

Key Points
A PI3K inhibitor with predominant α/δ activity, copanlisib, induces apoptosis and modulates BCL-xL and MCL-1 activity in BCR-dependent DLBCLs.
In BCR-dependent DLBCLs with genetic alterations of BCL2, the BCL-2 antagonist, venetoclax, and copanlisib are synergistic.

Abstract
Inhibition of the B-cell receptor (BCR) signaling pathway is a promising treatment strategy in multiple B-cell malignancies. However, the role of BCR blockade in diffuse large B-cell lymphoma (DLBCL) remains undefined. We recently characterized primary DLBCL subsets with distinct genetic bases for perturbed BCR/phosphoinositide 3-kinase (PI3K) signaling and dysregulated B-cell lymphoma 2 (BCL-2) expression. Herein, we explore the activity of PI3K inhibitors and BCL-2 blockade in a panel of functionally and genetically characterized DLBCL cell line models. A PI3K inhibitor with predominant α/δ activity, copanlisib, exhibited the highest cytotoxicity in all BCR-dependent DLBCLs. The proapoptotic effect of copanlisib was associated with DLBCL subtype-specific dysregulated expression of BCL-2 family members including harakiri (HRK) and its antiapoptotic partner BCL extra large (BCL-xL), BCL2 related protein A1, myeloid cell leukemia 1 (MCL-1), and BCL2 interacting mediator of cell death. Using functional BH3 profiling, we found that the cytotoxic activity of copanlisib was primarily mediated through BCL-xL and MCL-1–dependent mechanisms that might complement BCL-2 blockade. For these reasons, we evaluated single-agent activity of venetoclax in the DLBCLs and identified a subset with limited sensitivity to BCL-2 blockade despite having genetic bases of BCL-2 dysregulation. As these were largely BCR-dependent DLBCLs, we hypothesized that combined inhibition of PI3Kα/δ and BCL-2 would perturb BCR-dependent and BCL-2–mediated survival pathways. Indeed, we observed synergistic activity of copanlisib/venetoclax in BCR-dependent DLBCLs with genetic bases for BCL-2 dysregulation in vitro and confirmed these findings in a xenograft model. These results provide preclinical evidence for the rational combination of PI3Kα/δ and BCL-2 blockade in genetically defined DLBCLs.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoid malignancy in adults. Although ∼65% of patients with DLBCL can be cured with standard induction therapy, those with nonresponsive or relapsed disease have more limited options, highlighting the need for new targeted therapies. DLBCL is a genetically heterogeneous disease that is transcriptionally subclassified into germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.1 A subset of both GCB- and ABC-DLBCLs have known dependence on proximal B-cell receptor (BCR) signaling2,3 (reviewed in Bojarczuk et al4 ). Following phosphorylation of BCR-associated CD79α and β, downstream signaling is propagated via the spleen tyrosine kinase (SYK), phosphoinositide 3-kinase (PI3K)/AKT, and Bruton tyrosine kinase (BTK)/phospholipase C, γ 1 (PLCγ)/NF-κB.2 In contrast to GCB tumors, ABC-DLBCLs have high baseline NF-κB activity.5
In a previous study, we defined distinct BCR/PI3K-mediated survival pathways and subtype-specific apoptotic mechanisms in BCR-dependent DLBCLs.2 In BCR-dependent DLBCLs with low baseline NF-κB activity (GCB tumors), targeted inhibition or genetic depletion of proximal BCR/PI3K pathway components induced expression of the proapoptotic sensitizer protein, harakiri (HRK). In BCR-dependent DLBCLs with high NF-κB activity (ABC tumors), BCR/PI3K blockade decreased expression of the antiapoptotic B-cell lymphoma 2 (BCL-2) family member and NF-κB target, BCL2 related protein A1 (BFL-1).
Antiapoptotic BCL-2 family members such as BCL-2, myeloid cell leukemia 1 (MCL-1), BCL extra large (BCL-xL), and BFL-1 suppress apoptosis by 2 mechanisms. First, these proteins bind and prevent the oligomerization of the proapoptotic effector proteins, BAX and BAK, which initiate apoptosis by forming pores in the mitochondrial outer membrane that release cytochrome c (reviewed in Montero and Letai6 ). Additionally, the antiapoptotic BCL-2 family members bind and sequester the proapoptotic BH3-only activator proteins, BCL2 interacting mediator of cell death (BIM) and BID, which activate BAX and BAK.6 Proapoptotic sensitizers such as HRK, PUMA, BAD, or NOXA do not induce apoptosis on their own, but compete with BH3-only activators for binding to antiapoptotic BCL-2 family members, thereby releasing BIM or BID.6 Functional BH3 profiling can be used to measure apoptotic sensitivities and changes in apoptotic priming in response to targeted therapies.7,8
Our recent genetic analyses revealed structural bases for perturbed proximal BCR/PI3K signaling and defined poor-prognosis DLBCL subsets with discrete BCR/PI3K/Toll-like receptor pathway alterations (cluster 3 and cluster 5 DLBCLs).9 Cluster 3 DLBCLs (largely GCB-DLBCLs) exhibited frequent PTEN deletions and inactivating mutations and additional GNA13 and HVCN1 mutations that likely increase BCR/PI3K signaling.9 Cluster 5 DLBCLs (largely ABC-DLBCLs) had frequent activating MYD88L265P and CD79B mutations that often occurred together.9 Of interest, these DLBCL subtypes also had distinct genetic bases for dysregulated BCL-2–BCL2 translocations in cluster 3 and arm-level 18q or focal 18q21.33 (BCL2) copy-number gains in cluster 5.9 These observations prompted us to explore the activity of BCR/PI3K inhibitors and BCL-2 blockade in genetically defined DLBCLs.
Materials and methods
DLBCL cell lines
The 10 DLBCL cell lines SU-DHL2 (DHL2), SU-DHL4 (DHL4), SU-DHL6 (DHL6), HBL1, Karpas422 (K422), OCI-LY1 (LY1), OCI-LY7 (LY7), OCI-LY19 (LY19), TMD8, and TOLEDO were cultured as previously described10 and reported to be Epstein-Barr virus–negative by the respective repositories (ATCC, Manassas, VA and DSMZ, Braunschweig, Germany). The identities of the DLBCL cell lines used in this study were confirmed via the STR profiling PowerPlex 1.2 system from Promega (Madison, WI). These cell lines were characterized by whole-exome sequencing with expanded bait set as previously described.9
Reagents
The SYK inhibitor entospletinib (GS-9973), the BTK inhibitor ibrutinib (PCI-32765), the PI3Kδ inhibitor idelalisib (CAL-101), the PI3Kβ inhibitor AZD6482, and the pan-PI3K inhibitor pictilisib (GDC-0941) were purchased from Selleckchem (Houston, TX). The PI3Kα inhibitor alpelisib (BYL-719), the PI3Kβ/δ inhibitor AZD8186, the PI3Kα/δ-predominant inhibitor copanlisib (BAY 80-6946), and the BCL-2 inhibitor venetoclax (ABT-199) were obtained from MedChem Express (Monmouth Junction, NJ). The MCL-1 inhibitor (S63845) was purchased from Apexbio (Houston, TX).
Proliferation assay
Following 72-hour incubation with tested BCR/PI3K inhibitors, DLBCL cell proliferation was assayed with Alamar Blue (Invitrogen, Carlsbad, CA) as previously described.10 The 50% effective concentration (EC50) concentrations (micromolar) of tested inhibitors were calculated using GraphPad Prism 7.01 (GraphPad Software, La Jolla, CA) and log (inhibitor concentration) vs normalized response nonlinear least squares fit.
Apoptosis assay
The percentages of viable cells were determined 96 hours after treatment by flow cytometric analyses of cells stained with propidium iodide (PI) and annexin V–fluorescein isothiocyanate conjugate (BD Pharmingen, San Jose, CA) using FACSCanto II cytometer (BD). Double-negative cells were considered viable and all results were normalized to 100% viable cells (0% dead cells) in dimethyl sulfoxide (DMSO)-treated group. Annexin V/PI assessments were done using FlowJo software V7.6.1 for Windows (FlowJo LLC, Ashland, OR).
Immunoblotting
Immunoblotting was performed as previously published2 and as in supplemental Materials and methods (available on the Blood Web site). Unmodified raw images were arranged and labeled using CorelDRAW X7 (Corel Corporation, Ottawa, ON, Canada).
RT-qPCR
Reverse transcription–quantitative polymerase chain reaction (RT-qPCR) was performed as previously described2 with specific, intron-spanning primers for HRK, BCL2A1 (BFL-1), BCL2L11 (BCL-xL), BCL2L1 (BIM), and cyclophilin A (PPIA) genes, according to the manufacturer’s recommendations. Calculation of transcript abundance was performed with the comparative cycle threshold (Ct) method (2−ΔCt, where ΔCt = Ct HRK or BCL2A1 or BCL2L11, or BCL2L1 − Ct PPIA). Sequences of the primers for genes of interest were previously published.2 To measure the differences in expression of target genes in DMSO- vs copanlisib-treated groups, multiple Student t tests were performed with the 2-stage linear stepup procedure of Benjamini, Krieger, and Yekutieli; q < 0.1 was considered statistically significant.
BH3 profiling by intracellular staining
BH3 profiling of DLBCL cells was performed as previously described11 and in supplemental Materials and methods. The correlations between mitochondrial outer membrane permeabilization (MOMP) and copanlisib cytotoxicity were measured with the Pearson test, and 1-sided P values from all tested peptides were analyzed with the Benjamini-Hochberg procedure; q < 0.1 was considered statistically significant.
Assessment of copanlisib and venetoclax synergy
Combination indexes (CIs) for combinations of copanlisib and venetoclax were calculated using Compusyn (Combosyn Inc, Paramus, NJ) according to the Chou-Talalay algorithm.12 The median CIs for all assessed combinations are shown.
In vivo xenograft analyses
All murine studies were performed according to Dana-Farber Cancer Institute Institutional Animal Care and Use Committee–approved protocol. The DLBCL cell line LY1 was engineered for in vivo imaging as previously described.13 Subsequently, 5 × 106 viable Luc-mCherry–expressing lymphoma cells in 250 μL of sterile phosphate-buffered saline were injected via the lateral tail veins of 7-week-old female NOD SCID Il2rγnull mice (The Jackson Laboratory, Bar Harbor, ME). Three days following tumor inoculation, animals with established disease documented by imaging were divided into 4 cohorts with an average total flux bioluminescence (sum of prone and supine values) of 1.72 × 104 ± 1.73 × 103 photons (ph)/sec/cm2/steradian (sr) and treated with: (1) 12 mg/kg copanlisib IV, 2 days on/5 days off; (2) 100 mg/kg venetoclax orally, daily; (3) both drugs at the indicated doses; or (4) corresponding vehicles: 10% 0.1 N HCl and 90% saline for copanlisib (modified from Liu et al14 ) and 60% Phosal 50PG, 30% PEG400, 10% ethanol for venetoclax.15 We used previously reported doses of copanlisib14 and venetoclax15 that were judged to be equivalent to those administered in human clinical trials.16 After 21 days, all treatments were stopped, and the mice were observed for changes in total-body bioluminescence and survival. Disease burden was quantified using bioluminescence imaging as previously described,13 and data are presented as mean plus or minus standard error of the mean (SEM) with statistical significance determined by 1-sided t test. Differences in survival between the treatment groups were assessed with the log-rank test.
Results
Activity of multiple BCR/PI3K inhibitors in genetically and functionally defined DLBCL cell lines
We used a panel of 10 DLBCL cell lines that capture the previously characterized distinctions of BCR-dependent vs -independent and GCB vs ABC subtypes (Figure 1A).2 A subset of these cell lines exhibited hallmark genetic features of the recently described clusters 3 and 5 DLBCLs9 (Figure 1A). These include: (1) alterations that modulate BCR/PI3K signaling (inactivating mutations/deletions of PTEN and/or mutations of GNA13 or HVCN1) and BCL2 translocations (DHL4, DHL6, LY1 [BCR-dependent], K422 [BCR-independent], cluster 3) and (2) MyD88L265PCD79B mutations and arm-level 18q copy gains that encompass the BCL2 locus (HBL1, TMD8 [BCR-dependent], cluster 5) (Figure 1A). The DLBCL panel also includes additional BCR-independent GCB lines with BCL2 translocations (TOLEDO, LY19), a BCR-dependent GCB line with no BCL-2 expression (LY7) and a BCR-independent ABC line (DHL2) without genetic alterations of BCL2 (Figure 1A).
![Figure 1. Genomic characterization of DLBCL cell line models and prioritization of BCR/PI3K inhibitors. (A) Alterations in MYD88, CD79B, PTEN (mutation or copy loss), GNA13, HVCN1, and BCL-2 (translocation [structural variant (SV)] or copy-number gain) in a panel of 10 DLBCL cell lines. (B) Cellular proliferation after 72-hour exposure to specific inhibitors of BCR/PI3K signaling (as indicated in the figure). EC50 values in a colorimetric scale: very sensitive (<0.05 μM) in red, sensitive (=1 μM) in white, to resistant (>20 μM) in black. Results were averaged from 4 biological replicates.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/1/10.1182_blood-2018-08-872465/9/m_blood872465f1.png?Expires=1769084759&Signature=zIzx3IBR~pisjUkQ9~H8obEJzKiFDA5QSk3o2RM4HauKp~DXt-4jkUeigzB4Zw5NfrKEsOA92X9KAc4QFdDaMMAxLYWWkISVQLx2FvBxtxOfeJ1d2nWlsSQCO-UuqJAfu6uLiorpMUE0Ho8d1om0BMOVGBYgCDne8EKB7Vku~WsPDGly92CjZJjWBF6IlkwJPgtkvFFkzJmFpvd0jwbvimSmpw5jEs7qKVx81C4qoVCJjsPpmLilYTl3yFaP4Lf0A8upqnxJZtDj~rSE0GJ1Krpjcvu95NT7TcHk80woJwNrDNwQKpl6ZVpWUMwwyiLXcgSVlh4IoBfVRsqw0BmhrQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Genomic characterization of DLBCL cell line models and prioritization of BCR/PI3K inhibitors. (A) Alterations in MYD88, CD79B, PTEN (mutation or copy loss), GNA13, HVCN1, and BCL-2 (translocation [structural variant (SV)] or copy-number gain) in a panel of 10 DLBCL cell lines. (B) Cellular proliferation after 72-hour exposure to specific inhibitors of BCR/PI3K signaling (as indicated in the figure). EC50 values in a colorimetric scale: very sensitive (<0.05 μM) in red, sensitive (=1 μM) in white, to resistant (>20 μM) in black. Results were averaged from 4 biological replicates.
Genomic characterization of DLBCL cell line models and prioritization of BCR/PI3K inhibitors. (A) Alterations in MYD88, CD79B, PTEN (mutation or copy loss), GNA13, HVCN1, and BCL-2 (translocation [structural variant (SV)] or copy-number gain) in a panel of 10 DLBCL cell lines. (B) Cellular proliferation after 72-hour exposure to specific inhibitors of BCR/PI3K signaling (as indicated in the figure). EC50 values in a colorimetric scale: very sensitive (<0.05 μM) in red, sensitive (=1 μM) in white, to resistant (>20 μM) in black. Results were averaged from 4 biological replicates.
In previous studies, we found that BCR-dependent DLBCLs were sensitive to chemical inhibition or genetic depletion of SYK or chemical inhibition of PI3K.2 Others have described selective sensitivity of BCR-dependent ABC-DLBCLs to BTK blockade.3 In our earlier analyses of PI3K signaling in DLBCL, we used the pan-PI3K inhibitor and tool compound, LY294002. However, there are 4 isoforms of PI3K, 2 of which (α and δ) have defined roles in germinal B-cell biology17,18 (supplemental Figure 1A). All 4 PI3K isoforms are expressed in the 10 DLBCL cell lines (supplemental Figure 1B). Of note, PIK3CA (encoding PI3Kα) is a FOXO1 target that is upregulated following BCR/PI3K blockade and associated nuclear retention of FOXO1 (supplemental Figure 1C).2 For these reasons, we selected a range of PI3K inhibitors to profile in these DLBCLs, with the expectation that an optimal PI3K inhibitor might target both the PI3Kα and δ isoforms.
We first used a cell-proliferation assay to assess the sensitivity of the DLBCL cell lines to chemical inhibitors of SYK, PI3K, and BTK that are currently in clinical trials (Figure 1B). We confirmed selective activity (low micromolar EC50s) of the SYK inhibitor, entospletinib, in the majority of BCR-dependent GCB- or ABC-DLBCLs (Figure 1B). Additionally, we confirmed the previously described low nanomolar activity of the BTK inhibitor, ibrutinib, in BCR-dependent ABC-DLBCLs (Figure 1B).3 Of interest, the BCR-independent ABC cell line, DHL2, was insensitive to ibrutinib treatment.
We then analyzed the antiproliferative activity of isoform-specific, α/δ-predominant, dual β/δ and pan-PI3K inhibitors in the DLBCL panel (Figure 1B). The single isoform α-, β-, and δ-specific PI3K inhibitors and the dual PI3Kβ/δ inhibitor were largely ineffective in these assays (Figure 1B). In contrast, the PI3Kα/δ-predominant inhibitor, copanlisib, exhibited low nanomolar antiproliferative activity in BCR-dependent DLBCLs of both GCB and ABC subtypes and more modest activity in the BCR-independent cell lines (Figure 1B). The pan-PI3K inhibitor, pictilisib, closely mirrored the antiproliferative activity of copanlisib, although the EC50 concentrations were consistently higher than those of copanlisib (Figure 1B). These findings (lack of activity of α- or δ-isoform–selective inhibitors and potent activity of dual PI3Kα/δ blockade) led us to prioritize the PI3Kα/δ-predominant inhibitor, copanlisib, and the pan-PI3K inhibitor, pictilisib, in subsequent functional analyses.
Inhibitors of BCR/PI3K signaling differentially regulate expression of proapoptotic and antiapoptotic BCL-2 family members
We next assessed induction of apoptosis in the DLBCLs using doses of the respective BCR/PI3K inhibitors that captured the distinctions between sensitive and resistant cell lines in proliferation assays (Figure 2A top panel and key). The SYK inhibitor, entospletinib, did not induce >50% apoptosis in any of the examined DLBCL cell lines. The BTK inhibitor, ibrutinib, selectively induced >50% apoptosis in the BCR-dependent ABC-DLBCL cell lines (HBL1, TMD8), but not in the BCR-independent ABC-DLBCL (DHL2) or the GCB-DLBCLs. In contrast, the PI3Kα/δ-predominant inhibitor, copanlisib, induced >50% apoptosis in all of the BCR-dependent cell lines (Figure 2A). In the majority of these DLBCLs, copanlisib was more effective than pictilisib in inducing apoptosis (Figure 2A). Consistent with these findings, copanlisib was also the most active at inducing poly ADP ribose polymerase (PARP) cleavage in the BCR-dependent DLBCLs (Figure 2A bottom panel). Additionally, copanlisib was the most potent inhibitor in abolishing baseline and anti-immunoglobulin–induced phosphorylation of AKT (pAKT S473) (supplemental Figure 2).
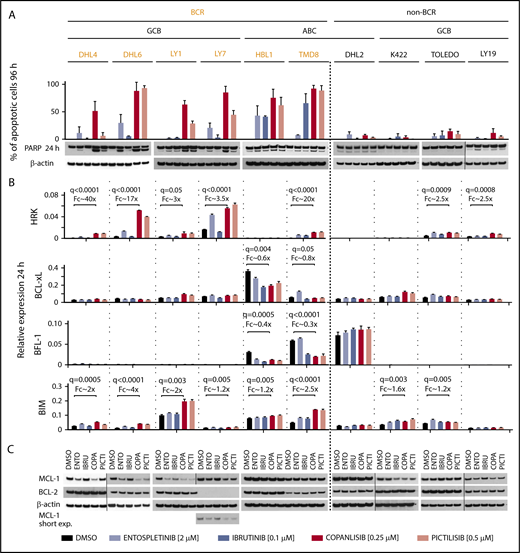
Inhibitors of BCR signaling differentially regulate expression of proapoptotic and antiapoptotic BCL2 family members. (A) Top panel, Induction of apoptosis after 96-hour exposure to DMSO, entospletinib (ENTO, 2 μM), ibrutinib (IBRU, 0.1 μM), copanlisib (COPA, 0.25 μM), or pictilisib (PICTI, 0.5 μM) shown as percentage of annexin V/PI-positive cells plus or minus SEM from at least 3 biological replicates. Bottom panel, Induction of PARP cleavage after 24-hour exposure to the drugs listed in the panel A description was assessed by immunoblotting, β-actin–loading control. One representative image of 3 independent replicates is shown. Vertical lines have been inserted to indicate repositioned gel lanes. (B) HRK, BCL-xL, BFL-1, or BIM transcript abundance after 24-hour exposure to DMSO, entospletinib (2 μM), ibrutinib (0.1 μM), copanlisib (0.25 μM), or pictilisib (0.5 μM) was determined with RT-qPCR relative to PPIA. Bars show the average of 3 technical replicates plus or minus SEM. The statistically significant differences between DMSO- and copanlisib-treated groups (q ≤ 0.05) are noted. (C) MCL-1 and BCL-2 protein abundance in DLBCL cells treated as in panel B was assessed by immunoblotting. β-actin–loading control. One representative image of 3 independent biological replicates is shown. Vertical lines have been inserted to indicate repositioned gel lanes.
Inhibitors of BCR signaling differentially regulate expression of proapoptotic and antiapoptotic BCL2 family members. (A) Top panel, Induction of apoptosis after 96-hour exposure to DMSO, entospletinib (ENTO, 2 μM), ibrutinib (IBRU, 0.1 μM), copanlisib (COPA, 0.25 μM), or pictilisib (PICTI, 0.5 μM) shown as percentage of annexin V/PI-positive cells plus or minus SEM from at least 3 biological replicates. Bottom panel, Induction of PARP cleavage after 24-hour exposure to the drugs listed in the panel A description was assessed by immunoblotting, β-actin–loading control. One representative image of 3 independent replicates is shown. Vertical lines have been inserted to indicate repositioned gel lanes. (B) HRK, BCL-xL, BFL-1, or BIM transcript abundance after 24-hour exposure to DMSO, entospletinib (2 μM), ibrutinib (0.1 μM), copanlisib (0.25 μM), or pictilisib (0.5 μM) was determined with RT-qPCR relative to PPIA. Bars show the average of 3 technical replicates plus or minus SEM. The statistically significant differences between DMSO- and copanlisib-treated groups (q ≤ 0.05) are noted. (C) MCL-1 and BCL-2 protein abundance in DLBCL cells treated as in panel B was assessed by immunoblotting. β-actin–loading control. One representative image of 3 independent biological replicates is shown. Vertical lines have been inserted to indicate repositioned gel lanes.
To characterize bases of apoptosis in BCR-dependent DLBCLs treated with proximal BCR/PI3K inhibitors, we first assessed the transcriptional modulation of BCL-2 family genes (Figure 2B). In BCR-dependent DLBCLs with low NF-κB activity (GCB tumors), there was highly significant induction of the proapoptotic sensitizer, HRK, following treatment with the PI3Kα/δ-predominant inhibitor, copanlisib. In BCR-dependent DLBCLs with high NF-κB activity (ABC tumors), the NF-κB target and antiapoptotic BCL-2 family member, BFL-1, was transcriptionally downregulated after copanlisib exposure (Figure 2B). In these DLBCLs, there was also significant transcriptional downregulation of BCL-xL, another NF-κB target and antiapoptotic partner of HRK, following copanlisib treatment (Figure 2B). Additionally, in all BCR-dependent DLBCLs, copanlisib increased the transcript abundance of the FOXO1 target and proapoptotic activator, BIM. In BCR-dependent GCB-DLBCLs, a potential basis for the increased efficacy of copanlisib, in comparison with entospletinib, was the more robust modulation of HRK and BIM (Figure 2B). In BCR-dependent ABC-DLBCLs, copanlisib was as effective as ibrutinib in decreasing BCL-xL and BFL-1 abundance and as or more effective in inducing BIM (Figure 2B). In the BCR-independent DLBCLs, there was less striking modulation of HRK and BIM and no changes in BCL-xL or BFL-1 transcript abundance following BCR/PI3K blockade (Figure 2B).
We also analyzed protein expression of another antiapoptotic BCL-2 family member, MCL-1, which is regulated posttranscriptionally by AKT19 (Figure 2C). The BCR-dependent DLBCL cell lines with low NF-κB activity (DHL4, DHL6, LY1, and LY7) exhibited MCL-1 downregulation following SYK or PI3Kα/δ inhibition (Figure 2C). An additional line (Karpas422) with genetic bases for increased PI3K/AKT activity (PTENdel, PTENmut, GNA13mut) had decreased MCL-1 levels following PI3K blockade. The MCL-1 data highlight an additional posttranscriptional mechanism of regulating the viability of BCR-dependent DLBCLs. In contrast to MCL-1, BCL-2 protein expression was not altered by BCR/PI3K inhibition (Figure 2C).
Copanlisib-induced apoptotic cell death is dependent on BCL-xL– and MCL-1–mediated mitochondrial priming
We next used an orthogonal approach, BH3 profiling, to define BCL-2 family member dependencies and correlate these data with sensitivity to copanlisib. With BH3 profiling, we analyzed the induction of MOMP by specific peptides derived from BH3 domains of proapoptotic BCL-2 family members11 (Figure 3A). The output of BH3 profiling is shown as the area under the curve (AUC) of cytochrome c release from mitochondria (indicative of MOMP) over the dose range of evaluated BH3 peptides (Figure 3B). For each DLBCL cell line, the AUC values (Figure 3C-D y-axes) were compared with the percentage of apoptosis induced by copanlisib (Figure 3C-D x-axes).
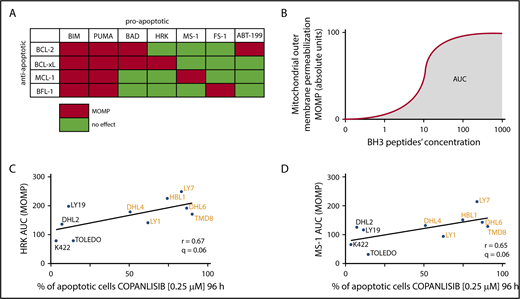
Copanlisib-induced apoptotic cell death is dependent on BCL-xL– and MCL-1–mediated mitochondrial priming. (A) Interaction pattern between proapoptotic BH3 peptides (or BH3 mimetic–ABT-199) and antiapoptotic BCL-2 family members. Red indicates high-affinity binding; green reflects undetectable binding. (B) Illustration of the determination of AUC as a function of cytochrome c release over the dose range of BH3 peptides. (C-D) Correlation between cytochrome c release (indicative of MOMP) induced by HRK (C) or MS-1 (D) peptides (y-axis) and induction of apoptosis after 96-hour exposure to copanlisib (x-axis). Each AUC is an average of 3 biological replicates. Note that BH3 profiling and copanlisib-induced apoptosis were assessed in separate experiments. One-sided P values from the correlation with all tested BH3 peptides were recalculated with the Benjamini-Hochberg procedure and q < 0.1 was considered statistically significant.
Copanlisib-induced apoptotic cell death is dependent on BCL-xL– and MCL-1–mediated mitochondrial priming. (A) Interaction pattern between proapoptotic BH3 peptides (or BH3 mimetic–ABT-199) and antiapoptotic BCL-2 family members. Red indicates high-affinity binding; green reflects undetectable binding. (B) Illustration of the determination of AUC as a function of cytochrome c release over the dose range of BH3 peptides. (C-D) Correlation between cytochrome c release (indicative of MOMP) induced by HRK (C) or MS-1 (D) peptides (y-axis) and induction of apoptosis after 96-hour exposure to copanlisib (x-axis). Each AUC is an average of 3 biological replicates. Note that BH3 profiling and copanlisib-induced apoptosis were assessed in separate experiments. One-sided P values from the correlation with all tested BH3 peptides were recalculated with the Benjamini-Hochberg procedure and q < 0.1 was considered statistically significant.
Of interest, the MOMP induction by the HRK peptide strongly correlated with the cytotoxic activity of copanlisib in the DLBCL cell lines (Figure 3C). These data align with our finding that copanlisib increased HRK transcript abundance in DHL4, DHL6, LY1, and LY7 or decreased transcript levels of the HRK partner, BCL-xL, in HBL1 and TMD8 (Figure 2B). Additionally, there was a significant correlation between MOMP induced by the MS-1 peptide (which binds to MCL-1) and apoptosis induced by copanlisib (Figure 3D). In DHL4, DHL6, LY1, and LY7, this likely reflects the decreased abundance of MCL-1 protein following copanlisib treatment (Figure 2B) and an associated reduction in the amount of MCL-1 complexed with the proapoptotic BIM protein (supplemental Figure 3A). In TMD8 and HBL1, the MS-1 data reveal an additional functional dependence on MCL-1, as well as other antiapoptotic BCL-2 family members, in copanlisib-treated cells (Figure 3D). The MOMP induced by BIM, PUMA, BAD, or FS-1 peptides (Figure 3A) did not correlate with apoptosis induced by copanlisib (data not shown).
Copanlisib sensitizes DLBCL cells to venetoclax
In this series of DLBCL cell lines, PI3Kα/δ inhibition did not alter BCL-2 expression (Figure 2C). Given the genetic bases for BCL-2 dysregulation in a subset of these DLBCLs, we assessed the activity of the BCL-2 inhibitor, venetoclax, by measuring the induction of apoptosis in vitro (Figure 4A). DLBCL cell lines treated with single-agent venetoclax showed a range of responses. DLBCL cell lines with wild-type BCL2 (DHL2) or no BCL-2 expression (LY7) were resistant to venetoclax. A subset of DLBCL cell lines with BCL2 genetic alterations was sensitive to venetoclax treatment (LY1, TOLEDO, LY19). An additional group of DLBCL cell lines was partially or completely resistant to venetoclax despite having genetic alterations of BCL2 and documented BCL-2 expression (Figure 2C; supplemental Figure 3B).
![Figure 4. Copanlisib sensitizes DLBCL cells to venetoclax. (A) Induction of apoptosis shown as percentage of viable (annexin V/PI–negative) cells after 96-hour exposure to ABT-199 (0-250 nM). Results show average viability plus or minus SEM from at least 3 biological replicates. Cell lines are ranked from the most resistant to the most sensitive to venetoclax. (B) Correlation between cytochrome c release induced by BAD peptide (BCL2 plus BCL-xL) minus that induced by the HRK peptide (BCL-xL) (BCL-2 dependency; y-axis) with induction of apoptosis after 96-hour exposure to ABT-199 (250 nM; x-axis). (C) Induction of apoptosis (percentage of viable [annexinV/PI–negative] cells) after 96-hour exposure to the combination of MCL-1 inhibitor (S63845, 0-250 nM) and ABT-199 (0-250 nM). Results show average viability plus or minus SEM from 2 biological replicates. Synergy visualized as median CI. CI < 1 indicates synergy.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/1/10.1182_blood-2018-08-872465/9/m_blood872465f4.png?Expires=1769084759&Signature=1qzGADsQQhF1~9fmKMIPnTO2AWRatB3uKRRnvOS97y2VTx5Eb~1loG6iEa2b8SetEZ~cIT79hfNqBRvyQTbndEWcdKUepqB2y~JtN4lPoW5pLR5WSJKTAgN22bYNzg9Yei7ygKrANDBACe3yt66g0W04rN5qhwaidnfMDXYBDfXDNGA-mCqa8am6rXiMGIJt25pUBYl7Tl8kzR6cZUmnGmy2RQJUEE~BzCH~Idsys5esOAPMAoReLO6FcatTjmjBMW5W4wf5ydHuB~N2AW74wDqOY2dGVtpbJ6ZHElb0SPUc1SEf8rgcM9ZrSJ88MVyAAQCaNRdHdx7LPqd33BQejg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Copanlisib sensitizes DLBCL cells to venetoclax. (A) Induction of apoptosis shown as percentage of viable (annexin V/PI–negative) cells after 96-hour exposure to ABT-199 (0-250 nM). Results show average viability plus or minus SEM from at least 3 biological replicates. Cell lines are ranked from the most resistant to the most sensitive to venetoclax. (B) Correlation between cytochrome c release induced by BAD peptide (BCL2 plus BCL-xL) minus that induced by the HRK peptide (BCL-xL) (BCL-2 dependency; y-axis) with induction of apoptosis after 96-hour exposure to ABT-199 (250 nM; x-axis). (C) Induction of apoptosis (percentage of viable [annexinV/PI–negative] cells) after 96-hour exposure to the combination of MCL-1 inhibitor (S63845, 0-250 nM) and ABT-199 (0-250 nM). Results show average viability plus or minus SEM from 2 biological replicates. Synergy visualized as median CI. CI < 1 indicates synergy.
Copanlisib sensitizes DLBCL cells to venetoclax. (A) Induction of apoptosis shown as percentage of viable (annexin V/PI–negative) cells after 96-hour exposure to ABT-199 (0-250 nM). Results show average viability plus or minus SEM from at least 3 biological replicates. Cell lines are ranked from the most resistant to the most sensitive to venetoclax. (B) Correlation between cytochrome c release induced by BAD peptide (BCL2 plus BCL-xL) minus that induced by the HRK peptide (BCL-xL) (BCL-2 dependency; y-axis) with induction of apoptosis after 96-hour exposure to ABT-199 (250 nM; x-axis). (C) Induction of apoptosis (percentage of viable [annexinV/PI–negative] cells) after 96-hour exposure to the combination of MCL-1 inhibitor (S63845, 0-250 nM) and ABT-199 (0-250 nM). Results show average viability plus or minus SEM from 2 biological replicates. Synergy visualized as median CI. CI < 1 indicates synergy.
To further characterize the BCL-2 dependency of the DLBCL cell lines, we compared their sensitivity to venetoclax with MOMP induction by BH3 profiling (Figure 4B). In these analyses, we assessed MOMP induced by the BAD peptide, which targets BCL-2 and BCL-xL, minus MOMP induced by HRK, which selectively targets BCL-xL (Figure 3A; Figure 4B y-axis). We found that BCL-2 dependency, as reflected by BH3 profiling (MOMP-induced by BAD minus that induced by HRK), was highly predictive of response to venetoclax (Figure 4B).
We postulated that DLBCLs with BCL2 alterations and more limited sensitivity to single-agent venetoclax depended on additional antiapoptotic BCL-2 family members. In BCR-dependent DLBCLs with low baseline NF-κB (GCB), copanlisib treatment decreased MCL-1 protein abundance and the interaction of MCL-1 with proapoptotic BIM (Figure 2C; supplemental Figure 3A). These findings prompted us to assess the additional antiapoptotic role of MCL-1 in BCR-dependent DLBCL cell lines with low baseline NF-κB, BCL-2 structural variants (SVs), and more limited sensitivity to single-agent venetoclax (DHL4 and DHL6). Of interest, pharmacologic inhibition or genetic depletion of MCL-1 synergized with venetoclax treatment in these DLBCLs (Figure 4C; supplemental Figure 3C). Taken together, these data suggested that BCR-dependent DLBCLs with structural alterations of BCL2 might exhibit increased sensitivity to combined PI3Kα/δ and BCL-2 blockade.
Copanlisib and venetoclax are synergistic in in vitro DLBCL models
For these reasons, we assessed the cytotoxic activity of combined PI3Kα/δ and BCL-2 inhibition in the representative DLBCL cell line panel using copanlisib (0-250 nM) and venetoclax (0-250 nM) (Figure 5). The combination therapy was synergistic in DLBCL cell lines that are BCR-dependent and have genetic bases for BCL-2 dysregulation (DHL4, DHL6, LY1, HBL1, and TMD8) (Figure 5). Synergy was also observed in Karpas422, which has BCR-independent genetic mechanisms of activating the PI3K/AKT pathway and BCL2 translocation. In addition, there was single-agent activity of venetoclax in BCR-independent cell lines with BCL2 SVs (TOLEDO, LY19) (Figure 5). Conversely, venetoclax and copanlisib were not synergistic in DLBCL cell lines without genetic BCL2 alterations (LY7, DHL2). This observation is concordant with the LY7 and DHL2 cell lines having the lowest BCL-2 dependency as measured by BH3 profiling (Figure 4B). Taken together, these in vitro findings set the stage for assessing the combination of copanlisib and venetoclax in BCR-dependent DLBCL models in vivo.
![Figure 5. Combination of copanlisib and venetoclax is synergistic in in vitro DLBCL models. Induction of apoptosis (percentage of viable [annexin V/PI–negative] cells) after 96-hour exposure to the combination of copanlisib (0-250 nM) and ABT-199 (0-250 nM). Results show average viability plus or minus SEM from 2 biological replicates. Synergy visualized as median CI. CI < 1 indicates synergy. (A-C,E-F,H) Synergy; (I-J) single agent venetoclax activity; (D,G) antagonism.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/1/10.1182_blood-2018-08-872465/9/m_blood872465f5.png?Expires=1769084759&Signature=rSmxP4L0-c7zW3aCvyQDUGKf9RtbWDWSdaBWWfk5PcOH4UAU-yW1q6-RX7~VsmH3Ba4KbJ2dGIizyXbWGzSlnhE24ckNYA~PHjDHaEmdWkFgq2l~IEWzSp7yEpkYIt2WRlS3e9GcFbk1jAMEtu3lgR7VVO5JfBOLVCtNcpMGI8GxDUQ4M2eM0J6UO4SMVPqst-jwon-sLmZJVKQ8V6ol7AEdhpZxRmz5zqKiP90cDGQTSvUL~FazmcAuUBhjS~Qf5GMx2DnVrjSIx0nIzGbtAYm1vpGnSNr19aYPswl2liRuFQ~KCRfc4oxuC8PljPG8RKgR4aJvR-iO6dQiCK9Smg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Combination of copanlisib and venetoclax is synergistic in in vitro DLBCL models. Induction of apoptosis (percentage of viable [annexin V/PI–negative] cells) after 96-hour exposure to the combination of copanlisib (0-250 nM) and ABT-199 (0-250 nM). Results show average viability plus or minus SEM from 2 biological replicates. Synergy visualized as median CI. CI < 1 indicates synergy. (A-C,E-F,H) Synergy; (I-J) single agent venetoclax activity; (D,G) antagonism.
Combination of copanlisib and venetoclax is synergistic in in vitro DLBCL models. Induction of apoptosis (percentage of viable [annexin V/PI–negative] cells) after 96-hour exposure to the combination of copanlisib (0-250 nM) and ABT-199 (0-250 nM). Results show average viability plus or minus SEM from 2 biological replicates. Synergy visualized as median CI. CI < 1 indicates synergy. (A-C,E-F,H) Synergy; (I-J) single agent venetoclax activity; (D,G) antagonism.
Combination of copanlisib and venetoclax is synergistic in an in vivo DLBCL model
We next assessed copanlisib and venetoclax activity in a xenograft model using a DLBCL cell line with PTENdel, GNA13mut, HVCN1mut, and BCL2 translocation (LY1; Figure 1A). Animals with established disease documented by imaging (supplemental Figure 4) were divided into 4 cohorts of 7 to 9 animals each. Thereafter, the animals were treated with vehicle alone; single-agent copanlisib on days 1, 2, 8, 9, 15, 16 (IV, 12 mg/kg); single-agent venetoclax on days 1 to 21 (orally, 100 mg/kg); or the combination of copanlisib and venetoclax. All treatments were stopped on day 21.
In this model, single-agent copanlisib did not delay tumor growth (Figure 6A-B) or improve survival (Figure 6C). In contrast, single-agent venetoclax delayed tumor growth (P < .0001) (Figure 6A-B) and improved median survival (27 vs 51 days; P = .0002) (Figure 6C). Most notably, we found that the combination of copanlisib and venetoclax delayed tumor growth significantly longer than single-agent venetoclax (P < .0001) (Figure 6A-B). Additionally, the combination of copanlisib and venetoclax extended the median survival of treated mice significantly longer than single-agent venetoclax (51 days vs not reached on day 139; P = .0013) (Figure 6C). At the end point of this experiment (139 days), 2 of 8 animals (25%) treated with the combination of copanlisib and venetoclax died of progressive disease and 6 of 8 mice (75%) remain disease-free (Figure 6A,C).
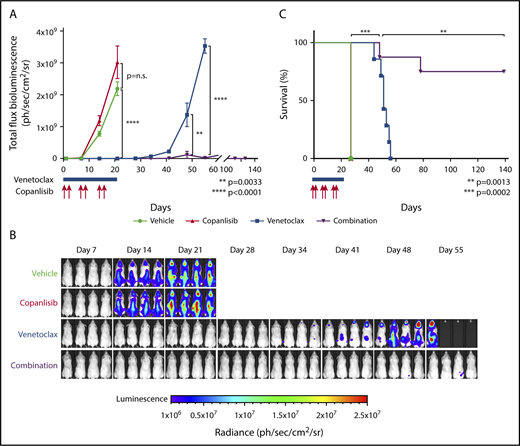
Combination of copanlisib and venetoclax is highly synergistic in a DLBCL xenograft model. (A) Bioluminescence of NSG mice xenotransplanted with luciferized mCherry+ LY1 DLBCL cells treated with vehicle (black; n = 8), copanlisib alone (red, 12 mg/kg IV, 2 days on/5 days off on days 1, 2, 8, 9, 15, 16; n = 9), ABT-199 alone (blue, 100 mg/kg orally, daily on days 1-21; n = 7), or combination copanlisib and ABT-199 (purple; n = 8). Treatment was stopped at day 21. Asterisks indicate P values <.05 in a 1-sided Student t test. Error bars represent SEM. (B) Representative prone bioluminescence images from panel A. (C) Kaplan-Meier survival plot of the treated cohorts. The P values were obtained by log-rank test.
Combination of copanlisib and venetoclax is highly synergistic in a DLBCL xenograft model. (A) Bioluminescence of NSG mice xenotransplanted with luciferized mCherry+ LY1 DLBCL cells treated with vehicle (black; n = 8), copanlisib alone (red, 12 mg/kg IV, 2 days on/5 days off on days 1, 2, 8, 9, 15, 16; n = 9), ABT-199 alone (blue, 100 mg/kg orally, daily on days 1-21; n = 7), or combination copanlisib and ABT-199 (purple; n = 8). Treatment was stopped at day 21. Asterisks indicate P values <.05 in a 1-sided Student t test. Error bars represent SEM. (B) Representative prone bioluminescence images from panel A. (C) Kaplan-Meier survival plot of the treated cohorts. The P values were obtained by log-rank test.
Discussion
We have characterized BCR/PI3K-dependent signaling pathways and associated therapeutic vulnerabilities in genetically defined DLBCL models. Dual inhibition of PI3Kα/δ was active in BCR-dependent DLBCLs with low and high baseline NF-κB activity (GCB and ABC, respectively). In these DLBCLs with characteristic genetic alterations of BCR-PI3K signaling pathway components, PI3Kα/δ blockade modulated distinct apoptotic pathways. In BCR-dependent NF-κB–low (GCB) DLBCLs, which often have inactivating mutations of PTEN and/or GNA13 (as in cluster 3), PI3Kα/δ blockade induced the proapoptotic HRK and BIM peptides and decreased the abundance of the antiapoptotic MCL-1 protein (Figure 7A). In BCR-dependent NF-κB–high (ABC) DLBCLs with frequent MYD88L265P and CD79B mutations (as in cluster 5), PI3Kα/δ inhibition decreased the abundance of NF-κB targets including the antiapoptotic BCL-xL and BFL-1 family members (Figure 7B). In DLBCLs with genetic alterations of BCL2 (translocations, cluster 3; copy gain, cluster 5) and functional BCL-2 dependency, BCL-2 blockade released proapoptotic BH3 peptides from the antiapoptotic BCL-2 protein (Figure 7). These findings prompted our evaluation of combined PI3Kα/δ and BCL-2 inhibition, with copanlisib and venetoclax, which was synergistic in multiple BCR-dependent DLBCL models.

Consequences of PI3Kα/δ and BCL-2 inhibition in BCR-dependent DLBCLs with functional BCL-2 dependency. In blue: genetically modulated components of BCR/PI3K/TLR pathway and BCL-2 family. In red: functional consequences of PI3Kα/δ inhibition. In green: antiapoptotic BCL-2 family members. In purple: proapoptotic BCL-2 family members. In BCR-dependent NF-κB–low (GCB) DLBCLs, which often have inactivating mutations of PTEN and/or GNA13 (as in cluster 3), PI3Kα/δ blockade induced the proapoptotic HRK and BIM peptides and decreased the abundance of the antiapoptotic MCL-1 protein (A). In BCR-dependent NF-κB–high (ABC) DLBCLs with frequent MYD88L265P and CD79B mutations (as in cluster 5), PI3Kα/δ inhibition decreased the abundance of NF-κB targets including the antiapoptotic BCL-xL and BFL-1 family members (B). In DLBCLs with genetic alterations of BCL2 (translocations, cluster 3; copy gain, cluster 5) and functional BCL-2 dependency, BCL-2 blockade released proapoptotic BH3 peptides from the antiapoptotic BCL-2 protein.
Consequences of PI3Kα/δ and BCL-2 inhibition in BCR-dependent DLBCLs with functional BCL-2 dependency. In blue: genetically modulated components of BCR/PI3K/TLR pathway and BCL-2 family. In red: functional consequences of PI3Kα/δ inhibition. In green: antiapoptotic BCL-2 family members. In purple: proapoptotic BCL-2 family members. In BCR-dependent NF-κB–low (GCB) DLBCLs, which often have inactivating mutations of PTEN and/or GNA13 (as in cluster 3), PI3Kα/δ blockade induced the proapoptotic HRK and BIM peptides and decreased the abundance of the antiapoptotic MCL-1 protein (A). In BCR-dependent NF-κB–high (ABC) DLBCLs with frequent MYD88L265P and CD79B mutations (as in cluster 5), PI3Kα/δ inhibition decreased the abundance of NF-κB targets including the antiapoptotic BCL-xL and BFL-1 family members (B). In DLBCLs with genetic alterations of BCL2 (translocations, cluster 3; copy gain, cluster 5) and functional BCL-2 dependency, BCL-2 blockade released proapoptotic BH3 peptides from the antiapoptotic BCL-2 protein.
The current preclinical studies highlight the importance of PI3K as a therapeutic target in DLBCL and emphasize the need to block both the PI3Kα and δ isoforms. These findings build on earlier work by our group2 and others who identified an ABC-DLBCL subset that depended on PI3K–NF-κB cross talk20 and a GCB-DLBCL subset with PTEN loss and sensitivity to pan-PI3K blockade.21
In our studies, BCR-dependent DLBCLs with low and high baseline NF-κB (GCB and ABC, respectively) were largely resistant to single-agent PI3Kα or PI3Kδ inhibition but were sensitive to the PI3Kα/δ-predominant inhibitor, copanlisib (Figure 1B). These findings align with previous studies in which PI3Kα was induced by proximal BCR/PI3K inhibition (supplemental Figure 1C)2 and PI3Kα was activated by PI3Kδ blockade22 in specific DLBCL subsets. Additionally, isoform-specific PI3Kδ inhibition was largely inactive in DLBCL in early clinical trials.23,24 Moreover, the combined application of isoform-specific PI3Kα and PI3Kδ inhibitors was more effective than either single agent in in vitro analyses of select ABC-DLBCL cell lines.25 In these studies, the PI3Kα/δ-predominant inhibitor, copanlisib, mimicked the increased efficacy of combined isoform-specific PI3Kα and PI3Kδ inhibitors in certain ABC-DLBCLs.25 These data informed the development of an ongoing phase 2 clinical trial of copanlisib in unselected patients with relapsed/refractory DLBCL.26 In this single-agent study, 19% of patients had an objective response, including 7.5% complete responses; the overall response rate was 31.6% in patients with ABC-DLBCL and 13.3% in those with GCB-DLBCL.26
Although previous studies highlight the role of PI3Kβ in PTEN-deficient solid tumor models,27,28 we found that PI3Kβ or PI3Kβ/δ inhibition was largely inactive in the examined DLBCLs (Figure 1B). However, copanlisib exhibits more modest PI3Kβ blockade in addition to predominant PI3Kα/δ inhibition.14 It is possible that the additional PI3Kβ blockade contributes to the efficacy of copanlisib in DLBCLs with specific alterations of PI3K pathway members.
Our recent comprehensive genomic analysis of DLBCLs identifies additional genetic heterogeneity in transcriptionally defined ABC and GCB subtypes, including structural alterations of BCL2 in select DLBCL subsets.9 These data suggest that it may be advantageous to block PI3Kα/δ-dependent survival pathways as well as structurally dysregulated BCL2 in genetically defined DLBCL groups (Figure 7). These findings are of particular importance because only 18% of unselected patients with relapsed/refractory DLBCL responded to single-agent venetoclax in an initial clinical trial.29
The concept of intrinsic and acquired resistance to BCL-2 antagonists mediated by other members of the BCL-2 family such as MCL-1, BCL-xL, or BFL-1 has been proposed by us30 and others.31,32 In our current study of genetically and functionally characterized DLBCL cell lines, we used BH3 profiling to correlate BCL-2 dependency with sensitivity to venetoclax. We identified DLBCL cell lines with wild-type BCL2 or BCL-2 loss and venetoclax resistance, lines with BCL2 structural variants and sensitivity to venetoclax, and additional lines with BCL2 alterations and more limited sensitivity to single-agent venetoclax. In BCR-dependent DLBCLs with genetic alterations of BCL2, PI3Kα/δ inhibition modulated additional BCL-2 family members and synergized with BCL-2 blockade in vitro and in vivo (Figures 5-7). These findings are of additional interest because venetoclax treatment was reported to decrease PTEN expression and increase AKT activation in select DLBCL cell lines.33
Taken together, our results provide in vitro and in vivo preclinical evidence for the rational combination of PI3Kα/δ and BCL-2 blockade in BCR-dependent DLBCLs and set the stage for clinical evaluation of copanlisib and venetoclax in patients with genetically defined tumors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Polish Ministry of Science and Higher Education within the “Mobility Plus” program (1261/MOB/IV/2015/0; K.B.), the Leukemia & Lymphoma Society and Rising Tide Foundation Patient-focused Research Grant for Blood Cancer (M.A.S.), a Dana-Farber Cancer Institute Medical Oncology Award (M.A.S.), Bayer research funding (M.A.S.), and National Institutes of Health, National Cancer Institute grant RO1 CA205967 (A.L.).
Authorship
Contribution: K.B., B.C., and M.A.S. designed the study; K.B., K.W., J.A.R., L.C., M.V.-O., E.M., J.S., and B.C. performed the experiments; K.B., A.L., L.N.L., B.C., and M.A.S. analyzed the data; K.B., L.N.L., B.C., and M.A.S. wrote the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: M.A.S. has received research funding from Bayer, Bristol-Myers Squibb, and Merck; served as a scientific advisor to Bristol-Myers Squibb; and received honoraria from AstraZeneca. A.L. reports sponsored research and consulting from AbbVie, Novartis, and Astra Zeneca and is a founder and equity holder in Flash Therapeutics and Vivid Bioscience. The remaining authors declare no competing financial interests.
The current affiliation for L.C. is Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA.
The current affiliation for J.S. is Department of Immunology, Medical University of Warsaw, Warsaw, Poland.
The current affiliation for B.C. is Department of Hematology and Oncology, University Medical Center, Göttingen, Germany.
Correspondence: Margaret A. Shipp, Department of Medical Oncology, Dana-Farber Cancer Institute, 450 Brookline Ave, Mayer 513, Boston, MA 02215; e-mail: margaret_shipp@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal