activation via caspase 8 isoform | ↑ Pyroptosis and β-catenin signaling | 24, 32, 40 | ||
| U2AF1 | ↑ DNA-RNA hybrids, ATG7 alternate splicing impairing autophagy | ↑ Pyroptosis and β-catenin signaling ; impaired autophagy leading to NLRP3 activation | 40, 43, 44 | ||
| Chromosomal abnormality | N/A | Deletion 5q | Haploinsufficiency: RPS14 ↑ S100A8/A9; miR-145/146 + TIFAB ↑ TRAF6/IRAK1 | ↑ Pyroptosis and β-catenin signaling | 8, 11, 22 |
| Genetic abnormality | Gene class | Mutant gene/ Chromosome alteration | Innate immune-signaling effect* | Inflammasome-signaling effect† | Ref. |
|---|---|---|---|---|---|
| Somatic mutations | Epigenetic modifiers | TET2 | ↑ IL-6 production via ↓ HDAC2 recruitment; ↑ IL-1β | ↑ Pyroptosis and β-catenin signaling | 28, 30, 40 |
| DNMT3A | ↑ Type 1 IFN production via ↑ HDAC9 expression | 29 | |||
| ASXL1 | ↑ NADPH oxidase ROS; ↑ TLR4, TICAM2 | ↑ Pyroptosis and β-catenin signaling | 31, 40 | ||
| EZH2 | ↑ S100A8/A9 via NF-κB derepression | 25 | |||
| Spliceosomes | SF3B1 | ↑ NF-κB activation via ↓ MAP3K7 | ↑ Pyroptosis and β-catenin signaling | 32, 40 | |
| SRSF2 | ↑ S100A8 and S100A9, DNA-RNA hybrids; ↑ NF-κB activation via caspase 8 isoform | ↑ Pyroptosis and β-catenin signaling | 24, 32, 40 | ||
| U2AF1 | ↑ DNA-RNA hybrids, ATG7 alternate splicing impairing autophagy | ↑ Pyroptosis and β-catenin signaling ; impaired autophagy leading to NLRP3 activation | 40, 43, 44 | ||
| Chromosomal abnormality | N/A | Deletion 5q | Haploinsufficiency: RPS14 ↑ S100A8/A9; miR-145/146 + TIFAB ↑ TRAF6/IRAK1 | ↑ Pyroptosis and β-catenin signaling | 8, 11, 22 |
HDAC, histone deacetylase; IFN, interferon; miR, microRNA; N/A, not applicable; NADPH, dihydronicotinamide-adenine dinucleotide phosphate; NLRP3, NOD-like receptor protein 3; Ref., reference(s); ROS, reactive oxygen species; RPS14, ribosomal protein small subunit 14; TICAM2, TIR domain-containing adapter molecule 2; TIFAB, TRAF-interacting protein with forkhead-associated domain B.
Arrows indicate (↑) increase and (↓) decrease.
Arrow (↑) indicates that pyroptosis would be driven by NLRP3 inflammasome activation.
Reciprocal interaction between the BM microenvironment and S100A8/A9 in MDS pathogenesis
MDS HSPCs are specifically susceptible to DAMPs by virtue of upregulation of PRRs, and, in some cases as in del(5q), genetic priming of receptor signaling. Critical soluble intermediaries in this process are the inflammatory proteins, S100A8 and S100A9. These cation-binding proteins preferentially heterodimerize to form calprotectin, which, like the monomeric forms, are endogenous ligands for TLR-4 and the receptor for advanced glycated end products.15 These specific myeloid-related proteins play a fundamental role in the sterile inflammation of both autoimmune and malignant disorders via autocrine and paracrine interactions.16 Receptor engagement by S100A9 activates and expands MDSCs in solid tumor models, which serve to induce immune tolerance. Notably, activated MDSCs also secrete S100A8/A9, thereby fueling both autocrine and paracrine stimulation of proximate bystander cells, and reinforcing inflammatory cytokine generation (Figure 1).17
In MDS in particular, MDSCs are key effectors of cytopenias, particularly in lower-risk disease (Figure 1). Specifically, MDSCs, immature myeloid cells characterized by a CD33+/Lin−/HLA-DR− surface phenotype, are profoundly expanded in the BM of MDS patients as a result of S100A9 engagement of TLRs, and its high-affinity sialic acid-binding immunoglobulin-type lectins (Siglec)-family receptor, CD33.18 CD33 activation, via its immunoreceptor tyrosine-based inhibition motif, triggers elaboration of the immunosuppressive cytokines IL-10 and transforming growth factor-β, and granzyme-B mobilization to directly suppress hematopoiesis. Indeed, in a S100A9 transgenic mouse model, MDSC accumulation over time was accompanied by development of an age-dependent MDS phenotype that was reversed by MDSC depletion or inhibition of the S100A9/CD33/TLR4 axis. More importantly, MDSCs in primary BM specimens lack somatic gene mutations found in the MDS clone, indicating that MDS-MDSCs are genetically distinct and not derived from the malignant clone. Additionally, other cellular components of innate and adaptive immunity are dysregulated in MDS including macrophages, natural killer cells, and T-lymphocyte subsets.19 However, the interaction of these immune cell populations with inflammatory signaling in MDS has not been defined.
Further evidence that the S100A8/9-TLR axis and BM stromal cells play an initiating oncogenic role in MDS was provided by a series of elegant studies by Raaijmakers et al.20 Specifically, Dicer1 gene deletion in mouse mesenchymal osteoprogenitors perturbed hematopoiesis and fostered emergence of a genetically distinct, MDS clone.20 Using a mouse model of the preleukemic disorder, Schwachman-Diamond syndrome (SDS), they later showed that Sbds gene deletion in the osteoid niche was sufficient to induce MDS transformation. Mechanistically, genetic alteration of the osteoid progenitors activated the p53-S100A8/9-TLR axis, inducing mitochondrial dysfunction and oxidative genotoxicity in HSPCs.21 Identification of S100A8/A9 overexpression was performed via unbiased whole transcriptome sequencing of purified CD271+ mesenchymal stromal cells in SDS and MDS patients. Furthermore, activation of the S100A8/9-TLR axis in primary MDS BM specimens was linked to leukemic evolution and progression-free survival. Together, these data suggest that the mesenchymal compartment plays a critical role in the disease phenotype and could be the initiating event in some patients. This innate immune circuit is similarly licensed by specific genetic events in MDS (Table 1). Schneider et al showed in a ribosomal protein small subunit 14 (RPS14) haploinsufficient mouse model that phenocopies the dyserythropoiesis in del(5q) MDS, that erythroid cell death was dependent upon activation of the p53-S100A8/9-TLR4 axis, while inhibition of this pathway restored hematopoiesis.22 Specifically, identification of innate immune activation was performed via quantitative proteomics of purified erythroid progenitor cells where S100A8/A9 were the proteins most highly enriched in haplodeficient cells. These observations were validated by immunohistochemistry and messenger RNA (mRNA) quantification, demonstrating increased S100A8 expression in erythroid progenitors as well as in monocytes and macrophages. Notably, the phenotypic impact of S100A8/A9-TLR axis activation may be dependent upon the spatial interaction with S100 protein-expressing stromal cells, secondary to local accumulation in extracellular matrices.23 Specifically, inflammatory mesenchymal niche cells, which are directly adjacent to CD34+ HSPCs, predominantly affected HSPCs whereas inflammatory monocytes/macrophages, present in the erythroblastic island, predominantly induced an erythroid differentiation block.21,22 Moreover, transcriptome analysis shows that S100A8/9 is highly upregulated by other common MDS somatic mutations such as Srsf2P95H.24 Additionally, recent investigations indicate that wild-type EZH2 represses NF-κB,25 suggesting that S100A9 upregulation with inactivating Ezh2 gene mutations is mediated by NF-κB derepression. Lastly, the epigenetic regulator KDM6B, which is overexpressed in HSPCs of MDS patients,26 is a transcriptional activator of S100A9 and other inflammatory response genes.27 Importantly, small molecule inhibition of KDM6B restored effective hematopoiesis.
Inactivating mutations of other genes in MDS that normally function in extinguishing transcription of TLR-induced proinflammatory cytokines reinforce the inflammatory microenvironment (Table 1). Ten-eleven translocation 2 (TET2) and DNA (cytosine-5)-methyltransferase 3A (DNMT3A) gene mutations impair resolution of inflammation and type I interferon production, respectively.28,29 Independent of its role in modifying DNA methylation, TET2 is necessary to extinguish the late-phase inflammatory response to TLR-4 agonists by repressing IL-6 transcription through recruitment of Hdac2 to repress IL-6 transcription by histone deacetylation.28 Indeed, TET2 inactivation results in upregulation of inflammatory cytokines in response to lipopolysaccharide (LPS) and a severe inflammatory phenotype in TET2-deficient mice, indicating that disruption of the TET2/NF-κB regulatory circuit by inactivating mutations fosters unrestrained inflammation and inflammasome priming in response to TLR-4 activation. Additionally, both TET1 and TET2 are negative regulators of IL-1β. Consequently, loss-of-function mutations in TET2 increase IL-1β elaboration, thereby augmenting the proinflammatory BM niche.30 Further supporting a role of epigenetic mutations in innate immune activation, TLR-4 and TIR domain-containing adapter molecule 2 (TICAM2) were both upregulated in an addition of sex combs like 1 (ASXL1) knockout mouse model.31 Lastly, mutations of the spliceosome machinery have recently been identified to result in activation of innate immune signaling.32 Although mutations in SF3B1 and SRSF2 impart distinct perturbations of mRNA splicing, both of these mutations functionally converge with hyperactivation of NF-κB. Specifically, NF-κB activation in SF3B1 mutant cells occurs via downregulation of MAP3K7 whereas in SRSF2 mutant cells, it is mediated by generation of a C-terminal truncated caspase-8 isoform.
NLRP3 inflammasome and pyroptosis drive the MDS phenotype
Although apoptosis, a noninflammatory caspase-3–mediated cell death, had long been implicated in the ineffective hematopoiesis of MDS, PRR engagement by DAMPs or PAMPs directs a proinflammatory, caspase-1–dependent lytic cell death, termed pyroptosis (Figure 2).3 Pyroptotic cell death is mediated by multiprotein inflammasome complexes initiated by nucleotide-binding domain and leucine-rich repeat (NLR) PRRs.33 NOD-like receptor protein 3 (NLRP3), a redox-sensitive NLR, recruits the adaptor apoptosis-associated speck like protein containing a caspase-recruitment domain (ASC) upon activation.33 This interaction triggers rapid ASC polymerization creating large, filamentous, helical clusters termed ASC specks that serve as a platform for the recruitment and autocatalytic cleavage of pro–caspase-1. Active caspase-1 converts pro–IL-1β and pro–IL-18 to their active forms, as well as the pore-forming protein gasdermin D, which serves as the executioner of cytolysis.34 The N-terminal p30 fragment of gasdermin D oligomerizes before binding to the plasma membrane inner leaflet to create nonselective membrane pores of 10 to 20 nm diameter.34-37 These pores serve as a conduit for cation entry causing cell swelling, and for passive release of soluble intracellular proteins such as mature IL-1β and IL-18.38 Notably, the alarmins S100A8/A9 and oxidized mitochondrial DNA are potent activators of the NLRP3 inflammasome.39
![Figure 2. S100A8/9-mediated inflammasome formation and pyroptosis defines MDS phenotype. (A) S100A8/A9 heterodimers bind both CD33 and TLR4, resulting in NLRP3 inflammasome assembly. Ligation of S100A8/A9 to TLR4 results in NF-κB–mediated transcription and subsequent production of proinflammatory cytokines such as pro-IL-1β and pro-IL-18, along with inflammasome components. (B) S100A8/A9 promotes activation of NADPH oxidase (NOX), which results in dual processes. First, NOX proteins generate ROS, with increasing levels leading to thioredoxin-interacting protein (TXNIP) dissociation from thioredoxin (TRX) with consequent binding to NLPR3 and inflammasome assembly. Second, NOX-derived ROS results in oxidation of nuceloredoxin (NRX), leading to its dissociation from Dishevelled (Dvl). Once dissociated, Dvl suppress the β-catenin destruction complex (glycogen synthase kinase 3 [GSKβ]/casein kinase 1 [CK1]/adenomatous polyposis coli [APC]/Axin/protein phosphatase 2A [PP2A]), resulting in stabilization of β-catenin. This allows β-catenin to enter the nucleus and induce transcription of T-cell factor (TCF) controlled genes, including cyclin-D1 and c-Myc. (C) Inflammasome assembly occurs through activation of NLRP3, triggering the recruitment of the apoptosis-associated speck like protein containing a caspase-recruitment domain (ASC), leading to the formation of cytosolic, heptameric complexes which serve as a platform for activation of pro-caspase-1. Once activated, inflammasomes mediate conversion of pro-caspase-1 to its mature and catalytically active form. Active caspase-1 cleaves pro-IL-1β and pro-IL-18 to their mature forms. (D) MDS-related gene mutations activate NF-κB and NLRP3 via NOX-generated ROS. Dotted line highlights potential mechanism that has not been definitively proven in MDS. β-Cat, β-Catenin; BCL, B-cell lymphoma; CARD, caspase activation and recruitment domain; CBP, CREB-binding protein; l-Arg, l-arginine; LRR, leucine-rich repeat; NOS, nitric oxide synthase; P, phosphate; PYD, pyrin domain.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/10/10.1182_blood-2018-10-844654/3/m_blood844654f2.png?Expires=1770015406&Signature=VvX7NnAex8G2pQI05Aq9ldkMAtd3a4RsxkEu3ICXi9Nge8I-GGYeXmJEF7BEtgOZBMfMfBrbagSoH69XamPszXkfwzmwSr-7qID0EbXY07SCZVVqAxNtC-rfPxs18Sc4hiKJZnX9A2ZkChMj89XnzTa06PN-ISCcoOzYWqiwSL1fWftUBqFz2-lRFy~zF8HQyvNBulHdXAic1msGwzzYwM9FexmkuncPzDneT83OnOl0DTBnxVsubluCgSdv9-ITejVEiJkjcfuhurEDH0hFQFDQ8LeELL4SH~BspOynw0ptrBkgv5Yq~dpgnlKUIKDSR3x234YZQYtb2cZveNVZAA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
S100A8/9-mediated inflammasome formation and pyroptosis defines MDS phenotype. (A) S100A8/A9 heterodimers bind both CD33 and TLR4, resulting in NLRP3 inflammasome assembly. Ligation of S100A8/A9 to TLR4 results in NF-κB–mediated transcription and subsequent production of proinflammatory cytokines such as pro-IL-1β and pro-IL-18, along with inflammasome components. (B) S100A8/A9 promotes activation of NADPH oxidase (NOX), which results in dual processes. First, NOX proteins generate ROS, with increasing levels leading to thioredoxin-interacting protein (TXNIP) dissociation from thioredoxin (TRX) with consequent binding to NLPR3 and inflammasome assembly. Second, NOX-derived ROS results in oxidation of nuceloredoxin (NRX), leading to its dissociation from Dishevelled (Dvl). Once dissociated, Dvl suppress the β-catenin destruction complex (glycogen synthase kinase 3 [GSKβ]/casein kinase 1 [CK1]/adenomatous polyposis coli [APC]/Axin/protein phosphatase 2A [PP2A]), resulting in stabilization of β-catenin. This allows β-catenin to enter the nucleus and induce transcription of T-cell factor (TCF) controlled genes, including cyclin-D1 and c-Myc. (C) Inflammasome assembly occurs through activation of NLRP3, triggering the recruitment of the apoptosis-associated speck like protein containing a caspase-recruitment domain (ASC), leading to the formation of cytosolic, heptameric complexes which serve as a platform for activation of pro-caspase-1. Once activated, inflammasomes mediate conversion of pro-caspase-1 to its mature and catalytically active form. Active caspase-1 cleaves pro-IL-1β and pro-IL-18 to their mature forms. (D) MDS-related gene mutations activate NF-κB and NLRP3 via NOX-generated ROS. Dotted line highlights potential mechanism that has not been definitively proven in MDS. β-Cat, β-Catenin; BCL, B-cell lymphoma; CARD, caspase activation and recruitment domain; CBP, CREB-binding protein; l-Arg, l-arginine; LRR, leucine-rich repeat; NOS, nitric oxide synthase; P, phosphate; PYD, pyrin domain.
S100A8/9-mediated inflammasome formation and pyroptosis defines MDS phenotype. (A) S100A8/A9 heterodimers bind both CD33 and TLR4, resulting in NLRP3 inflammasome assembly. Ligation of S100A8/A9 to TLR4 results in NF-κB–mediated transcription and subsequent production of proinflammatory cytokines such as pro-IL-1β and pro-IL-18, along with inflammasome components. (B) S100A8/A9 promotes activation of NADPH oxidase (NOX), which results in dual processes. First, NOX proteins generate ROS, with increasing levels leading to thioredoxin-interacting protein (TXNIP) dissociation from thioredoxin (TRX) with consequent binding to NLPR3 and inflammasome assembly. Second, NOX-derived ROS results in oxidation of nuceloredoxin (NRX), leading to its dissociation from Dishevelled (Dvl). Once dissociated, Dvl suppress the β-catenin destruction complex (glycogen synthase kinase 3 [GSKβ]/casein kinase 1 [CK1]/adenomatous polyposis coli [APC]/Axin/protein phosphatase 2A [PP2A]), resulting in stabilization of β-catenin. This allows β-catenin to enter the nucleus and induce transcription of T-cell factor (TCF) controlled genes, including cyclin-D1 and c-Myc. (C) Inflammasome assembly occurs through activation of NLRP3, triggering the recruitment of the apoptosis-associated speck like protein containing a caspase-recruitment domain (ASC), leading to the formation of cytosolic, heptameric complexes which serve as a platform for activation of pro-caspase-1. Once activated, inflammasomes mediate conversion of pro-caspase-1 to its mature and catalytically active form. Active caspase-1 cleaves pro-IL-1β and pro-IL-18 to their mature forms. (D) MDS-related gene mutations activate NF-κB and NLRP3 via NOX-generated ROS. Dotted line highlights potential mechanism that has not been definitively proven in MDS. β-Cat, β-Catenin; BCL, B-cell lymphoma; CARD, caspase activation and recruitment domain; CBP, CREB-binding protein; l-Arg, l-arginine; LRR, leucine-rich repeat; NOS, nitric oxide synthase; P, phosphate; PYD, pyrin domain.
Basiorka et al reported that pyroptosis, not apoptosis, drives cell death in MDS, serving as a common pathway licensed by cell-intrinsic genetic events such as somatic gene mutations and/or cell-extrinsic signals from alarmins (eg, S100A8/A9).40 Moreover, NLRP3 inflammasome activation was redox-dependent, and responsible for many of the hallmark features of MDS such as macrocytosis, β-catenin–instructed proliferation, and ineffective hematopoiesis (Figure 2).4,40 S100A9 protein content is profoundly increased in HSPCs of MDS patients as well as in BM plasma, and highest in lower-risk disease.40 Increased circulating S100A8 in MDS patients was also identified in a separate study in comparison with patients with aplastic anemia or healthy donors.41 S100A9 transgenic mice recapitulate these features, developing time-dependent progressive cytopenias mediated by NLRP3 inflammasome-induced pyroptosis.40 Recombinant human S100A9 treatment of normal BM mononuclear cells was sufficient to induce inflammasome assembly and caspase-1 activation. Lastly, S100A9 and/or gene mutations were able to induce ROS generation leading to β-catenin activation. Importantly, inhibition of multiple points of inflammasome signaling was sufficient to restore effective hematopoiesis. Both neutralization of S100A9 in MDS BM plasma or pharmacologic inhibition of inflammasome assembly suppressed pyroptosis and restored colony-forming capacity in vitro and effective hematopoiesis in the S100A9Tg murine MDS model. Furthermore, neutralization of S100A9 may also augment erythropoiesis by derepression of erythropoietin elaboration. Specifically, S100A9 significantly suppresses production of erythropoietin, which could be inhibited by treatment with lenalidomide.42
Notably, Basiorka et al found that the NLRP3-pyroptosis axis was activated in primary MDS specimens irrespective of functional classes of somatic gene mutation (Figure 2).40 Specifically, HSPCs from mouse or transduced cell line models of varied RNA splicing gene (U2AF1, SF3B1, and SRSF2) and epigenetic regulatory gene (ASXL1 and TET2) mutations similarly displayed high pyroptotic fractions, pore formation with cell volume expansion, and induction of nuclear β-catenin and corresponding target genes that was suppressed by inhibition of the NLRP3 inflammasome or NADPH-derived ROS. Interestingly, the proportion of pyroptotic erythroid progenitors in primary MDS BM specimens increased directly with somatic gene mutation allele burden and mutation complexity. Precisely how somatic mutations of disparate functional classes activate the NLRP3 inflammasome is not clear, but all share dependence on ROS induction. Chen et al recently reported that SRSF2 and U2AF1 splicing mutations trigger replication stress by ATR-Chk1–induced transcriptional pauses that generates excess genome-wide DNA:RNA hybrids.43 Importantly, impaired colony-forming capacity in mutant HSPCs was rescued partly by RNase H overexpression, indicating that excess DNA:RNA hybrids directly contribute to accelerated cell death. Inflammasome activation is normally restrained by autophagy through removal and recycling of damaged organelles (eg, mitochondria). Using RNA sequence analysis, Park et al recently found that U2AF1S34F mutations promote usage of a distal poly(A) site in autophagy-related factor 7 (Atg7) mRNA that yields a longer and poorly translated transcript.44 The consequent depletion of ATG7 protein disrupted autophagy and predisposed mutant cells to secondary somatic gene mutations. In a separate study, myeloid-specific ablation of either Atg7, Parkin, or the p62/Sqstm1 mitochondrial autophagy adapter in mice, each of which is indispensable for autophagy integrity, resulted in NLRP3 inflammasome activation and development of severe IL-1β–mediated immunopathologies.45
Given the importance of NLRP3 inflammasome-directed pyroptosis in MDS pathogenesis, recent studies have investigated its disease specificity and whether pathway components could serve as disease-specific biomarkers or an index of the extent of medullary pyroptosis. ASC specks formed during inflammasome assembly are released upon pyroptotic cytolysis and are resistant to proteolytic degradation.46 ASC-speck–bound caspase1 remains catalytically active, thereby augmenting IL-1β and IL-18 maturation in the extracellular space, and when engulfed or phagocytosed, induce pyroptosis.24,47 We recently reported that ASC specks of 1 to 3 μm in diameter are demonstrable in MDS primary BM specimens by immunofluorescence as a solitary paranuclear complex, and can be quantitated by flow cytometry in the peripheral blood and/or BM plasma from patients with MDS.48 Peripheral blood ASC-speck percentage directly correlated with plasma concentrations of the endogenous alarmins S100A8 and S100A9, confirming the relationship to pyroptosis. Notably, not only was ASC-speck percentage significantly higher in plasma from MDS patients compared with age-matched healthy controls, but ASC-speck levels were also highest in patients with lower-risk MDS, whereas levels in other lymphoid or myeloid malignancies were either normal or lower, findings that were confirmed in an independent validation cohort (P < .0001). The percentage of plasma specks significantly increased proportionate to somatic gene mutation number, underscoring the pathobiological relevance of the biomarker. The percentage of plasma ASC specks proved to be a robust marker for pyroptosis in MDS (area under the curve = 0.888) and, at a cutoff of 0.80, maximized both sensitivity (0.84; 95% confience interval [CI], 0.65-0.91) and specificity (0.87; 95% CI, 0.58-0.97). These findings emphasize the pathobiological relevance of ASC specks and their potential usefulness as a sensitive and specific biomarker of the index of medullary pyroptotic cell death, which may prove useful in future therapeutic investigations of inflammasome inhibition. Future investigations should discern whether ASC specks may have utility as a marker of MDS progression in patients with clonal hematopoiesis of indeterminate potential (CHIP). Whether “inflammaging” is a driver of clonal hematopoiesis or whether clonal hematopoiesis directly leads to a proinflammatory state is not clear, although both mechanisms can be supported by studies to date.
Novel therapeutic approaches targeting innate/inflammatory signaling in MDS
Management of patients with MDS is based upon clinical prognostic risk assessment by models such as the International Prognostic Scoring System (IPSS) and the revised IPSS (IPSS-R), generally dichotomizing patients into lower- or higher-risk disease.49,50 To date, approved disease-modifying treatments for MDS include lenalidomide for lower-risk MDS with del(5q) and hypomethylating agents (HMAs; ie, azacitidine and decitabine) for higher-risk MDS.51,52 To date, HMAs have not been shown to be disease modifying in lower-risk MDS patients. These studies have identified a fundamental role of innate/inflammatory signaling that could be exploited therapeutically, particularly in lower-risk MDS patients (Figure 1; Table 2).
Novel therapeutic agents targeting innate/inflammatory signaling for the treatment of MDS
| Therapeutic class | Agent | Novel features | Patient population | Stage of development | Preliminary results | NCT no. | Ref. |
|---|---|---|---|---|---|---|---|
| TLR signaling inh | OPN-305 | Anti-TLR2 monoclonal antibody | HMA failure lower-risk MDS | Phase 1-2 | 50% HI (6/12); 17% TI (2/12) | NCT02363491 | 53 |
| Bortezomib | TRAF6 inh via bortezomib-mediated autophagy | R/R lower and higher-risk MDS and AML; also in combination with Len, HMA, or HDACi | Phase 1-2 | 20% HI (3/15) | NCT01891968 | 55 | |
| MDSC elimination | BI 836858 | Anti-CD33 monoclonal antibody (reduce MDSCs by ADCC) | R/R lower-risk MDS (HMA naïve and HMA failure) | Phase 1-2 | N/A | NCT02240706 | 56 |
| AMV564 | CD33/CD3 BiTE | HMA failure higher-risk MDS | Phase 1 | N/A | NCT03516591 | ||
| 161533 | CD16/IL-15/CD33 TriKE | R/R higher-risk MDS, AML, or advanced SM | Phase 1-2 | N/A | NCT03214666 | ||
| NLRP3 inflammasome inhibitors | PF-06650833 | IRAK 4 inh (↓ NLRP3 transcription); synergy with BCL-2 inh | No current MDS trial | Phase 1 | N/A | NCT02996500 | |
| MCC950 | Specific NLRP3 inh (used as prototypic inh in laboratory models) | No current MDS trial | N/A | N/A | 59 | ||
| Ibrutinib | BTK inh; directly binds ASC and NLRP3; inh ASC speck formation | HMA failure higher-risk MDS; in combination with Len or AZA | Phase 1 | N/A | NCT03359460; NCT02553941 | 62 | |
| IL-1β inhibitors | Canakinumab | IL-1β neutralizing monoclonal antibody; ↓ MDSC | Planned R/R lower-risk MDS; approved for CAPS | Phase 1-2 | N/A | 75 | |
| Rilonacept | Soluble decoy receptor that binds IL-1β and IL-1α | No current MDS trial; approved for CAPS | N/A | N/A | |||
| Anakinra | Direct antagonist of the IL-1R | No current MDS trial; approved for CAPS | N/A | N/A |
| Therapeutic class | Agent | Novel features | Patient population | Stage of development | Preliminary results | NCT no. | Ref. |
|---|---|---|---|---|---|---|---|
| TLR signaling inh | OPN-305 | Anti-TLR2 monoclonal antibody | HMA failure lower-risk MDS | Phase 1-2 | 50% HI (6/12); 17% TI (2/12) | NCT02363491 | 53 |
| Bortezomib | TRAF6 inh via bortezomib-mediated autophagy | R/R lower and higher-risk MDS and AML; also in combination with Len, HMA, or HDACi | Phase 1-2 | 20% HI (3/15) | NCT01891968 | 55 | |
| MDSC elimination | BI 836858 | Anti-CD33 monoclonal antibody (reduce MDSCs by ADCC) | R/R lower-risk MDS (HMA naïve and HMA failure) | Phase 1-2 | N/A | NCT02240706 | 56 |
| AMV564 | CD33/CD3 BiTE | HMA failure higher-risk MDS | Phase 1 | N/A | NCT03516591 | ||
| 161533 | CD16/IL-15/CD33 TriKE | R/R higher-risk MDS, AML, or advanced SM | Phase 1-2 | N/A | NCT03214666 | ||
| NLRP3 inflammasome inhibitors | PF-06650833 | IRAK 4 inh (↓ NLRP3 transcription); synergy with BCL-2 inh | No current MDS trial | Phase 1 | N/A | NCT02996500 | |
| MCC950 | Specific NLRP3 inh (used as prototypic inh in laboratory models) | No current MDS trial | N/A | N/A | 59 | ||
| Ibrutinib | BTK inh; directly binds ASC and NLRP3; inh ASC speck formation | HMA failure higher-risk MDS; in combination with Len or AZA | Phase 1 | N/A | NCT03359460; NCT02553941 | 62 | |
| IL-1β inhibitors | Canakinumab | IL-1β neutralizing monoclonal antibody; ↓ MDSC | Planned R/R lower-risk MDS; approved for CAPS | Phase 1-2 | N/A | 75 | |
| Rilonacept | Soluble decoy receptor that binds IL-1β and IL-1α | No current MDS trial; approved for CAPS | N/A | N/A | |||
| Anakinra | Direct antagonist of the IL-1R | No current MDS trial; approved for CAPS | N/A | N/A |
ADCC, antibody-dependent cellular cytotoxicity; AML, acute myeloid leukemia; ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain; AZA, azacitidine; BiTE, bispecific T-cell engager; BTK, Bruton tyrosine kinase; CAPS, cryopyrin-associated periodic syndrome; HDACi, histone deacetylase inhibitor; HI, hematologic improvement; HMA, hypomethylating agent; inh, inhibitor; Len, lenalidomide; NCT, National Clinical Trial; R/R, relapse/refractory; SM, systemic mastocytosis; TI, transfusion independent; TriKE, trispecific killer cell engager. Other abbreviations are explained in Table 1.
One approach intended to antagonize TLR signaling is OPN-305, a fully humanized IgG4 κ monoclonal antibody TLR-2 antagonist. In a phase 1 trial performed in patients with lower-risk MDS, 50% of patients experienced hematologic improvement and 2 achieved red blood cell transfusion independence (NCT02363491).53 Additionally, the TLR-signaling intermediate TRAF6 can be degraded through bortezomib-mediated lysosomal degradation of TRAF6 and consequent induction of cell death.54 As proof in principle, bortezomib treatment resulted in hematologic improvement in 20% (3 of 15) of lower-risk MDS patients, with accompanying reduction in NF-κB as evidenced by reduction in pp65 levels.55 Novel strategies to deplete the key innate immune cellular effectors, MDSCs, are currently in development. In March 2017, the US Food and Drug Administration (FDA) gave orphan drug approval for clinical investigation of the monoclonal antibody BI 836858, which is engineered for improved antibody-dependent cell-mediated cytotoxicity (ADCC) in lower-risk MDS (NCT02240706).56 Additional CD33-directed therapeutics under active investigation in MDS include novel technologies such as bispecific T-cell engager (BiTE) antibodies, trispecific killer cell engagers (TriKE), the quadrivalent CD33/CD3 T-cell engager AMV564, and chimeric antigen receptor engineered T cells (CARs).
Inhibition of the NLRP3 inflammasome is not only an attractive therapeutic strategy for MDS, but also a wide array of chronic inflammatory diseases.57 Activation of NLRP3 is tightly regulated with multiple candidate approaches for inflammasome inhibition. Small molecule inhibitors of IRAK1 and IRAK4 show selective cytotoxicity to MDS HSPCs and display synergy with BCL-2 antagonists.14 An additional benefit in MDS of IRAK4 inhibition is the suppression of NLRP3 transcriptional priming by virtue of suppression of NF-κB activation. PF-06650833 is a first-in-class small molecule inhibitor of IRAK4, which was found to be safe in normal volunteers, and is now undergoing investigation in rheumatoid arthritis (NCT02996500). Type 1 interferons can initially suppress IL-1β production via dual mechanisms including STAT1-mediated repression of NLRP3 activation as well as increased IL-10 leading to decreased pro–IL-1β, however, at the risk of furthering NF-κB inflammatory cytokine generation.58 Additional potential mechanisms to inhibit NLRP3 activation and inflammasome assembly include adenylyl cyclase–activating G-protein–coupled receptor activators, targeting the trimeric adenosine triphosphate (ATP)-gated cation channel P2X7 receptor, and inhibitors of the lysosomal damage pathway via cathepsin inhibition.57
The best-characterized NLRP3 inflammasome inhibitor is the sulfonylurea derivative MCC950 (also known as CP-456 773), which inhibits NLRP3 activation at nanomolar concentrations and is highly specific for both canonical and noncanonical activation pathways.59 In an inflammatory colitis model, MCC950 significantly attenuated colitis activity and reduced IL-1β and IL-18 maturation.60 Despite its encouraging activity in preclinical models, clinical development of MCC950 was limited by hepatotoxicity in early phase trials. Novel NLRP3 inhibitors are in clinical development and offer significant therapeutic promise in MDS. Selective inhibition of NLRP3 may be advantageous in regards to less immune-suppression toxicity than IL-1β inhibition although potentially limited by alternative pathway compensation.
Bruton tyrosine kinase (BTK) was identified as a key regulator of the NLRP3 inflammasome through direct interaction with ASC and NLPR3.61 Using phosphoproteomics, Liu et al showed that BTK is a potent regulator of NLRP3 inflammasome activation, and that BTK inhibition using ibrutinib prevented ASC-speck formation and caspase-1 activation.62 Notably, peripheral blood mononuclear cells from ibrutinib-treated cancer patients had significant suppression of IL-1β maturation and caspase-1 activation ex vivo in response to inflammasome activators. Ibrutinib is currently undergoing investigation in MDS in combination with lenalidomide (NCT03359460) and azacitidine (NCT02553941).
The identification of NLRP3-activating mutations in cryopyrin-associated periodic syndromes (CAPS) has ushered in the first proof of concept for clinical efficacy of pharmacologic inhibition of the NLRP3 pathway. Specifically, targeting IL-1β signaling represents a potentially promising approach with agents approved for treatment of autoinflammatory diseases including canakinumab (an IL-1β–neutralizing monoclonal antibody), anakinra (a direct antagonist of the IL-1 receptor), and rilonacept (a soluble decoy receptor that binds IL-1β and IL-1α).63 Notably, sustained elevations in IL-1β lead to chronic NF-κB and MAPK signaling and corresponding excess IL-6 generation.64,65 Moreover, IL-1β has been shown to directly promote MDSC accumulation.66 Further supporting this, IL-1 receptor–deficient mice have delayed accumulation of MDSCs leading to decreased tumor progression.67 Together, these data, in addition to the success of IL-1β neutralization in CV protection (see next section), strongly support investigation of IL-1β–signaling antagonists in lower-risk MDS.
Sterile inflammation in hematopoietic cells and CV risk
Recent population studies have shown that CHIP is a common age-related phenomenon characterized by silent clonal acquisition of somatic gene mutations common to MDS in hematologically normal individuals. The most frequent mutations include loss-of-function mutations in epigenetic regulatory genes such as DNMT3A, TET2, and ASXL1.68-70 Whereas CHIP is rare in patients <50 years of age, the frequency of CHIP in patients >60 years of age approaches 10%, with a rising frequency with advancing age or utilization of a deeper level of sequencing.71 Notably, these mutations can be identified years prior to onset of hematologic malignancy (eg, median 9.6 years in the study of the Women’s Health Initiative), with clone size and specific mutations (ie, IDH1/2 and TP53) portending the highest risk.72 Importantly, all of these studies showed that individuals with CHIP have an increase in all-cause mortality relating to risk of hematologic malignancy (10-fold increase), CV disease, and type 2 diabetes. Notably, the increased CV risk was independent of well-known CV factors such as diabetes, hyperlipidemia, hypertension, and smoking.
Large case-control studies involving 4726 participants confirm the CV risk associated with CHIP, demonstrating a twofold greater risk of coronary heart disease and fourfold higher risk of early onset myocardial infarction, respectively.73 Specifically, mutations in DNMT3A, TET2, ASXL1, and JAK2 all carried increased risk of coronary heart disease as well as increased atherosclerosis, with a direct correlation between clone size and atherosclerotic burden (ie, variant allele frequency >10% at highest risk). To date, the mechanism underlying the increased risk of TET2 mutation–associated CV disease risk has been ascribed to inflammasome activation in affected macrophages. In a murine TET2 knockout model, Tet2 deletion increased the size of atherosclerotic plaques accompanied by increased maturation of IL-1β in inflammatory macrophages although the role of NLRP3 was not evaluated in this study.73 Similar findings were observed in a TET2-deficient model while mechanistically demonstrating NLRP3-inflammasome directed IL-1β secretion in plaque macrophages.74 More importantly, NLRP3 inhibition suppressed acceleration of atherosclerosis.74 These data provide the initial causal link behind somatic gene mutations in epigenetic regulatory genes with inflammatory processes central to the NLRP3 inflammasome. Most importantly, the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) provides proof of concept that inhibiting proinflammatory cytokines can attenuate risk of CV events.75 This large-scale, randomized, double-blind phase 3 trial of 10 061 patients with a history of myocardial infarction and high C-reactive protein (CRP) ≥2 mg/L randomized patients to receive canakinumab (at 50, 150, or 300 mg) vs placebo. The results of this study revealed a 15% reduction in the primary CV end point of nonfatal myocardial infarction, nonfatal stroke, or CV death in the 150-mg arm. Canakinumab led to dose-dependent reductions in CRP levels, indicating that IL-1β neutralization effectively suppresses inflammatory activity.
In the CANTOS study, IL-6 and high CRP levels were significantly higher at baseline in patients who had a subsequent diagnosis of lung cancer, and lung cancer frequency was lower in canakinumab-treated patients. Total cancer mortality was significantly reduced in patients treated with canakinumab, an effect that was dose-dependent wherein the highest dose (300 mg) was the only arm independently associated with lower cancer mortality (hazard ratio, 0.49 [95% CI, 0.31-0.75]; P = .0009) as well as lung cancer–specific mortality (hazard ratio, 0.23 [95% CI, 0.10-0.54]; P = .0002).76 These findings agree with experimental data relating IL-1β to progression and invasiveness of select solid tumors, particularly lung cancer.77 Whether IL-1β inhibition may be disease modifying in lower-risk MDS is unknown, but it is of particular interest given its role in disease pathobiology.
Conclusions and future directions
Critical insight into the pathogenesis of the hallmark features of MDS has emerged from understanding the role of innate immune and proinflammatory signaling. Notably, NLRP3 inflammasome activation and consequent pyroptosis represent a central cell death pathway in lower-risk MDS that is driven by cell-extrinsic alarmins such as S100A8/A9 and cell-intrinsic somatic gene mutations. Additionally, recent studies provide a causal link between clonal hematopoiesis and CV morbidity/mortality that is driven by mutant gene licensing of the NLRP3 inflammasome. Together, these findings not only may aid in cases of diagnostic uncertainty, but may also provide strong rationale for future biologically rational therapeutic strategies that may alter the natural history of disease in MDS.
Authorship
Contribution: D.A.S. and A.L. wrote the paper.
Conflict-of-interest disclosure: D.A.S and A.L. have received institutional research support from Celgene.
Correspondence: Alan List, Department of Malignant Hematology, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: alan.list@moffitt.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal