

Abstract
The heterogeneity of myelodysplastic syndromes (MDSs) has made evaluating patient response to treatment challenging. In 2006, the International Working Group (IWG) proposed a revision to previously published standardized response criteria (IWG 2000) for uniformly evaluating clinical responses in MDSs. These IWG 2006 criteria have been used prospectively in many clinical trials in MDSs, but proved challenging in several of them, especially for the evaluation of erythroid response. In this report, we provide rationale for modifications (IWG 2018) of these recommendations, mainly for “hematological improvement” criteria used for lower-risk MDSs, based on recent practical and reported experience in clinical trials. Most suggestions relate to erythroid response assessment, which are refined in an overall more stringent manner. Two major proposed changes are the differentiation between “procedures” and “criteria” for hematologic improvement–erythroid assessment and a new categorization of transfusion-burden subgroups.
Introduction
Myelodysplastic syndromes (MDSs) are disorders in which hematopoietic stem cells do not mature into healthy blood cells, leading to cytopenias, most commonly anemia. Anemia-related symptoms such as fatigue are generally the most commonly reported symptoms in MDSs1-3 and ∼50% of patients with MDSs require regular red blood cell (RBC) transfusions.4 Although RBC transfusions provide short-term symptomatic benefit, they cannot correct most aspects of health-related quality of life (QoL), they contribute to iron overload, and they consume substantial human as well as financial resources.5
Anemia associated with MDSs is therefore routinely treated by erythropoiesis-stimulating agents (ESAs), or lenalidomide in lower-risk MDSs with isolated del(5q), and by hypomethylating agents (HMAs) in higher-risk disease, whereas novel compounds are currently under clinical development. Thrombocytopenia and neutropenia are less frequent than anemia in lower-risk MDSs and treatment of these cytopenias has proven more difficult. Thrombopoietic agents are currently undergoing evaluation in patients with thrombocytopenia at risk for bleeding events or platelet transfusions.
Given the development of new treatments for MDSs, it is important to standardize response evaluation to allow optimal clinical decision-making as well as robust comparison of clinical trial data across studies. Currently, response evaluation is based on criteria proposed by the International Working Group (IWG) in 2000,6 which were modified in 20067 and have thus far been widely accepted (Table 1).
Suggested procedures for supporting accurate response evaluation: HI-E
| Item | Suggested IWG 2018 procedures | IWG 2006 procedures |
|---|---|---|
| Baseline assessment procedures | ||
| Screening period for the evaluation of transfusion burden and baseline Hb levels* | 16 wk but only in lower-risk MDSs, when anemia is the predominant or only cytopenia; patients should be off any active treatment during this period | 8 wk |
| Transfusions: Patients with unusual or abnormal changes of their transfusion rate during the 16-wk observation period should be evaluated carefully for confounding factors (ie, bleeding, hemolysis, EPO levels, iron metabolism), including a potential extension of the evaluation period | ||
| Baseline Hb: For the determination of the baseline Hb level, we suggest using the mean of all available Hb measurements during the 16-wk screening period; to avoid bias, measurements prior to transfusions should be used in this calculation for TD patients and the measurements should be at least 7 d apart | ||
| No./frequency of Hb measurements prior therapy | Hb measurements for the determination of baseline Hb values should be performed (or retrospective results should be available) at least every 2 wk, if possible, during the 16 wk screening period | NA |
| Blood count device/method and laboratory | Investigators should be aware of potential fluctuations in Hb measurements due to different blood count devices or laboratories | NA |
| To avoid any ambiguities in Hb levels, investigators should check when using several devices/methods or laboratories whether they yield similar Hb levels; in case of different values, baseline Hb level (as well as subsequent response and response duration) should be assessed based on measurements from only 1 device/method or laboratory, especially at key time points of a clinical trial | ||
| Baseline Hb level | Hb < 10 g/dL as prerequisite for patients in need of therapy | Hb < 11 g/dL |
| Response evaluation procedures | ||
| Response evaluation period | 24 wk | 8 wk |
| No./frequency of Hb measurements | Hb measurement should be performed (or results be available) at least every 2 wk during the first 16 wk of therapy | NA |
| Blood count device/method and laboratory | Investigators should be aware of potential fluctuations in Hb measurements due to different blood count devices or laboratories | NA |
| To avoid any ambiguities in Hb levels, investigators should check when using several devices/methods or laboratories whether they yield similar Hb levels; in case of different values, baseline Hb level, response, and response duration should be assessed based on measurements from only 1 device/method or laboratory, especially at key time points of a clinical trial | ||
| Dose adjustment policy for high Hb levels | Treatment should be continued at a lower dose level (ie, increased intervals between doses or administration of lower dose level) rather than stopped when 2 subsequent Hb measurements exceed a predefined threshold | NA |
| If the drug under investigation is being reduced in dose, stopped, or its administration delayed in a responding patient for protocol-defined reasons leading to a loss of response, this should not be counted as such, if reintroduction of the drug at the same or lower dose induces a new response | ||
| If the reintroduction of the drug at a lower dose does not reinduce a response, this should be documented as such | ||
| When the investigational drug is being reduced in dose, stopped, or its administration delayed, blood counts are required continuously to monitor subsequent blood levels |
| Item | Suggested IWG 2018 procedures | IWG 2006 procedures |
|---|---|---|
| Baseline assessment procedures | ||
| Screening period for the evaluation of transfusion burden and baseline Hb levels* | 16 wk but only in lower-risk MDSs, when anemia is the predominant or only cytopenia; patients should be off any active treatment during this period | 8 wk |
| Transfusions: Patients with unusual or abnormal changes of their transfusion rate during the 16-wk observation period should be evaluated carefully for confounding factors (ie, bleeding, hemolysis, EPO levels, iron metabolism), including a potential extension of the evaluation period | ||
| Baseline Hb: For the determination of the baseline Hb level, we suggest using the mean of all available Hb measurements during the 16-wk screening period; to avoid bias, measurements prior to transfusions should be used in this calculation for TD patients and the measurements should be at least 7 d apart | ||
| No./frequency of Hb measurements prior therapy | Hb measurements for the determination of baseline Hb values should be performed (or retrospective results should be available) at least every 2 wk, if possible, during the 16 wk screening period | NA |
| Blood count device/method and laboratory | Investigators should be aware of potential fluctuations in Hb measurements due to different blood count devices or laboratories | NA |
| To avoid any ambiguities in Hb levels, investigators should check when using several devices/methods or laboratories whether they yield similar Hb levels; in case of different values, baseline Hb level (as well as subsequent response and response duration) should be assessed based on measurements from only 1 device/method or laboratory, especially at key time points of a clinical trial | ||
| Baseline Hb level | Hb < 10 g/dL as prerequisite for patients in need of therapy | Hb < 11 g/dL |
| Response evaluation procedures | ||
| Response evaluation period | 24 wk | 8 wk |
| No./frequency of Hb measurements | Hb measurement should be performed (or results be available) at least every 2 wk during the first 16 wk of therapy | NA |
| Blood count device/method and laboratory | Investigators should be aware of potential fluctuations in Hb measurements due to different blood count devices or laboratories | NA |
| To avoid any ambiguities in Hb levels, investigators should check when using several devices/methods or laboratories whether they yield similar Hb levels; in case of different values, baseline Hb level, response, and response duration should be assessed based on measurements from only 1 device/method or laboratory, especially at key time points of a clinical trial | ||
| Dose adjustment policy for high Hb levels | Treatment should be continued at a lower dose level (ie, increased intervals between doses or administration of lower dose level) rather than stopped when 2 subsequent Hb measurements exceed a predefined threshold | NA |
| If the drug under investigation is being reduced in dose, stopped, or its administration delayed in a responding patient for protocol-defined reasons leading to a loss of response, this should not be counted as such, if reintroduction of the drug at the same or lower dose induces a new response | ||
| If the reintroduction of the drug at a lower dose does not reinduce a response, this should be documented as such | ||
| When the investigational drug is being reduced in dose, stopped, or its administration delayed, blood counts are required continuously to monitor subsequent blood levels |
NA, not available.
The 16-wk screening period applies mainly to lower-risk MDSs where anemia is the major cytopenia. Patients with higher-risk MDSs or with severe thrombocytopenia may require earlier treatment and an 8-wk screening period is acceptable in that situation.
Clinical experience has shown limitations of those criteria. For example, studies of HMAs in higher-risk MDSs showed that restarting HMA cycles on day 29 of the previous cycle usually prevented complete recovery from cytopenias (especially neutropenia) and therefore precluded fulfillment of complete remission (CR) criteria, even if the bone marrow blast count had normalized. More recently, the present authors have been involved in the steering committees of various industry and academic sponsored clinical trials in lower-risk MDSs focusing mainly on the treatment of anemia (and, in 1 case, thrombocytopenia) as well as in independent reviews of investigators’ response assessment in MDS trials. In the context of these assessments, challenges emerged with regard to evaluating erythroid or platelet response when stringently applying IWG 2006 criteria.8-10 A few illustrative examples of clinical cases will be presented and discussed within this manuscript. They all highlight potential pitfalls in determining a clinical response in a given patient undergoing disease-specific treatment. On the other hand, treatment of anemia, including its accurate monitoring and assessment, is crucial for both the general improvement of the QoL of the patients as well as disease-specific outcomes.11-21
As a panel of international experts in MDSs, we provide a proposal to improve and further develop IWG criteria, focusing on “hematological improvement,” that is, improvement of cytopenias in lower-risk MDSs, especially anemia. The concept of this IWG 2018 consensus is not to define narrow criteria but rather to describe challenges associated with the current response assessment and guidelines applicable for most patients included in clinical trials.
Methods
A task force of experts in the field of MDS research in the European Union (European LeukemiaNet [ELN] and European Myelodysplastic Syndromes Cooperative Group [EMSCO]) and North America followed the procedures used for the development of the current ELN guidelines on MDSs.22 The consensus development was based on review of recent literature on the topic as well as recent clinical experience, especially in 3 recent randomized trials9,10,23 using drugs (ESA or lenalidomide) to treat anemia of lower-risk MDSs, which served as a platform to identify pitfalls of current IWG 2006 guidelines.7 Different clinical scenarios were prepared, shared and discussed within the panel. A consensus was considered only when agreement was reached by more than two-thirds of the experts.
Results
1. Assessment of the erythroid lineage in MDS patients
1.1. Baseline assessment procedures
1.1.1. Current obstacles in defining RBC transfusion dependence and independence
A reliable characterization of the RBC transfusion burden of MDS patients and its evolution during treatment is crucial, especially in the context of clinical research.
Using current IWG 2006 criteria, anemic MDS patients are categorized in a binary fashion as “transfusion dependent” (TD) and “transfusion independent” (TID); TD being defined as having received at least 4 U of RBCs within 8 weeks for hemoglobin (Hb) of < 9 g/dL. Hematologic improvement–erythroid (HI-E) in the TID patients requires an Hb improvement of at least 1.5 g/dL lasting for at least 8 weeks. HI-E response in patients in the TD category is defined by IWG 2006 criteria as a reduction of at least 4 U per 8 weeks transfused for the same Hb threshold (eg, from 8 to 4 U, 6 to 2 U, or 4 to 0 U) (Table 1) and also lasting for at least 8 weeks (2 months).
This definition is problematic in 2 ways:
The screening period for the assessment of transfusion dependence is not well defined; it is often considered to be 8 weeks given the above definition for TD. However, 8 weeks is a short period for the objective assessment of Hb levels, as certain natural or event-driven fluctuations are not adequately reflected. If fluctuations are, for instance, caused by intercurrent infections or bleeding events, they are not influential for the long-term transfusion needs of the patients. However, for the short-term, nonrecognized bleeding could lead to wrongly assessed transfusion need.
The categorization into the 2 current groups (TD/TID) does not take into account a large number of patients with low transfusion burden, that is, receiving transfusions but <4 U of RBCs within 8 weeks, for whom transfusion elimination is considered an appropriate therapeutic goal, which is not reflected by current HI-E criteria.
Therefore, we suggest the following clarification and revisions within the IWG criteria.
1.1.1.a. Screening period
To illustrate the potential issues generated by the current screening period of 8 weeks, Figure 1 reflects 5 potential patients having all received 4 U of RBCs for an Hb of <9 g/dL within 8 weeks (56 days) prior to a putative day of onset of a therapeutic agent. According to the IWG 2006 criteria, these patients would all be considered TD.
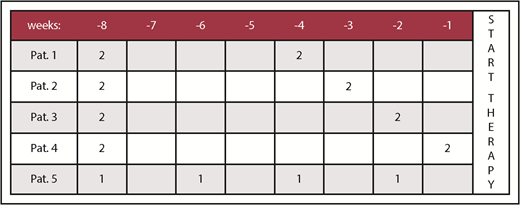
Transfusion history of 5 hypothetical MDS patients within 8 weeks. All receiving RBC support for an Hb of <9 g/dL for up to 8 weeks (56 days) prior to day 0, that is, start for a subsequent therapy, for example, within a clinical trial. The total number of RBC U within 8 weeks is 4 in all patients (Pat.).
Transfusion history of 5 hypothetical MDS patients within 8 weeks. All receiving RBC support for an Hb of <9 g/dL for up to 8 weeks (56 days) prior to day 0, that is, start for a subsequent therapy, for example, within a clinical trial. The total number of RBC U within 8 weeks is 4 in all patients (Pat.).
However, when the retrospective analysis is extended to 16 weeks (112 days), the transfusion burden in these 5 patients may differ meaningfully, as illustrated in Figure 2. Assessing the 16-week period across these 5 exemplary patients, only patients 1 and 5 fulfill IWG 2006 criteria of an average number of at least 2 U of RBCs within 28 days (or 8 U within 16 weeks). We thus consider having a history of RBC transfusions during the 16 weeks (which could be also part of the screening period) preceding onset of treatment in a clinical trial mandatory. This recommendation can, of course, only apply to lower-risk MDSs, when anemia is the predominant or only cytopenia. Indeed, significantly delaying treatment of higher-risk MDSs or in patients with severe thrombocytopenia would not be acceptable.
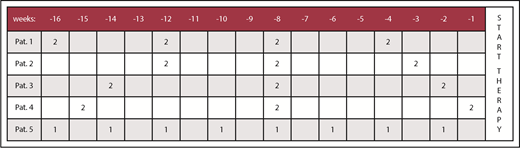
Transfusion history of 5 hypothetical MDS patients within 16 weeks. Transfusion history of the same 5 hypothetical MDS patients (Figure 1) but with an extended period of 16 weeks (112 days) prior to the start of a putative therapy. The total number of RBC U within 8 weeks is <4 in patients 2, 3, and 4, respectively.
Transfusion history of 5 hypothetical MDS patients within 16 weeks. Transfusion history of the same 5 hypothetical MDS patients (Figure 1) but with an extended period of 16 weeks (112 days) prior to the start of a putative therapy. The total number of RBC U within 8 weeks is <4 in patients 2, 3, and 4, respectively.
As in IWG 2006 criteria, only RBC transfusions administered for a mean Hb level below 9 g/dL are to be considered (see also comment in Table 2), which takes into account that a certain variation (see “1.2.3 Transfusion policy”) can occur. Transfusions for intercurrent diseases (bleeding, surgical procedure, etc) should not be considered.
Panel recommendation
A screening period of 16 weeks before the anticipated treatment start is mandatory and should be added as a prerequisite for subsequent IWG response determination in patients with lower-risk MDSs where anemia is the predominant or only cytopenia. Patients should be off of any active treatment during this period.
Note: Patients with unusual or abnormal changes of their transfusion rate during the 16-week screening period should be evaluated carefully for confounding factors (ie, bleeding, hemolysis, erythropoietin [EPO] levels, iron metabolism), including a potential extension of the evaluation period.
Suggested modified IWG 2018 HI-E criteria for response evaluation
| Item | Suggested IWG 2018 criteria | IWG 2006 criteria |
|---|---|---|
| Baseline criteria | ||
| Definition of transfusion-burden categories | 3 groups: NTD (0 RBCs in 16 wk)* LTB (3-7 RBCs in 16 wk in at least 2 transfusion episodes, maximum 3 in 8 wk)* HTB (≥8 RBCs in 16 wk, ≥4 in 8 wk) | 2 groups: TD (at least 4 U of RBC with 8 wk for Hb < 9 g/dL) TID (<4 U of RBC with 8 wk for Hb < 9 g/dL) |
| Pretreatment RBC transfusion policy | Transfusion policy for the individual patient prior to therapy should be maintained on treatment† | Transfusion threshold of 9 g/dL, no exception for clinical indication |
| Response evaluation criteria: HI-E | ||
| NTD (0 RBCs in 16 wk)* | At least 2 consecutive Hb measurements ≥1.5 g/dL for a period of minimum 8 wk in an observation period of 16 to 24 wk compared with the lowest mean of 2 Hb measurements (apart from any transfusion) within 16 wk before treatment onset‡; only a response duration of at least 16 wk, however, is considered clinically meaningful | Hb increase by 1.5 g/dL and/or relevant reduction of U of RBC transfusions by an absolute number of at least 4 RBC transfusions/8 wk compared with the pretreatment transfusion number in the previous 8 wk; only RBC transfusions given for an Hb of ≤9.0 g/dL pretreatment will count in the RBC transfusion response evaluation |
| LTB (3-7 RBCs in 16 wk in at least 2 transfusion episodes, maximum 3 in 8 wk)* | HI-E in LTB patients corresponds to transfusion independence, defined by the absence of any transfusions for at least 8 wk in an observation period of 16-24 wk with the same transfusion policy (defined below) compared with 16 wk prior to treatment; only a response duration of at least 16 wk, however, is considered clinically meaningful | |
| HTB (≥8 RBCs in 16 wk, ≥4 in 8 wk) | Major response: Major HI-E response in HTB patients corresponds to transfusion independence, defined by the absence of any transfusions over a period of minimum 8 wk in an observation period of 16-24 wk with the same transfusion policy (defined below) compared with 16 wk prior to treatment; only a response duration of at least 16 wk, however, is considered clinically meaningful | |
| Minor response: Minor HI-E response in HTB patients is defined as a reduction by at least 50% of RBCs over a minimum of 16 wk with the same transfusion policy (defined below) compared with 16 wk prior to treatment | ||
| On-treatment RBC transfusion policy§ | Transfusion policy for the individual patient prior to therapy should be maintained on treatment if not otherwise clinically indicated (documentation by the treating physician required); we suggest a maximum variation between pre- and on-study practice of 1 g/dL (or 0.6 mmol/L) in terms of transfusion threshold | Transfusion threshold of 9 g/dL, no exception for clinical indication |
| Dose adjustment thresholds for high Hb levels | If the drug under investigation is stopped or its dose reduced in a responding patient for protocol-defined reasons leading to a loss of response, this should not be counted as such if reintroduction at the same or lower dose of the drug induces a new response; if reintroduction of the drug at a lower dose does not reinduce a response, this should be documented as such | NA |
| Item | Suggested IWG 2018 criteria | IWG 2006 criteria |
|---|---|---|
| Baseline criteria | ||
| Definition of transfusion-burden categories | 3 groups: NTD (0 RBCs in 16 wk)* LTB (3-7 RBCs in 16 wk in at least 2 transfusion episodes, maximum 3 in 8 wk)* HTB (≥8 RBCs in 16 wk, ≥4 in 8 wk) | 2 groups: TD (at least 4 U of RBC with 8 wk for Hb < 9 g/dL) TID (<4 U of RBC with 8 wk for Hb < 9 g/dL) |
| Pretreatment RBC transfusion policy | Transfusion policy for the individual patient prior to therapy should be maintained on treatment† | Transfusion threshold of 9 g/dL, no exception for clinical indication |
| Response evaluation criteria: HI-E | ||
| NTD (0 RBCs in 16 wk)* | At least 2 consecutive Hb measurements ≥1.5 g/dL for a period of minimum 8 wk in an observation period of 16 to 24 wk compared with the lowest mean of 2 Hb measurements (apart from any transfusion) within 16 wk before treatment onset‡; only a response duration of at least 16 wk, however, is considered clinically meaningful | Hb increase by 1.5 g/dL and/or relevant reduction of U of RBC transfusions by an absolute number of at least 4 RBC transfusions/8 wk compared with the pretreatment transfusion number in the previous 8 wk; only RBC transfusions given for an Hb of ≤9.0 g/dL pretreatment will count in the RBC transfusion response evaluation |
| LTB (3-7 RBCs in 16 wk in at least 2 transfusion episodes, maximum 3 in 8 wk)* | HI-E in LTB patients corresponds to transfusion independence, defined by the absence of any transfusions for at least 8 wk in an observation period of 16-24 wk with the same transfusion policy (defined below) compared with 16 wk prior to treatment; only a response duration of at least 16 wk, however, is considered clinically meaningful | |
| HTB (≥8 RBCs in 16 wk, ≥4 in 8 wk) | Major response: Major HI-E response in HTB patients corresponds to transfusion independence, defined by the absence of any transfusions over a period of minimum 8 wk in an observation period of 16-24 wk with the same transfusion policy (defined below) compared with 16 wk prior to treatment; only a response duration of at least 16 wk, however, is considered clinically meaningful | |
| Minor response: Minor HI-E response in HTB patients is defined as a reduction by at least 50% of RBCs over a minimum of 16 wk with the same transfusion policy (defined below) compared with 16 wk prior to treatment | ||
| On-treatment RBC transfusion policy§ | Transfusion policy for the individual patient prior to therapy should be maintained on treatment if not otherwise clinically indicated (documentation by the treating physician required); we suggest a maximum variation between pre- and on-study practice of 1 g/dL (or 0.6 mmol/L) in terms of transfusion threshold | Transfusion threshold of 9 g/dL, no exception for clinical indication |
| Dose adjustment thresholds for high Hb levels | If the drug under investigation is stopped or its dose reduced in a responding patient for protocol-defined reasons leading to a loss of response, this should not be counted as such if reintroduction at the same or lower dose of the drug induces a new response; if reintroduction of the drug at a lower dose does not reinduce a response, this should be documented as such | NA |
Abbreviations are explained in Table 1.
The coauthors did not fully agree on whether patients who received only 1 or 2 RBC concentrates during the 16-wk screening period should be categorized in the NTB or LTB group. If such patients are included in clinical trials evaluating HI-E, it is recommended that HI-E achievement requires not only transfusion independence but also an increase of Hb by at least 1.5 g/dL (= 0.9 mmol/L).
As in IWG 2006 criteria, only RBC transfusions administered for an Hb level below 9 g/dL are taken into account. Exceptions to this rule may be accepted in cases of well-documented moderate or severe angina pectoris, cardiac or pulmonary insufficiency, or ischemic neurologic diseases. In these cases, a higher transfusion trigger level may be established for an individual patient. These patients may require special attention when analyzing responses within clinical trials. Transfusions for intercurrent diseases (bleeding, surgical procedure, etc) are not considered.
Oscillations (eg, natural or due to drug intervals) within this period are accepted as long as the patient remains off any transfusions and the same transfusion policy has been maintained. We suggest accepting 1 drop to an increase of between 1.0 and 1.5 g/dL over a period of 8 wk. We recommend that intervals between blood counts do not exceed 2 wk.
Exceptions to this rule may be accepted in cases of well-documented moderate or severe angina pectoris, cardiac or pulmonary insufficiency, or ischemic neurologic diseases. In these cases, a higher transfusion trigger level may be established for an individual patient. These patients may require special attention when analyzing responses within clinical trials. Transfusions for intercurrent diseases (bleeding, surgical procedure, etc) should not be taken into account.
1.1.1.b. Transfusion-burden categories
Another issue with the current IWG 2006 criteria, as stated previously, is that patients transfused <4 U within 8 weeks (but receiving regular transfusions) cannot be considered as responders if they become free of transfusions but without an Hb improvement of at least 1.5 g/dL after receiving therapy on a clinical trial. This highlights the particular problem of baseline Hb-level definition, which fluctuates relative to the time to previous transfusions. In addition, an important part of this patient group reaches transfusion independence with new therapeutic agents compared with patients being transfused >4 U every 8 weeks.24-26
Panel recommendation
Refinement of the RBC transfusion burden by dividing patients into 3 categories (nontransfused [NTD], low transfusion burden [LTB], and high transfusion burden [HTB]) as defined in the following sections.
• NTD patients
This category includes patients having received no transfusions within the screening period of 16 weeks. In these patients, an increase of Hb (by at least 1.5 g/dL = 0.9 mmol/L) is the main goal of therapeutic approaches, as already defined by IWG 2006 criteria. This threshold is based on several studies linking an improvement of QoL and disease-specific outcomes with Hb levels.11-21
• LTB patients
This category includes patients who received 3 to 7 RBCs within the screening period of 16 weeks, on at least 2 different dates/points in time. In addition, in those patients, we consider achieving transufsion independence a clinically meaningful end point even in the absence of an increase of Hb by at least 1.5 g/dL (as defined by the current IWG 2006 criteria) if there is no modification of the transfusion policy, especially of the transfusion threshold.
• HTB patients
This third category includes patients with HTB, which is overlapping the current IWG 2006 TD cohort, that is, defined by receiving ≥8 U of RBCs in 16 weeks (rather than just ≥4 RBCs in 8 weeks), in at least 2 transfusion episodes.
The coauthors did not fully agree on whether patients who received only 1 or 2 RBC concentrates during this 16-week period should be categorized in the NTD or LTB group. If such patients are included in clinical trials evaluating HI-E, it is recommended, however, that HI-E achievement require not only transfusion independence but also an increase of Hb by at least 1.5 g/dL (= 0.9 mmol/L) in these patients.
1.1.2. Determination of baseline Hb levels
1.1.2.a. Hb-level threshold
Apart from the definition of transfusion dependence and independence, another major criterion for the assessment of the erythroid response is the baseline Hb level, which according to IWG 2006 has to be <11 g/dL before treatment in NTD patients.
For the determination of the baseline Hb level, we suggest using the mean of all available Hb measurements during the 16-week screening period. To avoid bias, measurements prior to transfusions should be used in this calculation for TD patients and the measurements should be at least 7 days apart.
Panel recommendation
In terms of clinical threshold for a given therapy, we suggest reducing this from the current <11 g/dL according to IWG 2006 to <10 g/dL as patients considered candidates for clinical trials or specific treatments are rarely symptomatic above 10 g/dL. This is supported by the fact that the Revised International Prognostic Scoring System (IPSS-R) also considers 10 g/dL as the threshold having a prognostic impact.27
1.1.2.b. Number and frequency of baseline Hb measurements
To adequately determinate baseline Hb values, it is important to have a sufficient number of Hb measurements. Statistically validated data in patients with renal disease suggest that weekly Hb measurements provide the most reliable monitoring of anemia.28 From this evidence, we recommend frequent monitoring at regular intervals and believe that, for MDS patients, a biweekly interval is frequent enough without imposing an unreasonable burden on patients and investigators.
Panel recommendation
For the determination of baseline Hb levels, Hb measurements should be performed (or retrospective results should be available) at least every 2 weeks, if possible, during the 16-week screening period.
1.1.2.c. Blood count device/method or laboratory for Hb determination
On a more technical level, the experience of some of the members of the steering committee involved in recent clinical trials in lower-risk MDSs was that different hemocytometer devices sometimes gave different Hb-level results. Similar challenges are observed when comparing other evaluations in MDSs: for instance, marrow blast counts in the context of morphological assessments. Results for the same patient and time point can vary significantly between reviewers. This is why many trials dictate central morphologic evaluations, at least for the key measurements, such as baseline levels and response-assessment visits. We recommend that Hb levels should be treated similarly based on the inconsistency sometimes observed between devices.
Panel recommendation
To avoid any ambiguities in Hb levels when using several devices/methods or laboratories, investigators should check whether they yield similar Hb levels. In case of different values, baseline Hb level, response, and response duration should be assessed based on measurements from only 1 device/method or laboratory, especially at key time points of a clinical trial.
1.2. Response evaluation
Based on the comments made in the previous sections, we suggest the following amendments to IWG 2006 criteria for “hematological improvement.”
1.2.1. Response-evaluation period
Similarly, as we propose a prolonged screening period of 16 weeks to adequately determine the transfusion status of a given patient before starting an investigational treatment, we suggest that the erythroid response should be assessed on an observation period of at least 16 weeks. Because erythroid response, with any drug, is often only observed after a certain delay, we consider that, in these cases, a minimum 24-week period of observation after treatment onset is required.
In addition, onset of response is not clearly determined using IWG 2006 criteria. We suggest that response starts when the Hb level rises by at least 1.5 g/dL in non-TD patients and from the time point of the anticipated next transfusion (if the patient remains NTD) in TD patients.
Panel recommendation
We recommend defining an observation period to assess response of at least 16 to 24 weeks starting directly after study treatment initiation at week 0. We consider response to start when the Hb level rises by at least 1.5 g/dL in non-TD patients and from the date of the anticipated next transfusion (if the patient remains NTD) in TD patients.
1.2.2. Assessment of HI-E response depending on pretreatment transfusion burden
The 3 newly proposed transfusion-burden category groups (NTD, LTB, HTB) lead to a need for a more discriminate response evaluation. In addition, although we acknowledge that a response duration of at least 8 weeks is sufficient to consider a patient as responder (especially in early phase clinical trials), we feel that only a response duration of at least 16 weeks is clinically meaningful. Response duration should therefore always be reported.
1.2.2.a. NTD patients (screening period: no RBC transfusion in 16 weeks)
Response in those patients is assessed solely based on an increase in the Hb level.
Panel recommendation
In terms of required Hb increase to reach HI-E, we do not suggest modifying the current threshold (Hb increase by at least 1.5 g/dL). However, we would recommend defining an HI-E by at least 2 consecutive Hb measurements ≥1.5 g/dL, maintained over at least 8 weeks during an observation period of 16 to 24 weeks (as defined in “1.2.1. Response-evaluation period”) compared with the mean of 2 Hb measurements within 16 weeks before treatment onset. Only a response duration of at least 16 weeks, however, is considered clinically meaningful.
Also, importantly, we acknowledge that, thereafter, natural fluctuations of Hb levels might be observed irrespective of continuation of therapy. Therefore, a patient with a drop of previous Hb level increase below 1.5 g/dL compared with pretreatment (but showing an increase of at least 1.0 g/dL) would still count as a responder. However, drops to levels between 1.0 and 1.5 g/dL are only admitted on a maximum of 2 occasions over a period of 16 weeks. We recommend that intervals between blood counts do not exceed 2 weeks.
1.2.2.b. LTB patients (screening period: 3-7 RBCs in 16 weeks)
Panel recommendation
HI-E in LTB patients is defined as follows. Response in LTB patients corresponds to transfusion independence, defined by the absence of any transfusions in a period of at least 8 weeks during an observation period of 16 to 24 weeks with the same transfusion policy (defined in “1.2.4. Transfusion policy”) compared with 16 weeks prior to treatment. Only response duration of at least 16 weeks, however, is considered clinically meaningful.
Again, we want to highlight that these responses can occur in the absence of an increase of Hb by at least 1.5 g/dL = 0.9 mmol/L (former IWG 2006) but must not be associated with a decrease in Hb from baseline.
1.2.2.c. HTB patients (screening period: ≥8 RBCs in 16 weeks)
Panel recommendation
HI-E in HTB patients is defined as follows.
Major response: Major HI-E response in HTB patients corresponds to transfusion independence, defined by the absence of any transfusions over a period of a minimum of 8 weeks within an observation period of 16 to 24 weeks with the same transfusion policy (defined in “1.2.4. Transfusion policy”) compared with 16 weeks prior to treatment. Only a response duration of at least 16 weeks, however, is considered clinically meaningful.
Minor response: Minor HI-E response in HTB patients is defined as a reduction by at least 50% of RBCs over a minimum of 8 weeks with the same transfusion policy (defined in “1.2.4. Transfusion policy”) compared with 16 weeks prior to treatment. Only a response duration of at least 16 weeks, however, is considered clinically meaningful.
Again, we want to highlight that the above responses can occur in the absence of an increase of Hb by at least 1.5 g/dL = 0.9 mmol/L (former IWG 2006) but must not be associated with a decrease in Hb from baseline.
In LTB and HTB patients who achieve transfusion independence, the duration of transfusion independence (eg, number of patients with 8 weeks vs 24 weeks of continuous transfusion independence) as well as the magnitude of increase in mean Hb values should also be reported (by dose, by drug, etc) to better assess the therapeutic potential of a specific treatment.
1.2.2.d. Patients having received 1 or 2 RBC concentrates during the screening period
As stated in “HTB patients,” some coauthors did not fully agree on whether patients who received only 1 or 2 RBC concentrates during the 16-week screening period should be categorized in the NTB or LTB group. If such patients are included in clinical trials evaluating HI-E, it is recommended that HI-E achievement requires not only transfusion independence but also an increase of Hb by at least 1.5 g/dL (=0.9 mmol/L).
1.2.3. Number and frequency of Hb measurements for response determination
As mentioned in “1.1.2. b. Number and frequency of baseline Hb measurements,” we suggest biweekly intervals for the assessment of response and its duration.
Panel recommendation
Hb measurement should be performed or results should be available at least every 2 weeks during the first 16 weeks of therapy.
1.2.4. Transfusion policy
The proposed more stringent definitions of transfusion-burden categories may lead to the fact that changes in transfusion policy can have an even more important impact on response compared with current IWG 2006 criteria. Especially in LTB patients, where transfusion independence is the criterion for response, predefined transfusion policies (eg, in terms of Hb threshold for transfusing) must be maintained to avoid bias and distorted response evaluations.
Panel recommendation
We therefore recommend incorporating, as a new prerequisite for an unbiased response evaluation, the maintenance of a comparable transfusion policy on therapy as compared with the 16-week screening period. For example, patients transfused below a certain threshold (eg, 8.5 g/dL) before therapy should receive transfusions at the same threshold on study, unless an exceptional circumstance requires this threshold to be punctually changed (documentation by the treating physician will be required). We suggest a maximum variation between pre- and on-study practice of 1 g/dL (or 0.6 mmol/L) in terms of transfusion threshold.
Exceptions to this rule may be accepted in cases of well-documented moderate or severe angina pectoris, cardiac or pulmonary insufficiency, or ischemic neurologic diseases. In these cases, a higher transfusion trigger level may be established for an individual patient. These patients may require special attention when analyzing responses within clinical trials. Transfusions for intercurrent diseases (bleeding, surgical procedure, etc) should not be taken into account.
1.2.5. Impact of blood count device/method or laboratories on response evaluation
Response evaluation for a given patient should be based on only 1 device if discrepancies for the Hb level are seen between different laboratories or devices (Figure 3). This is also mandatory for determining responses in other lineages (hematologic improvement platelet [HI-P], hematologic improvement neutrophil [HI-N]).
Panel recommendation
Investigators should be aware of potential fluctuations in Hb measurements due to different blood count devices/methods or laboratories. Responses and their duration should be evaluated based on blood counts performed in the same laboratory or with the same technique if such fluctuations are observed.
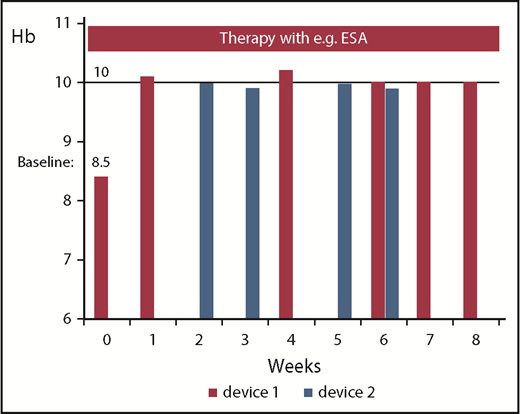
Hb measurements with 2 different devices or laboratories at different points in time in 1 patient treated, for example, with ESA. The figure illustrates that certain fluctuations of Hb values might be simply a result of differences in the accuracy of blood devices/methods. An Hb increase by ≥1.5 g/dL is achieved constantly only with device 1, which was also used during screening.
Hb measurements with 2 different devices or laboratories at different points in time in 1 patient treated, for example, with ESA. The figure illustrates that certain fluctuations of Hb values might be simply a result of differences in the accuracy of blood devices/methods. An Hb increase by ≥1.5 g/dL is achieved constantly only with device 1, which was also used during screening.
1.2.6. Dose-adjustment policy for blood counts on treatment
Mainly based on the indication in chronic renal failure, avoiding increases of Hb levels above 12 g/dL29 during treatment with ESA is recommended in many countries (especially in Europe). In most clinical trials, this rule is strictly enforced and, once a patient presents with Hb ≥ 12 g/dL, therapy has to be stopped and restarted at a lower dose. Figure 4 illustrates a clinical example.

Undesired effects of dosing interruption. The figure shows the undesired effects of dosing interruption followed by dose reduction in a patient responding to ESA therapy and reaching an Hb ≥ 12 g/dL (starting from a baseline Hb of 8.5 g/dL). The lower line marks the IWG 2006 response (8.5-10 g/dL) and upper line the general Hb cutoff for dose interruption in many clinical trials (12 g/dL).
Undesired effects of dosing interruption. The figure shows the undesired effects of dosing interruption followed by dose reduction in a patient responding to ESA therapy and reaching an Hb ≥ 12 g/dL (starting from a baseline Hb of 8.5 g/dL). The lower line marks the IWG 2006 response (8.5-10 g/dL) and upper line the general Hb cutoff for dose interruption in many clinical trials (12 g/dL).
Deciding on drug interruption and subsequent dose reduction based on 1 Hb measurement ≥ 12 g/dL might lead to undesired Hb decrease, which can hardly be regained by continuation of the treatment at a lower dose level. The authors believe that dose reductions should be performed only if 2 subsequent measurements exceed the predefined threshold, which may vary according to different protocols. To maintain response, treatment should not be stopped but continued at a lower dose level (ie, increased intervals between doses or lower dose level). A maximum of 2 consecutive Hb values below the response threshold are allowed in cases of protocol-defined dose adjustments or delays. On the contrary, further increase of the Hb levels, even with dose reduction, should lead to further dose reduction to avoid potential thrombotic events.
Panel recommendation
If the drug under investigation is stopped or its dose reduced in a responding patient for protocol-defined reasons leading to a loss of response, this should not be counted as such if reintroduction of the drug at the same or lower dose induces a new response. If reintroduction of the drug at a lower dose does not reinduce a response, this should be documented as such.
2. Response evaluation for platelets and neutrophils
Current response definitions for other lineages than the erythroid are provided in Table 3. The authors agree that these reflect clinically meaningful end points. For platelet response, we suggest leaving the current criteria with additional modifications for bleeding symptoms and dose reductions. The latter should only be performed if the platelet count exceeds the normal range (450 × 109/L) in 2 consecutive blood counts in order to avoid sharp decreases of platelet counts thereafter. Furthermore, we suggest reporting increments of neutrophils and platelets also for patients with a pretreatment absolute neutrophil count (ANC) of <1.0 × 109/L and pretreatment platelet counts of <100 × 109/L. This is because responses in patients with an ANC of ≥1.0 × 109/L and a platelet count of ≥100 × 109/L might reflect a multilineage response of certain drugs (eg, an agent considered to target anemia only, like luspatercept or sotatercept) and also translate into a better outcome for patients due to improvement of neutrophil and platelet number and function.
Suggested modified IWG 2018 HI-N and HI-P criteria for response evaluation
| Newly suggested evaluations: IWG 2018 | IWG 2006 criteria | ||
|---|---|---|---|
| Type of response | Criteria | Type of response | Criteria |
| Platelet response (pretreatment, <100 × 109/L), HI-P | • Absolute increase of 30 × 109/L for patients starting with >20 × 109/L PLTs or | Platelet response (pretreatment, <100 × 109/L), HI-P | • Absolute increase of 30 × 109/L for patients starting with >20 × 109/L PLTs or • Increase from <20 × 109/L to >20 × 109/L and by at least 100% |
| • Increase from <20 × 109/L to >20 × 109/L and by at least 100% | |||
| In addition, | |||
| • Evolution of bleeding symptoms is to be taken into account | |||
| • Increments of platelets also for patients with a pretreatment PLT count of >100 × 109/L are to be reported | |||
| Dose-adjustment policy for PLT counts on treatment | • If the drug under investigation is being stopped or its dose is being reduced in a responding patient for protocol-defined reasons leading to a loss of response, this should not be counted as such, if reintroduction at the same or lower dose of the drug induces a new response | None | |
| • When the investigational drug is stopped or reduced in dose, weekly blood counts are required to monitor the PLT levels | |||
| • 2 subsequent PLT counts >450 × 109/L are a sufficient reason for treatment discontinuation in the case of treatment with TPO agonists | |||
| Neutrophil response (pretreatment, all patients), HI-N | At least 100% increase and an absolute increase >0.5 × 109/L (pretreatment, <1.0 × 109/L) | Neutrophil response (pretreatment, <1.0 × 109/L), HI-N | At least 100% increase and an absolute increase >0.5 × 109/L |
| Increments of neutrophils also for patients with a pretreatment ANC of >1.0 × 109/L are to be reported | |||
| Newly suggested evaluations: IWG 2018 | IWG 2006 criteria | ||
|---|---|---|---|
| Type of response | Criteria | Type of response | Criteria |
| Platelet response (pretreatment, <100 × 109/L), HI-P | • Absolute increase of 30 × 109/L for patients starting with >20 × 109/L PLTs or | Platelet response (pretreatment, <100 × 109/L), HI-P | • Absolute increase of 30 × 109/L for patients starting with >20 × 109/L PLTs or • Increase from <20 × 109/L to >20 × 109/L and by at least 100% |
| • Increase from <20 × 109/L to >20 × 109/L and by at least 100% | |||
| In addition, | |||
| • Evolution of bleeding symptoms is to be taken into account | |||
| • Increments of platelets also for patients with a pretreatment PLT count of >100 × 109/L are to be reported | |||
| Dose-adjustment policy for PLT counts on treatment | • If the drug under investigation is being stopped or its dose is being reduced in a responding patient for protocol-defined reasons leading to a loss of response, this should not be counted as such, if reintroduction at the same or lower dose of the drug induces a new response | None | |
| • When the investigational drug is stopped or reduced in dose, weekly blood counts are required to monitor the PLT levels | |||
| • 2 subsequent PLT counts >450 × 109/L are a sufficient reason for treatment discontinuation in the case of treatment with TPO agonists | |||
| Neutrophil response (pretreatment, all patients), HI-N | At least 100% increase and an absolute increase >0.5 × 109/L (pretreatment, <1.0 × 109/L) | Neutrophil response (pretreatment, <1.0 × 109/L), HI-N | At least 100% increase and an absolute increase >0.5 × 109/L |
| Increments of neutrophils also for patients with a pretreatment ANC of >1.0 × 109/L are to be reported | |||
ANC, absolute neutrophil count; PLT, platelet; TPO, thrombopoietin.
Challenges in terms of achieving response under treatment with HMA need to be taken into account as it is known that HMAs can prevent recovery from cytopenias (especially neutropenia).
3. Progression or relapse after hematologic improvement
In the current IWG 2006 criteria, progression or relapse after HI are defined as at least a 50% decrement from maximum response levels in neutrophils or platelets, a reduction in Hb by ≥1.5 g/dL, and/or recurrence of transfusion dependence. The authors agree with these criteria but suggest some modifications in platelet and neutrophil response. We believe that a patient with an HI-P of, for example, 30 × 109/L to 300 × 109/L and a subsequent dip to 140 × 109/L, reflecting at least 50% decrement, is still a responder. The same is true for neutrophils (eg, from 4 × 109/L to 1.5 × 109/L).
Panel recommendation
We suggest that the current definition of loss of response (at least a 50% decrement from maximum response levels in neutrophils or platelets) is amended by the wording that the patient should also not meet IWG response criteria anymore (eg, ANC response from 0.5 to 1.2 and loss of response to 0.6 or platelet response from 30 to 70 and loss of response to 35) all in the absence of dose interruptions and in the absence of any infection, hemorrhagic events, and concomitant medications. The loss of erythroid response is currently well defined but should be amended by the comment that, for example, a response from Hb 8.5 to 13 and a consecutive decline to 11.5 g/dL (ie, by >1.5 g/dL) is not a loss of response whereas a decline to 9.5 or transfusion dependence is a loss of response because the patient does not meet response criteria anymore when levels are compared with baseline. Furthermore, some HTB patients will not become completely TID (eg, just a reduction by 50% of transfusion burden). As a result, progression should be defined by an increase in transfusion burden by at least 50% (eg, prior therapy 16 U within 16 weeks, reduction to 8 U during therapy as a response, and subsequent increase to 12 U).
As discussed in “1.2. Response evaluation,” we also emphasize that dose adjustments and subsequent loss of hematologic response need to be carefully evaluated (Table 1). In fact, if a drug is stopped or its dose is reduced in a responding patient for protocol-defined reasons (such as an adverse event) and if this leads to a loss of response, this should not be counted as relapse or progression, if reintroduction at the same or lower dose of the drug induces a new response.
Conclusions
The IWG criteria modified in 2006 have been adopted in many clinical trials and served as a valuable tool for the standardization of clinically meaningful response measures in MDSs. Recent experiences in lower-risk MDSs, however, show that there are still some pitfalls when adopting these criteria, which can lead to misinterpretation of outcome especially with regards to the erythroid assessment. To overcome the pitfalls in some of the IWG 2006 criteria as described herein, we suggest a review of the current IWG response criteria, especially for HI-E response evaluation. We have categorized this proposal into 3 parts: (1) baseline assessments, (2) response evaluation, and (3) evaluation of progression or relapse after HI. Tables 1-3 summarize novel considerations by the steering committee with regard to response and progression assessment. We believe that the modifications (IWG 2018) presented here will allow for more individualized pre- and on-study assessment and therefore provide the MDS community with more accurate results in terms of response evaluation. Although this work addresses “hematological improvement” criteria, mainly concerning lower-risk MDSs, we realize that CR, PR, and marrow CR may also have to be reviewed in the future, especially for higher-risk MDS patients treated with potentially disease-modifying treatments like HMAs. Furthermore, future work will focus on the integration of patient-reported outcomes including QoL and fatigue in response evaluation. This might be based on currently investigated models, such as the recently published FA-IPSS(h).30
Acknowledgment
This study was not supported by a pharmaceutical company or any other institution.
Authorship
Contribution: U.P. and P.F. designed the study; U.P., S.G., and P.F. collected, analyzed, and interpreted the data and wrote the paper; and L.A., A.G., V.S., A.A.v.d.L., D.B. T.d.W., G.G.-M., E.H.-L., U.G., R.S., L.M., M.A.S., and D.P.S. provided intellectual support and edited the paper.
Conflict-of-interest disclosure: U.P. and P.F. have received renumeration and research funding from Celgene Corporation. The remaining authors declare no competing financial interests.
Correspondence: P. Fenaux, Hôpital Saint-Louis, Assistance Publique Hôpitaux de Paris and Université Paris 7, Paris, France; e-mail: pierre.fenaux@aphp.fr; or U. Platzbecker, Medical Clinic and Policlinic 1, Hematology and Cellular Therapy, Leipzig University Hospital, Leipzig, Johannisallee 32 A, 04103 Leipzig, Germany; e-mail: uwe.platzbecker@medizin.uni-leipzig.de.
